

Abstract
Rationale:
TAFRO syndrome is a systemic inflammatory disease characterized by a constellation of symptoms: Thrombocytopenia, Anasarca, MyeloFibrosis, Renal dysfunction, and Organomegaly. Progressive renal insufficiency is a predominant symptom; however, the mechanism of acute kidney injury (AKI) remains unclear, probably because severe thrombocytopenia prevents kidney biopsy. We report a rare case of TAFRO syndrome with histologically confirmed renal involvement.
Patients concerns:
A 70-year-old man developed fever, anasarca, AKI, thrombocytopenia, and hepatosplenomegaly.
Diagnoses:
Plasma vascular endothelial growth factor and serum interleukin-6 levels were significantly elevated. The diagnosis of TAFRO syndrome was made based on his clinical and laboratory findings. Kidney biopsy was performed for the evaluation of AKI and provided a diagnosis of membranoproliferative glomerulonephritis–like lesions due to endothelial injury. Glomerular capillary lumens were extremely narrowed or occluded by endothelial swelling, and marked widening of the subendothelial space by electron-lucent material resulted in mesangiolysis and a double-contoured glomerular basement membrane with no immune complex deposits.
Interventions and Outcomes:
The patient required temporary hemodialysis due to oliguric AKI, but steroid therapy rapidly improved renal function.
Lessons:
Typically, patients with progressive renal involvement in TAFRO syndrome rapidly develop oliguric or anuric AKI. This report suggests that the reduction of glomerular perfusion by glomerular endothelial injury might be a primary factor in the progressive AKI of TAFRO syndrome. Our case and the literature review indicate that steroid and/or biological therapies result in highly favorable renal outcomes in patients with progressive AKI in TAFRO syndrome.
Keywords: endothelial injury; kidney biopsy; membranoproliferative glomerulonephritis; thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly syndrome; thrombotic microangiopathy
1. Introduction
Thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly (TAFRO) syndrome was first described in 2010 as a systemic inflammatory disorder characterized by thrombocytopenia, anasarca, fever, reticulin myelofibrosis, and organomegaly.[1] In the current accumulated data, progressive renal insufficiency occurred frequently; hence, renal insufficiency is listed among the latest criteria.[2] However, there were only 2 cases of TAFRO syndrome in which kidney biopsy was done, and the pathogenesis of renal involvement is still unknown. Here we describe a histologically confirmed case of progressive renal involvement in TAFRO syndrome. Previously published cases of progressive acute kidney injury (AKI) requiring hemodialysis in TAFRO syndrome are briefly summarized.
2. Case report
A 70-year-old Japanese man with no relevant medical history presented with a 3-week history of cough, fever, 8-kg weight gain, anasarca, and renal insufficiency. On admission, his temperature was 37.2°C and blood pressure was 136/74 mm Hg. Physical examination revealed pitting edema and abdominal distention due to ascites. No peripheral lymph nodes were palpable, and skin lesions suggestive of POEMS syndrome were not observed. Neurologic examination was unremarkable.
Urinalysis revealed a protein level of 2+ and microscopic hematuria with no casts. Twenty-four-hour urine protein excretion was 0.33 g/day. His serum creatinine (Cre) level was elevated from the baseline value of 0.80 to 1.28 mg/dL (reference range, 0.50–0.80 mg/dL). Liver function test results were normal except for an alkaline phosphatase (ALP) level of 868 U/L (reference range, 102–330 U/L). He had a serum albumin level of 2.5 g/dL (reference range, 3.4–5.2 g/dL), calcium level of 8.0 mg/dL (reference range, 8.4–10.4 mg/dL), C-reactive protein (CRP) level of 9.85 mg/dL (reference range, < 0.3 mg/dL), and IgG level of 701 mg/dL (reference range, 861–1747 mg/dL). Complete blood count revealed thrombocytopenia with a platelet count of 60,000/μL, increased white blood cell count of 11,900/μL, and normal hemoglobin level. There were no signs of intravascular hemolysis including fragmented erythrocytes on peripheral blood smear, elevated lactate dehydrogenase (LDH), or lower haptoglobin levels. ADAMTS13 activity was slightly decreased and we did not detect ADAMTS13 inhibitors. Connective tissue disease screening including antiphospholipid antibodies revealed negative results and serum complement levels were within the normal range. Urine electrophoresis revealed lambda-type Bence Jones protein (BJP); however, there were no monoclonal bands in serum. Serum-free light chains assay showed kappa chains at 32.9 mg/L and lambda chains at 126.0 mg/L, with a normal ratio of 0.26. Human immunodeficiency virus and Epstein-Barr virus screenings were negative. The test for human herpes virus 8 was not done.
Computed tomography showed pleural effusion, ascites, and mild hepatosplenomegaly. Apparent lymphadenopathy was not detected. Upper and lower endoscopy revealed early esophageal cancer. Bone marrow examination showed a normocellular marrow with normal plasma cells (1.5%) and increased megakaryocytes (6–8 per high-power field). Myelofibrosis was not observed. Based on his clinical and laboratory findings, a diagnosis of TAFRO syndrome was made. Further workup revealed higher levels of plasma vascular endothelial growth factor (VEGF) of 126 pg/mL (reference range, ≤38.3 pg/mL) and serum interleukin-6 (IL-6) of 33 pg/mL (reference range, <8.0 pg/mL).
Kidney biopsy was performed with platelet transfusion for the evaluation of AKI on hospital day 14. Of the 55 glomeruli examined, 5 glomeruli were globally sclerosed. Glomeruli were diffusely lobulated with a double contour of the glomerular basement membrane (GBM) and mesangiolysis (Fig. 1A, B). Glomerular capillary lumens were narrowed or occluded by endothelial cell swelling. Neither crescents nor glomerular thrombosis was observed. The interstitium was edematous without significant inflammatory cell infiltration (Fig. 1A). The arteries and arterioles were unremarkable. Immunofluorescence studies were all negative including lambda chains. Electron microscopy showed glomerular endothelial swelling and electron-lucent widening of the subendothelial space (Fig. 1C, D). No electron-dense deposits were present. These findings suggest a diagnosis of membranoproliferative glomerulonephritis (MPGN)-like lesions due to endothelial injury.
Figure 1.
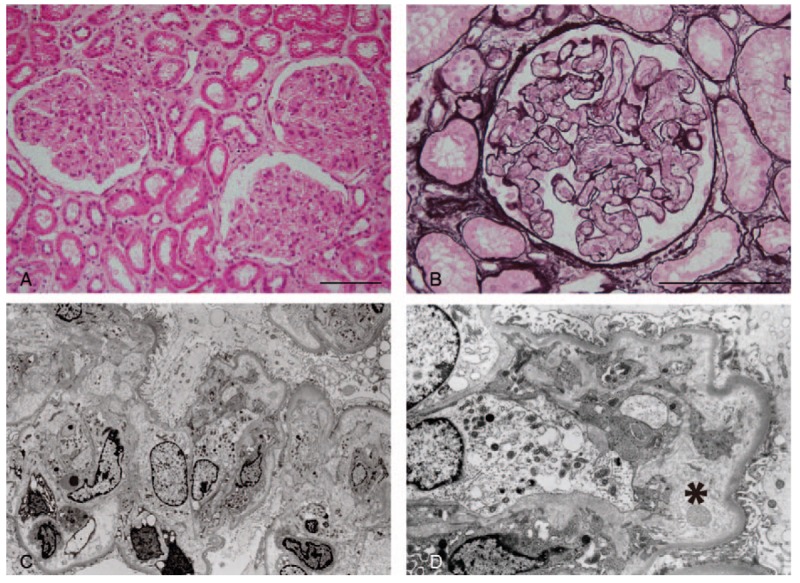
Renal biopsy findings were consistent with membranoproliferative glomerulonephritis (MPGN)-like lesions with glomerular endothelial injury. A, Glomerular tufts show lobular accentuation. Diffuse interstitial edema is noted. Tubules show normal architecture (hematoxylin and eosin stain, scale bar = 100 μm). B, Prominent swelling of glomerular endothelial cells occlude the capillary lumens with severe mesangiolysis. Glomerular thrombosis is not present (periodic acid-methenamine-silver stain, scale bar = 100 μm). C, Electron microscopy shows swelling of the endothelial cells and widening of the subendothelial space with no electron-dense deposits. (original magnification, ×2760). D, The subendothelial space is extremely widened by electron-lucent material (asterisk). A thin layer of newly formed glomerular basement membrane is observed. Podocyte foot processes are relatively preserved. (Original magnification, ×6900).
The patient developed oliguric prerenal failure several days after the kidney biopsy (Cre: 5.5 mg/dL; blood urea nitrogen: 133 mg/dL; fraction excretion of sodium: 0.2%). He did not respond to albumin infusions and required hemodialysis. Methylprednisolone pulse therapy (1 g daily for 3 days) followed by 55 mg/day of prednisolone was started on hospital day 19. Steroid therapy rapidly improved the systemic inflammation, and renal function was returned to normal within 1 month. However, his platelet count continued to decrease to a nidus of 8000/μL. He showed no response to repeated platelet transfusions; therefore, a thrombopoietin (TPO)-receptor agonist (eltrombopag) was administered on hospital day 57. Platelet levels increased gradually and normalized within 1 month. TPO-receptor agonist was discontinued at 2 months without recurrence. Six months after his first admission, surgery for esophageal cancer was performed successfully under treatment with 5 mg prednisolone. Steroid therapy was tapered and stopped at 1 year after surgery with no recurrent episodes.
This case report did not involve any human trials, and ethical review and approval were not necessary. Written informed consent was obtained from the patient for the publication of his clinical data.
3. Discussion
TAFRO syndrome was first reported as a form of atypical multicentric Castleman disease (MCD).[1] Most cases of TAFRO syndrome have been reported in Japan, and more recently also in Europe,[3,4] North America,[5,6] South America,[7] and Western Asia.[8] As case reports have accumulated, its distinct clinical characteristics have become more obvious and in 2015, updated diagnostic criteria were proposed.[2] According to these criteria, the presence of all 3 major and at least 2 of the 4 minor criteria is necessary for diagnosis of TAFRO syndrome. The major criteria are anasarca (pleural effusion, ascites, or general edema), thrombocytopenia (≤100,000/μL), and systemic inflammation (fever above 37.5°C and/or serum CRP ≥2 mg/dL). Minor criteria include MCD-like findings on lymph node biopsy, reticulin myelofibrosis and/or increased megakaryocytes in bone marrow, mild organomegaly, and progressive renal insufficiency. Additional features that support the diagnosis of TAFRO syndrome, which are uncommon in typical MCD, include acute clinical course, elevated level of ALP, absence of polyclonal hypergammopathy, normal LDH level, and small or unclear lymphadenopathy. Serum VEGF and IL-6 levels are frequently elevated. In the present case, a lymph node biopsy was not performed because of the lack of apparent lymphadenopathy. Therefore, our patient met the diagnostic criteria, satisfying all of the major and 3 of the 4 minor criteria (increased megakaryocytes in bone marrow, mild organomegaly, and progressive renal insufficiency), as well as almost all of the additional features. According to the severity classification for TAFRO syndrome,[2] the score of this patient was 11 out of 12, classified as grade 5 (the most severe form).
The differential diagnosis of TAFRO syndrome includes POEMS syndrome and malignancies including myeloma and solid tumor.[2] In the present case, BJP was observed in his urine; however, it appeared not to be associated with malignant plasma cell disorders, because the results of bone marrow and kidney biopsies ruled out the possibility of myeloma, amyloidosis, or light-chain deposition disease. All symptoms and signs of TAFRO syndrome completely resolved with steroid and TPO-receptor agonist therapies, although the esophageal cancer remained untreated. Therefore, the esophageal cancer also seemed unrelated to TAFRO syndrome in this patient. Although polyneuropathy is a mandatory criterion of POEMS syndrome,[9] our patient showed no neurological abnormalities. Moreover, he presented with none of the endocrinopathy, skin and bone lesions, and thrombocytosis that are important features of POEMS syndrome. Collectively, we determined that the diagnostic possibility of POEMS syndrome could be excluded.
Renal involvement is common and reported in about 55% of cases of TAFRO syndrome.[2] The mechanism of progressive AKI in TAFRO syndrome remains unclear, probably because severe thrombocytopenia prevents kidney biopsy. Only 1 case underwent kidney biopsy after the recovery of renal function (3 months after the episode of AKI)[10]; however, histopathological findings in the acute phase of renal involvement in TAFRO syndrome are unknown. We were able to perform kidney biopsy just before the onset of severe thrombocytopenia. In the present case, glomeruli showed lobular accentuation, mesangiolysis, and double contoured GBM due to prominent endothelial swelling and the widened subendothelial space with no deposits, which was compatible with the MPGN-like lesions due to glomerular endothelial injury. These lesions are seen in renal thrombotic microangiopathy (TMA); however, fibrin thrombi in glomerular capillaries or arterioles, another typical features of renal TMA, were not observed, and biological signs of TMA were absent in the present case. More recently, José et al[7] also reported a case of TAFRO syndrome that underwent kidney biopsy at an early stage of disease and their pathological findings were very similar to ours. Moreover, in 1 case mentioned above, kidney biopsy performed after the recovery of AKI showed mesangial cell proliferation and double contours of GBM without endothelial swelling.[10] These findings were consistent with the late phase of MPGN-like lesions due to endothelial injury. Based on our case and others, glomerular endothelial injury appears to play the major role in the development of AKI in TAFRO syndrome. Prominent glomerular endothelial injury might rapidly reduce glomerular perfusion, leading to subsequent oliguric or anuric AKI.
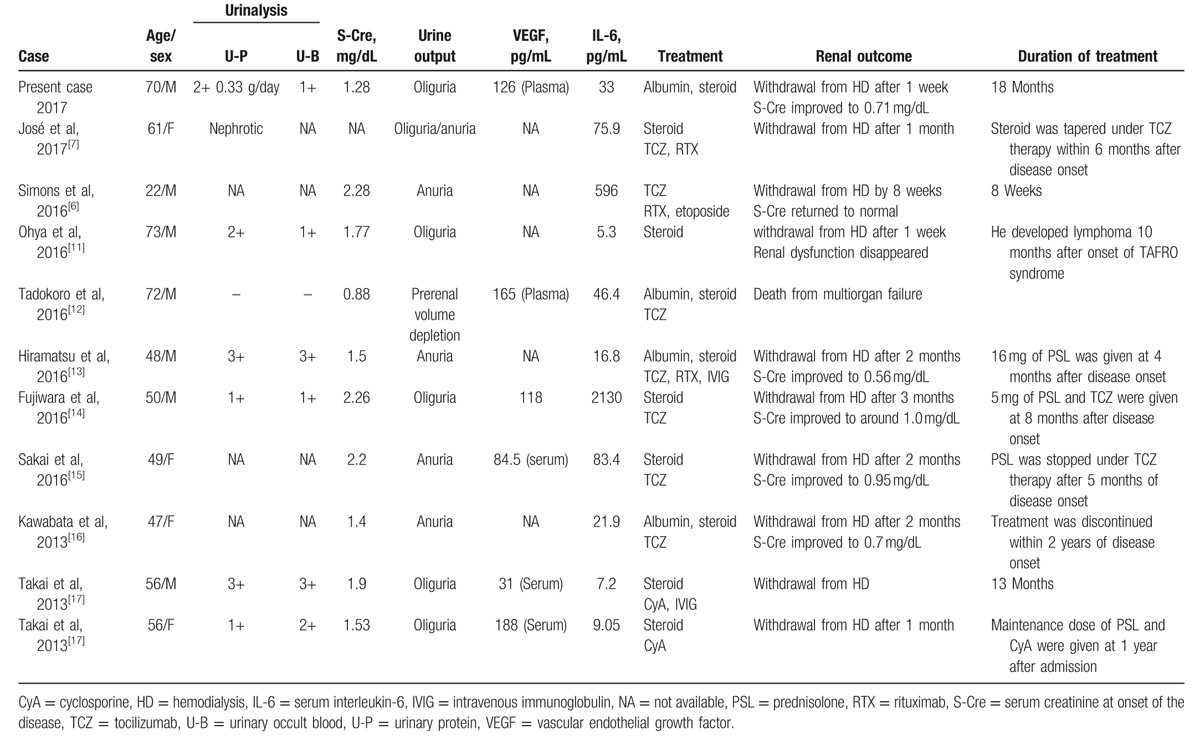
We identified 10 previous cases of TAFRO syndrome requiring hemodialysis due to progressive AKI. Clinical characteristics of these cases and the present case are summarized in Table 1. We found several typical clinical features of renal involvement among these cases. Overall, proteinuria or hematuria tended to be mild, and nephrotic-range proteinuria was only reported in 1 case. All cases developed oliguria or anuria rapidly. Albumin infusions were attempted in some cases, but without improvement in urine output. All cases were treated with steroid therapy and/or other agents including IL-6 receptor antagonist, anti-CD20 monoclonal antibody, and cyclosporine. Renal prognosis was favorable: all cases except 1, who died of multiorgan failure, recovered from AKI and withdrew from hemodialysis generally within 1 to 2 months after starting treatment.
Table 1.
Case reports of TAFRO syndrome requiring hemodialysis due to progressive renal involvement.
Similar renal lesions have been observed occasionally in patients with POEMS syndrome[18] and more rarely in patients with MCD.[19] Serum VEGF and IL-6 levels are usually elevated in both diseases and considered likely pathogenic factors. However, serum VEGF levels did not correlate with glomerular lesions in POEMS syndrome,[18] and whether higher levels of these molecules are causative in glomerular endothelial injury remains controversial. Systemic administration of VEGF to rats did not result in glomerular morphological changes.[20] In contrast, MPGN-like lesions with endothelial injury occurred in the clinical settings of inhibition of VEGF signaling, including anti-VEGF therapy[21] and preeclampsia.[22] Moreover, in a mouse model of podocyte-specific VEGF deletion, similar renal lesions also developed; VEGF secreted by podocytes has been considered essential for the maintenance of endothelial cells.[21] Although we were unable to examine the glomerular VEGF expression, podocyte VEGF expression was shown to be reduced in MPGN-like lesions in patients with MCD.[23] Further studies are required to evaluate whether the reduction of podocyte-derived VEGF rather than higher levels of serum VEGF might be a greater contributor to the renal involvement in TAFRO syndrome.
This case report is notable for confirming the histological diagnosis in the acute phase of renal involvement in TAFRO syndrome. Our findings suggest that severe glomerular endothelial injury might be a primary factor in the progressive AKI. As TAFRO syndrome is becoming increasingly common, it is important to clearly recognize the clinical and histological renal manifestations of this novel clinical entity.
Footnotes
Abbreviations: AKI = acute kidney injury, ALP = alkaline phosphatase, BJP = Bence Jones protein, Cre = creatinine, CRP = C-reactive protein, GBM = glomerular basement membrane, IL-6 = interleukin-6, LDH = lactate dehydrogenase, MCD = multicentric Castleman disease, MPGN = membranoproliferative glomerulonephritis, TAFRO = thrombocytopenia, anasarca, myelofibrosis, renal dysfunction, and organomegaly, TMA = thrombotic microangiopathy, TPO = thrombopoietin, VEGF = vascular endothelial growth factor.
HT and MT contributed equally to this work.
The authors report no conflicts of interest.
References
- [1].Takai K, Nikkuni K, Shibuya H, et al. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly [in Japanese]. Rinsho Ketsueki 2010;51:320–5. [PubMed] [Google Scholar]
- [2].Masaki Y, Kawabata H, Takai K, et al. Proposed diagnostic criteria, disease severity classification and treatment strategy for TAFRO syndrome, 2015 version. Int J Hematol 2016;103:686–92. [DOI] [PubMed] [Google Scholar]
- [3].Tedesco S, Postacchini L, Manfredi L, et al. Successful treatment of a Caucasian case of multifocal Castleman's disease with TAFRO syndrome with a pathophysiology targeted therapy—a case report. Exp Hematol Oncol 2015;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Allegra A, Rotondo F, Russo S, et al. Castleman-Kojima disease (TAFRO syndrome) in a Caucasian patient: a rare case report and review of the literature. Blood Cells Mol Dis 2015;55:206–7. [DOI] [PubMed] [Google Scholar]
- [5].Jain P, Verstovsek S, Loghavi S, et al. Durable remission with rituximab in a patient with an unusual variant of Castleman's disease with myelofibrosis-TAFRO syndrome. Am J Hematol 2015;90:1091–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Simons M, Apor E, Butera JN, et al. TAFRO syndrome associated with EBV and successful triple terapy treatment: case report and review of the literature. Case Rep Hematol 2016;2016:4703608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Jose FF, Kerbauy LN, Perini GF, et al. A life-threatening case of TAFRO syndrome with dramatic response to tocilizumab, rituximab, and pulse steroids: the first case report in Latin America. Medicine (Baltimore) 2017;96:e6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Alhoulaiby S, Ahmad B, Alrstom A, et al. Castleman's disease with TAFRO syndrome: a case report from Syria. Oxf Med Case Reports 2017;2017:omx021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dispenzieri A. POEMS syndrome: update on diagnosis, risk-stratification, and management. Am J Hematol 2015;90:951–62. [DOI] [PubMed] [Google Scholar]
- [10].Kawashima M, Usui T, Okada H, et al. TAFRO syndrome: 2 cases and review of the literature. Mod Rheumatol 2015;1–5. [DOI] [PubMed] [Google Scholar]
- [11].Ohya E, Mizutani M, Sakaguchi H, et al. Diffuse large B-cell lymphoma during corticosteroid therapy for TAFRO syndrome. Intern Med 2016;55:2861–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tadokoro A, Kanaji N, Hara T, et al. An uncharted constellation: TAFRO syndrome. Am J Med 2016;129:938–41. [DOI] [PubMed] [Google Scholar]
- [13].Hiramatsu S, Ohmura K, Tsuji H, et al. Successful treatment by rituximab in a patient with TAFRO syndrome with cardiomyopathy. Nihon Rinsho Meneki Gakkai Kaishi 2016;39:64–71. [DOI] [PubMed] [Google Scholar]
- [14].Fujiwara S, Mochinaga H, Nakata H, et al. Successful treatment of TAFRO syndrome, a variant type of multicentric Castleman disease with thrombotic microangiopathy, with anti-IL-6 receptor antibody and steroids. Int J Hematol 2016;103:718–23. [DOI] [PubMed] [Google Scholar]
- [15].Sakai K, Maeda T, Kuriyama A, et al. TAFRO syndrome successfully treated with tocilizumab: a case report and systematic review. Mod Rheumatol 2016;1–6. [DOI] [PubMed] [Google Scholar]
- [16].Kawabata H, Kotani S, Matsumura Y, et al. Successful treatment of a patient with multicentric Castleman's disease who presented with thrombocytopenia, ascites, renal failure and myelofibrosis using tocilizumab, an anti-interleukin-6 receptor antibody. Intern Med 2013;52:1503–7. [DOI] [PubMed] [Google Scholar]
- [17].Takai K, Nikkuni K, Momoi A, et al. Thrombocytopenia with reticulin fibrosis accompanied by fever, anasarca and hepatosplenomegaly: a clinical report of five cases. J Clin Exp Hematop 2013;53:63–8. [DOI] [PubMed] [Google Scholar]
- [18].Nakamoto Y, Imai H, Yasuda T, et al. A spectrum of clinicopathological features of nephropathy associated with POEMS syndrome. Nephrol Dial Transplant 1999;14:2370–8. [DOI] [PubMed] [Google Scholar]
- [19].Xu D, Lv J, Dong Y, et al. Renal involvement in a large cohort of Chinese patients with Castleman disease. Nephrol Dial Transplant 2012;27(suppl 3):iii119–25. [DOI] [PubMed] [Google Scholar]
- [20].Webb NJ, Watson CJ, Roberts IS, et al. Circulating vascular endothelial growth factor is not increased during relapses of steroid-sensitive nephrotic syndrome. Kidney Int 1999;55:1063–71. [DOI] [PubMed] [Google Scholar]
- [21].Eremina V, Jefferson JA, Kowalewska J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 2008;358:1129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kincaid-Smith P. The renal lesion of preeclampsia revisited. Am J Kidney Dis 1991;17:144–8. [DOI] [PubMed] [Google Scholar]
- [23].El Karoui K, Vuiblet V, Dion D, et al. Renal involvement in Castleman disease. Nephrol Dial Transplant 2011;26:599–609. [DOI] [PubMed] [Google Scholar]