

Abstract
Rationale:
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infancy that can cause permanent brain damage. Consequently, optimal management is extremely important. Current pharmacologic and surgical treatment were available that included diazoxide and octreotides.
Patient concerns:
A 4 month old Saudi male patient diagnosed at our hospital as CHI, treated with near total pancreatectomy and octreotide therapy of 30 mcg/kg/day presented with severe abdominal distension, vomiting and bloody diarrhea.
Diagnoses:
The patient was diagnosed as necrotising enterocolitis (NEC) associated with Rota virus infection which played together with octeriotides as risk factors for NEC.
Interventions:
Radiological investigations and multidisciplinary team management with endocrinologist, neonatologist, pediatric surgeon, and gastroenterologist.
Outcomes:
Resolution of NEC with conservative medical management and was discharged after 1 month of hospital stay with follow up with all concerned sub specialties.
Lessons:
NEC can develop in patients treated with octreotides especially when associated with another risk factor such as rotavirus infection.
Keywords: ABCC8, hyperinsulinism, hypoglycemia, necrotizing enterocolitis, octreotide, rotavirus, Usher syndrome
1. Introduction
Congenital hyperinsulinism (CHI) is a rare disease characterized by persistent hypoglycemia accompanied by inappropriately elevated circulating insulin concentration.[1] The worldwide estimated incidence is 1 in 50,000 live births. In Saudi Arabia, the incidence is 1 in 2675 live births, with 51% of them having parents who were first or second cousins.[2] CHI is caused by mutations in the gene responsible for the regulation of the release of insulin, which is produced by the beta cells of pancreas. Mutations in at least 9 genes have been found to cause CHI with ABCC8 gene mutations being the most common.[3] Current pharmacologic treatments are available that include diazoxide. However, in cases with diazoxide unresponsiveness, alternative medical and surgical approaches may be required to decrease the risk of hypoglycemia.[4] Octreotide, a somatostatin analog, often has a role in the management of those children. It acts on the somatostatin receptors SSTR2 and SSTR5 with inhibition of secretions of a variety of hormones, including insulin.[5]
Octreotide has been used for nearly 20 years for both short and long-term control of diazoxide-unresponsive CHI patients.[5] Octreotides have a variety of adverse effects, including a dose-dependent reduction in splanchnic blood flow[4] and an increase in the risk of bacterial and rarely viral infections. All the aforementioned drawbacks of Octreotides contribute to the pathogenesis of necrotizing enterocolitis (NEC). NEC is a serious condition affecting mostly preterm infants with high mortality rates owing to the fact that it results in both inflammation and bacterial invasion of the bowel wall.[6,7]
We report a Saudi male infant with homozygous deletion of the ABCC8 gene of CHI who had diazoxide-unresponsiveness and was treated with Octreotides preceded by a nearly total pancreatectomy, which precipitated him to NEC in association with ROTA virus infection.
2. Patient's presentation
Our patient is a Saudi term male infant who was a product of consanguinity marriage of first-degree cousins. His mother had an uneventful pregnancy. He was born by spontaneous vaginal delivery and was large for his gestational age. His APGAR score of 6 (2 points for the heart rate 123/min, 1 point for his irregular breathing, 1 point for his extremities blue color, 1 point for his weak tone and 1 point for his reflexes) and 8 (2 points for the heart rate 143/min, 1 point for his irregular breathing, 2 points for his pink color, 2 points for his activity and 1 point for the reflexes) at 1 and 5 min. Within few minutes, he started to develop respiratory distress and was admitted to the neonatal intensive care unit (NICU) with initial capillary blood gas of pH: 7.44, PCO2: 42, PO2:44, and HCO3: 28.
While being treated in NICU with noninvasive ventilation, he started to suffer from frequent attacks of supraventricular tachycardia (SVT), which was very difficult to treat with failure of adenosine infusion of 250 μg/kg and was finally controlled by DC cardioversion and Amidarone infusion of 5 mg/kg followed by maintenance infusion. Moreover, he was then shifted to Esmolol 100 mcq/min and Propranolol 1 mg/kg/dose/6 hours. Echocardiogram revealed septal hypertrophy and pulmonary stenosis.
After 2 hours of birth, not only did the baby had low blood glucose level that was down to 0.7 mmol/L, but he also suffered from frequent hypoglycemic episodes that were characterized by persistency and difficulty to control despite Dextrose 10% boluses and high glucose infusion rate reaching up to 25 mg/kg/min which raised the attention for the possibility of hyperinsulinism. Even though Diazoxide has been given with a maximum dose of 15 mg/kg/day, no response has been documented. As an aftermath, the patient started on Octreotide 20 μg/kg/day. Professional help by an Endocrinologist was sought who advised to send for serum critical sample for insulin, C-peptide, nonesterified free fatty acids, and adrenocorticotropic hormone (ACTH). It came out with very high insulin level of 2280.0 pmol/L (17.8–173 pmol/L) and very high C peptide of 7.19 pmol/L (0.37–1.47 pmol/L), while nonesterified free fatty acids and ACTH were normal. Not to mention the fact that the urine was negative for ketone bodies. After a gene study for CHI has been sent, homozygous deletion of ABCC8 exons 1–22 was revealed documenting the diagnosis of CHI.
At the age of 1 month after failure of medical therapy to control his hypoglycemic attacks, near total pancreatectomy with resection of up to 95% of his pancreas was done.
Frequent attacks of hypoglycemia have been documented after the operation, which were controlled by octreotide dose of 30 μg/kg/day.
At the age of 4 months, the patient was readmitted with abdominal distention, vomiting, and bloody diarrhea and was diagnosed as acute hemorrhagic gastroenteritis complicated with NEC. Not only did a positive abdominal X-ray uncover a suggestion of NEC (Fig. 1) but also isolated Rota virus was identified in a stool sample. Medical management was initiated with meropenem, metronidazole, and targocid with nothing per oral for 2 weeks. Total parenteral nutrition (TPN) was started and no surgical intervention has been required. He was discharged after 1 month with good condition, but delayed developmental milestones have been observed with neither being able to focus his eyes nor to support his head or roll over supine to prone position. He was not able to sit at the age of 7 months, nor able to stand or say any word at the age of 10 months.
Figure 1.
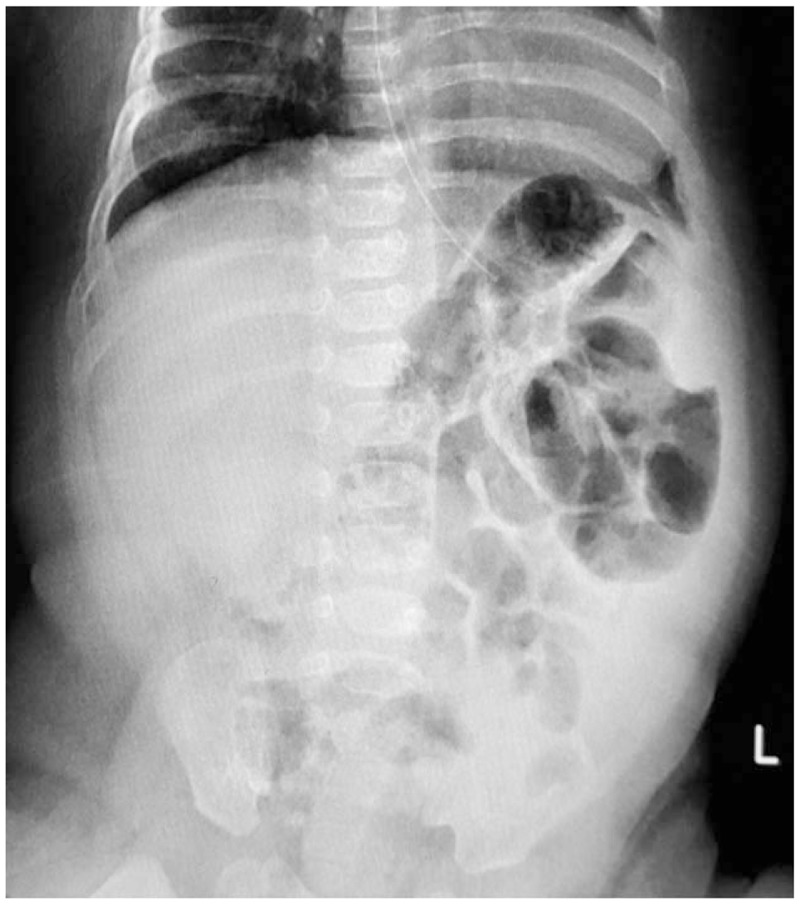
Abdominal X-ray, supine position demonstrating thick intestinal wall.
Auditory assessment revealed bilateral sensorineural hearing loss. On the contrary, ophthalmology examination was normal.
Magnetic resonant imaging was done and showed cystic hygroma with thin corpus callosum (Fig. 2).
Figure 2.
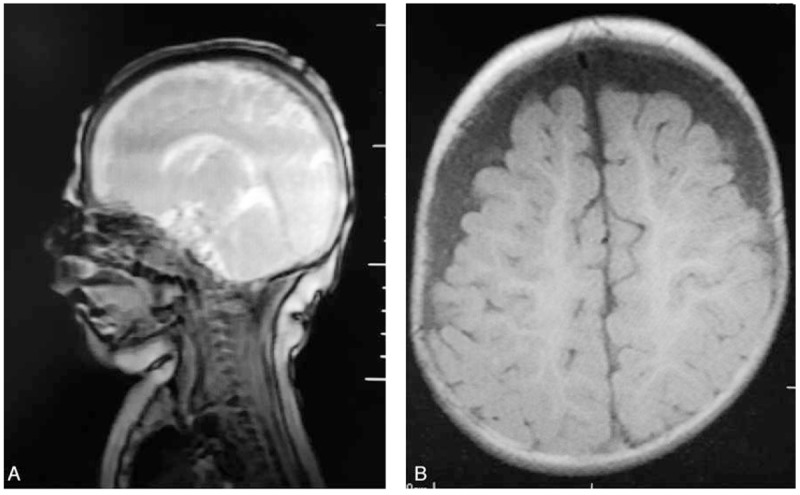
(A) Sagittal scan of the brain showing thin corpus collusum. (B) Coronal scan of the brain showing bilateral hygroma.
Our patient is currently 10 months old with multidisciplinary care and follows-up by pediatric endocrinologist, neurologist, gastroenterologist, and clinical nutritionist.
3. Discussion
CHI (MIM; 256450) is a heterogeneous disease with hyperinsulinemic hypoglycemia. Most frequently, the disorder is caused by a mutation in the ABCC8 gene (600508),[8] which encode the SUR1 subunit of the pancreatic beta cell potassium channel, on chromosome 11p15.[2] Other known etiologies include inactivating mutation of KCNJ11, HNF4A, HNF1A, HADH, and UCP2-activating mutations of GLUD1, GCK, and SLC16A1.[5]
Severe hypoglycemia in infancy can cause permanent brain damage; therefore, optimal management is extremely important[5] including Diazoxide or octrotide as an alternative, for those unresponsive to diazoxide.
Octreotide is a somatostatin analogue that was used for the first time in the treatment of CHI in 1986. Since then, it is commonly used in the management of CHI. Despite the fact that the medication has a wide variety of potential adverse reactions, octreotide is generally well tolerated. It has been extensively demonstrated that octreotide reduces the splanchnic blood flow in dose-dependent manner; as a result, not only does it affect the entire gastrointestinal tract,[9] but it also contributes to the development of NEC which seems to be not dose dependent as NEC wasn’t reported in a cohort 28.
Patients with CHI receiving high dose of octreotide up to 30 μg/kg,[10] on the contrary, NEC had developed in patients on relatively lower doses of 8 μg/kg/day in coexistence of other risk factors like total anomalous pulmonary venous connection, coarctation of the aorta , others.[4,9,11] It is likely that NEC in patients receiving octreotides needs an association needs coexisting other risk factors. For instance, our patient developed NEC in association with Rota virus as an additional risk factor. Although several outbreaks of NEC associated with Rota virus have been described previously in neonates in NICU,[12] no cases in term infants have been reported.
We believe that our patient is the first case reported in term infant with CHI on Octreotide therapy to develop NEC in association with Rota virus.
We also suspect that our patient suffers Usher-CHI syndrome owing to the fact that CHI is associated with symptoms of Usher syndrome, that is, hearing loss and childhood-onset retinitis pigmentosa.[8] This association is caused by a homozygous contiguous deletion, including USH1C (605242), ABCC8 (600509), and KCNJ11 (600937) on chromosome 11p15-p14 (MIM: 606528).[13]
Acknowledgment
The authors acknowledge Dr Ahmed A. Azab, Consultant Neonatologist, Alhada Military Hospital, for his great help in treatment of the patient.
Footnotes
Abbreviations: ABCC8 genes = ATP-binding cassette subfamily C member 8, ACTH = adrenocorticotropic hormone, CHI = congenital hyperinsulinism, NEC = necrotizing enterocolitis, PHHI = persistent hyperinsulinemic hypoglycemia of infancy, SSTR2 and SSTR5 = somatostatin receptor 2-5, SUR1 = the sulphonylurea receptors.
Authorship: AAA diagnosed the patient, did investigations, followed up him. AAA, AAB, NMK, GA reviewed literature, drafted the manuscript, and reviewed the manuscript final publication.
Funding/support: This research did not receive any grant from any funding agency.
The study was approved by Alhada Armed Forces Hospital Research and Ethical Committee. Written informed consent was obtained from the patient's father for contribution of his child in the study.
Personal information was not mentioned in a way that can lead to identification of the patient or his family.
All data generated or analyzed during this study are included in this published article.
The authors declare no conflicts of interest.
References
- [1].Glaser B, Phillip M, Carmi R, et al. Persistent hyperinsulinemic hypoglycemia of infancy (“nesidioblastosis”): autosomal recessive inheritance in 7 pedigrees. Am J Med Genet 1990;37:511–5. [DOI] [PubMed] [Google Scholar]
- [2].Familial hyperinsulinemic hypoglycemia 1. Available at: https://www.omim.org/entry/256450. Accessed September 30, 2016. [Google Scholar]
- [3].Congenital hyperinsulinism. Available at: https://ghr.nlm.nih.gov/condition/congenital-hyperinsulinism. Accessed September 30, 2016. [Google Scholar]
- [4].Hawkes CP, Adzick NS, Palladino AA, et al. Late presentation of fulminant necrotizing enterocolitis in a child with hyperinsulinism on octreotide therapy. Horm Res Paediatr 2016;86:131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yorifuji T. Congenital hyperinsulinism: current status and future perspectives. Ann Pediatr Endocrinol Metab 2014;19:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Thompson AM, Bizzarro MJ. Necrotizing enterocolitis in newborns: pathogenesis, prevention and management. Drugs 2008;68:1227–38. [DOI] [PubMed] [Google Scholar]
- [7].Zvizdic Z, Heljic S, Popovic N, et al. Contributing factors for development of necrotizing enterocolitis in preterm infants in the neonatal intensive care unit. Mater Sociomed 2016;28:53–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Al Mutair AN, Brusgaard K, Bin-Abbas B, et al. Heterogeneity in phenotype of usher-congenital hyperinsulinism syndrome: hearing loss, retinitis pigmentosa, and hyperinsulinemic hypoglycemia ranging from severe to mild with conversion to diabetes. Diabetes Care 2013;36:557–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Laje P, Halaby L, Adzick NS, et al. Necrotizing enterocolitis in neonates receiving octreotide for the management of congenital hyperinsulinism. Pediatr Diabetes 2010;11:142–7. [DOI] [PubMed] [Google Scholar]
- [10].Demirbilek H, Shah P, Arya VB, et al. Long-term follow-up of children with congenital hyperinsulinism on octreotide therapy. J Clin Endocrinol Metab 2014;99:3660–7. [DOI] [PubMed] [Google Scholar]
- [11].Reck-Burneo CA, Parekh A, Velcek FT. Isoctreotide a risk factor in necrotizing enterocolitis? J Pediatr Surg 2008;43:1209–10. [DOI] [PubMed] [Google Scholar]
- [12].Sızmaz E, Satar M, Ozlü F, et al. The coincidence of necrotizing enterocolitis and rotavirus infections and potential associations with cytokines. Can J Infect Dis Med Microbiol 2012;23:e103–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Homozygous 11p15-p14 Deletion Syndrome. Available at: https://www.omim.org/entry/606528. Accessed September 30, 2016. [Google Scholar]