

Graphical abstract

Abbreviations: BSA, bovine serum albumin; DCC, N,N’-dicyclohexylcarbodiimide; DIBAH, diisobutylaluminium hydride; DMF, N,N-dimethylformamide; ELISA, enzyme-linked immunosorbent assay; GAR, goat anti-rabbit antibody; GAR-Po, peroxidase labelled goat anti-rabbit antibody; LFIA, lateral flow immunoassay; LOD, limit of detection; NBS, N-bromosuccinimide; NHS, N-hydroxysuccinimide; NPS, new psychoactive substances; PEG, polyethylene glycol; RSA, rabbit serum albumin; RSD, relative standard deviation; SCs, synthetic cannabinoids; THC, thin layer chromatography
Keywords: Hapten synthesis, Synthetic cannabinoid, JWH-200, Immunomethods, ELISA, LFIA
Highlights
-
•
A hapten with [(morpholin-4-yl)alkyl]indole moiety is used in the detection of JWH-200.
-
•
The hapten is synthesized via Rosenmund–von Braun and Friedel-Crafts reactions.
-
•
ELISA and LFIA are developed and used for quantification of JHW-200 in saliva samples.
-
•
Our one-step LFIA provides a visual evidence of JWH-200 presence within 15 min.
Abstract
In recent years, the use of synthetic cannabinoids (SCs) as drugs of abuse has greatly increased. SCs are associated with a risk of severe poisoning or even death. Therefore, more rapid, cost effective and reliable methods are needed, especially for the screening of drivers after traffic accidents and for detailed toxicological analysis in forensic laboratories. In this study, we developed a lateral flow immunoassay (LFIA) and an enzyme linked immunosorbent assay (ELISA) for the detection of JWH-200 in oral fluids. For this purpose a new hapten was prepared using a ten-step synthetic route. The developed immuno methods are based on antibodies obtained from rabbit immunized with synthesized hapten conjugated to carrier protein. The proposed methods are highly sensitive (LODLFIA = 0.08 ± 0.04 ng mL−1; LODELISA = 0.04 ± 0.02 ng mL−1). They were applied to the quantification of JHW-200 in spiked oral fluids. The recoveries ranged from 82 to 134% for both methods. The results correlated excellently with results obtained using UHPLC–MS/MS (R2LFIA = 0.99; R2ELISA = 0.99). Our developed methods could be an important tool for analyses of JWH-200 in human oral fluids. The one-step LFIA is particularly suitable for roadside and on-site monitoring due to the rapid qualitative results it delivers, while the ELISA is especially useful for laboratory quantitative analyses of positive samples captured by LFIA.
1. Introduction
The frequent appearance of new psychoactive substances (NPS) as drugs of abuse is a matter of concern with the public. Synthetic cannabinoids (SCs) are the largest group of NPS monitored in Europe by the Early Warning System. These substances – commonly called ‘Spice’ are sold as ‘legal’ alternative to cannabis and may be marketed as ‘herbal incense blends’ or ‘herbal mixtures’ and usually labelled ‘not for human consumption’ in order to circumvent consumer protection and the law [1]. Their easy accessibility (especially via online shops), and impossible detectability using routine screening tests for cannabis contribute to an expansion in their abuse.
There have been numerous reports that abuse of SCs can cause a wide range of serious harms to human health (acute ischemic stroke, kidney damage, pulmonary and cardiovascular effects, and psychiatric symptoms) [[1], [2], [3], [4], [5], [6]]. Therefore, development of simple methods that could be used for rapid determination of SCs is needed.
JWH-200, systematically named 1-[2-(morpholin-4-yl)ethyl]-3-(naphthalene-1-carbonyl)-1H-indole, is considered to be one of the most widely known SCs. A seizure of Spice adulterated with JWH-200 was first reported in 2009 by Europol but the drug soon spread out throughout Europe, North America and Japan [[5], [7]]. JWH-200 became one of few SCs added to the list of controlled substances. That resulted in the great interest of state authorities to develop an effective analysis of this substance intended for use in the field.
Current methods used for the analysis of SCs in human fluids are mainly based on high performance liquid chromatography or gas chromatography coupled to various selective detectors [[8], [9]]. However, these techniques are relatively demanding with respect to costs, sample preparation, analysis times, and highly trained personnel and are unsuitable for screening analysis. On the other side, immunoassays provide an attractive alternative for rapid screening of samples. These days, enzyme-linked immunosorbent assay (ELISA) carried out in a microtiter plate is the most common technique used for immunoassays. The possibility of analysing liquid samples without any purification is one of the most outstanding advantages the immunoassays have over commonly used instrumental methods. ELISA has been successfully applied for the analysis of selected SCs mainly in urine [[9], [10], [11], [12], [13], [14]]. Its main advantages are the possibility to analyse multiple samples simultaneously, sensitivity and the relative simplicity. However, the performance of the necessary operations including repeated incubation and washing steps and enzyme reaction for final signal generation is laborious for laboratories that are not specialized for this process. Lateral flow immunoassay (LFIA) is considered to be one of the simplest methods, which fits perfectly for on-site and roadside monitoring [15]. It combines several benefits including primarily rapidity, user-friendly format and cost-effectiveness [[16], [17]]. On the other hand, LFIA gives only preliminary results, so it could be useful to have both, rapid and simple LFIA and also a sensitive method for quantification such as ELISA.
Herein we report the synthesis of a new hapten structurally derived from JWH-200 itself which will serve for the development of immunomethods. To the best of our knowledge, this is the first hapten bearing 1-[2-(morpholin-4-yl)ethyl]-1H-indole moiety used as an immunogen precursor in the detection of synthetic cannabinoids. The aims of the study are to provide the sensitive LFIA that would be as simple as possible to be applied by the state authorities for rapid roadside and on-site monitoring of JWH-200 in oral fluids and ELISA for toxicological quantitative analyses of positive samples captured by LFIA.
2. Material and methods
2.1. Material and reagents
Bovine serum albumin (BSA), rabbit serum albumin (RSA), Tween 20, polyethylene glycol (PEG), Triton X-100, N-hydroxysuccinimide (NHS), indole, naphthalen-1-ol, 4-(2-hydroxyethyl) morpholine, benzyl bromide, methanesulfonyl chloride, N-bromosuccinimide (NBS), ammonium formate, 10% palladium on carbon, sodium hydride, copper cyanide, silver nitrate and zirconium tetrachloride were purchased from Sigma-Aldrich Inc., USA. Ethyl 2-bromoacetate was obtained from Merck and N,N’-dicyclohexylcarbodiimide (DCC) was obtained from Fluka. Diisobutylaluminium hydride (DIBAH) solution in hexane, oxalyl chloride and N,N-dimethylformamide (DMF) extra dry were purchased from Acros. All the other solvents were obtained from Penta. Thin layer chromatography (TLC) was performed on Merck aluminium backed sheets coated with 60F 254 silica gel. Artificial saliva (1700-0305) was purchased from Pickering Laboratories, USA. Gold colloid nanoparticles (an average diameter of 40 nm) were purchased from BB international, UK. Goat anti-rabbit antibody (GAR) and peroxidase labelled goat anti-rabbit antibody (GAR-Po) were obtained from Nordic Immunological Laboratories, Netherlands. Nitrocellulose membranes (PRIMA 85; AE 99; AE 100) were supplied from Whatman GmbH, Germany. Other nitrocellulose membranes (HiFlow Plus HF135; HiFlow Plus HFB180), laminated card (HF000MC100), glass fiber conjugate pad (CFCP03000), cellulose fiber sample pad (CFSP173000) and absorbent pad (CFSP) were purchased from Millipore Corp., USA.
JWH-200 standard and all of the other drug standards used for cross-reactivity studies (Table 2) were obtained from Alfarma s.r.o., Czech Republic or Cayman Pharma, Czech Republic. Individual stock standard solutions containing 1 mg mL−1 of each compound were prepared by dissolving accurately weighted amounts in 96% ethanol and stored at −20 °C.
Table 2.
Cross-reactivity data for Anti-JWH-200 used in ELISA.
| Compound | Structure | CR (%) | Compound | Structure | CR (%) |
|---|---|---|---|---|---|
| JWH-200 | 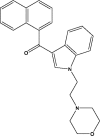 |
100.0 | JWH-018 | 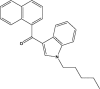 |
0.7 |
| 5F-PB-22 | 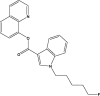 |
0.2 | JWH-081 |  |
0.9 |
| AM-1220 | 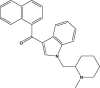 |
4.7 | JWH-122 |  |
1.0 |
| AM-2233 |  |
5.6 | PB-22 |  |
0.1 |
| AB-PINACA |  |
0.1 | pravadoline |  |
2.6 |
| JWH-030 |  |
0.4 | RCS-4 |  |
0.2 |
| JWH-073 |  |
1.5 |
Other tested cannabinoids (AB-FUBINACA, cannabidiol, cannabidivarin, canabigerol, cannabinol, dihydrocannabidiol, tetrahydrocannabinol) – all with cross-reactivity ˂ 0.01% in developed ELISA.
96-well polystyrene microtiter plates Costar 9018 were purchased from Corning Inc., USA.
2.2. Instrumentation
NMR spectra were recorded on a Varian Gemini 300 (300 MHz for 1H; 75 MHz for 13C) or Agilent 400-MR DD2 (400 MHz for 1H; 100 MHz for 13C) spectrometers. High resolution mass spectra were measured on a LTQ Orbitrap XL (Thermo Fischer Scientific) spectrometer using ESI ionization technique. Mass spectra of hapten-protein conjugates were measured on a Bruker Autoflex Speed MALDI-TOF/TOF spectrometer. Automated reverse phase chromatography was carried out using a CombiFlash Rf 200 apparatus (Teledyne ISCO) with prepacked Redisep Rf Gold C18 columns (packed with C18-reverse phase silica gel).
Microplate reader uQuant BIO-TEK was from Inc. Winooski, USA. Linomat V (Camag AG, Switzerland) and a strip cutter (Economic Cutter ZQ2000, Shanghai Kinbio Tech Co., China) were used for the preparation of immunostrips.
2.3. Buffers and solutions
2.3.1. LFIA buffers
Coating buffer (0.01 M carbonate/bicarbonate buffer pH 9.6); assay buffer (0.1 M borate buffer pH 8.8 containing Triton X-100 (1% v/v); conjugate pad buffer (0.2 M borate buffer pH 8.8 containing BSA (1% w/v); sucrose (1% w/v) and Tween 20 (1%)).
2.3.2. ELISA buffers
Coating buffer (0.01 M carbonate/bicarbonate buffer pH 9.6); assay buffer (0.01 M phosphate buffered saline (PBS), pH 7.4); wash buffer (0.01 M PBS pH 7.4 containing Tween 20 (0.05% v/v)); substrate solution for enzyme (9 mL 0.05 M citrate/phosphate buffer pH 5.0, 1 mg of TMB, 1 mL of DMSO, and 2 μL of 30% H2O2 (v/v)); stopping solution (2 M sulphuric acid in distilled water).
2.4. Synthesis of hapten (derivative of JWH-200)
The structure of the hapten was derived from JWH-200. The synthesis of the hapten bearing the linker with carboxylic functional group in the position 4 of the naphthalene ring was proposed and carried out (Fig. 1). Spectral data of intermediates and the final product are provided in Table 1.
Fig. 1.

Synthetic route leading to the derivative of JWH-200.
NBS: N-bromosuccinimide, DMF: N,N-dimethylformamide, DIBAH: diisobutylaluminium hydride, THF: tetrahydrofuran.
Table 1.
Spectral data of synthesized compounds.
| Compound | Spectral data |
|---|---|
| 2 | 1H NMR (300 MHz, CDCl3): δ 8.34–8.38 (m, 1H, ArH), 7.80 − 7.83 (m, 1H, ArH), 7.35–7.56 (m, 9H, ArH), 6.90 (d, J = 7.6 Hz, 1H, ArH), 5.27 (s, 2H, CH2). |
| 13C NMR (75 MHz, CDCl3): δ 154.61, 137.28, 134.67, 128.72, 128.05, 127.59, 127.50, 126.57, 125.96, 125.88, 125.37, 122.33, 120.61, 105.28, 70.18. | |
| 3 | 1H NMR (300 MHz, CDCl3): δ 8.36 (d, J = 8.1 Hz, 1H, ArH), 8.18 (d, J = 8.1 Hz, 1H, ArH), 7.36–7.66 (m, 8H, ArH), 6.76 (d, J = 8.1 Hz, 1H, ArH), 5.24 (s, 2H, CH2). |
| 13C NMR (75 MHz, CDCl3): δ 154.40, 136.81, 132.68, 129.55, 128.81, 128.24, 127.96, 127.52, 127.14, 127.04, 126.17, 122.77, 113.69, 106.08, 70.46. | |
| 4 | 1H NMR (300 MHz, CDCl3): δ 8.40 (dd, J = 8.5, 1.2 Hz, 1H, ArH), 8.18 (dd, J = 8.7, 1.2 Hz, 1H, ArH), 7.85 (d, J = 8.1 Hz, 1H, ArH), 7.71 (td, J = 7.6, 1.2 Hz, 1H, ArH), 7.60 (td, J = 7.6, 1.2 Hz, 1H, ArH), 7.39–7.53 (m, 5H, Ph-H), 6.91 (d, J = 8.1 Hz, 1H, ArH), 5.32 (s, 2H, CH2). |
| 13C NMR (75 MHz, CDCl3): δ 158.48, 135.89, 134.10, 133.67, 129.13, 128.92, 128.55, 127.57, 126.91, 125.46, 125.06, 123.06, 118.54, 104.71, 102.21, 70.72. | |
| MS (ESI) m/z: 260.1 [M + H+], 282.1 [M + Na+]. | |
| HRMS (ESI) m/z: calculated for C18H13ON + H+: 260.10699 [M + H+]; found: 260.10705. | |
| 5 | 1H NMR (300 MHz, CDCl3): δ 10.21 (s, 1H, COH), 9.32 (d, J = 8.7 Hz, 1H, ArH), 8.42 (d, J = 8.4 Hz, 1H, ArH), 7.90 (d, J = 8.1 Hz, 1H, ArH), 7.71 (t, J = 7.8 Hz, 1H, ArH), 7.51–7.61 (m, 3H, ArH), 7.39–7.48 (m, 3H, ArH), 6.98 (d, J = 8.1 Hz, 1H, ArH), 5.34 (s, 2H, CH2). |
| 13C NMR (75 MHz, CDCl3): δ 192.36, 159.94, 139.60, 136.07, 132.11, 129.71, 128.91, 128.51, 127.58, 126.59, 125.81, 125.30, 125.02, 122.68, 104.30, 70.70. | |
| 6 | 1H NMR (300 MHz, CD3OD): δ 9.04 (d, J = 8.2 Hz, 1H, ArH), 8.36 (d, J = 8.5 Hz, 1H, ArH), 8.26 (d, J = 8.4 Hz, 1H, ArH), 7.48–7.60 (m, 5H, ArH), 7.35–7.45 (m, 3H, ArH), 7.04 (d, J = 8.4 Hz, 1H, ArH), 5.37 (s, 2H, CH2). |
| 13C NMR (100 MHz, DMSO-d6): δ 168.27, 157.37, 136.55, 132.41, 132.28, 128.59, 128.13, 128.06, 127.65, 125.68, 125.64, 125.14, 122.01, 119.17, 104.68, 69.84. | |
| MS (ESI negative) m/z: 186.0 [M-C7H7−], 233.1 [M-COOH−], 277.0 [M-H−], 577.1 [2M-2H + Na−]. | |
| HRMS (ESI negative) m/z: calculated for C18H14O3—H−: 277.08702 [M-H−]; found: 277.08693. | |
| 8 | 1H NMR (300 MHz, CDCl3): δ 7.63 (d, J = 7.5 Hz, 1H, ArH), 7.36 (dd, J = 8.6, 0.8 Hz, 1H, ArH), 7.19–7.26 (m, 1H, ArH), 7.15 (d, J = 3.2 Hz, 1H, ArH), 7.08–7.13 (m, 1H, ArH), 6.50 (dd, J = 3.2, 0.9 Hz, 1H, ArH), 4.26 (t, J = 6.9 Hz, 2H, N-CH2), 3.71 (dd, J = 4.7, 4.7 Hz, 4H, CH2-O-CH2), 2.76 (t, J = 6.9 Hz, 2H, N-CH2-CH2), 2.50 (dd, J = 4.7, 4.7 Hz, 4H, CH2-N-CH2). |
| 13C NMR (75 MHz, CDCl3): δ 136.00, 128.65, 128.11, 121.55, 121.09, 119.45, 109.26, 101.37, 67.02, 58.26, 53.95, 44.04. | |
| MS (ESI) m/z: 231.1 [M + H+]. | |
| 9 | 1H NMR (300 MHz, CDCl3): δ 8.47–8.50 (m, 1H, ArH), 8.42 − 8.45 (m, 1H, ArH), 8.28–8.31 (m, 1H, ArH), 7.65 (d, J = 8.1 Hz, 1H, ArH), 7.34–7.58 (m, 11H, ArH), 6.91 (d, J = 8.1 Hz, 1H, ArH), 5.34 (s, 2H, O-CH2), 4.19 (t, J = 6.5 Hz, 2H, N-CH2), 3.59 (dd, J = 4.5, 4.5 Hz, 4H, CH2-O-CH2), 2.72 (t, J = 6.5 Hz, 2H, N-CH2-CH2), 2.42 (dd, J = 4.5, 4.5 Hz, 4H, CH2-N-CH2). |
| 13C NMR (75 MHz, CDCl3): δ 191.85, 156.22, 138.36, 137.12, 136.87, 132.43, 131.91, 128.83, 128.26, 127.72, 127.58, 127.51, 127.25, 126.08, 126.00, 125.89, 123.67, 123.12, 122.89, 122.43, 118.07, 109.73, 103.61, 70.43, 66.99, 57.69, 53.79, 44.32. | |
| MS (ESI) m/z: 261.2 [BnO-C10H6-CO+], 491.4 [M + H+], 513.4 [M + Na+]. | |
| HRMS (ESI) m/z: calculated for C32H30O3N2 + H+: 491.23309 [M + H+]; found: 491.23292. | |
| 10 | 1H NMR (300 MHz, CDCl3) δ ppm: 8.48 − 8.51 (m, 1H, ArH), 8.21 − 8.29 (m, 2H, ArH), 7.46 − 7.51 (m, 4H, ArH), 7.34 − 7.38 (m, 3H, ArH), 6.72 (d, J = 7.8 Hz, 1H, ArH), 4.19 (t, J = 6.5 Hz, 2H, N-CH2), 3.61 (dd, J = 4.8, 4.8 Hz, 4H, CH2-O-CH2), 2.73 (t, J = 6.5 Hz, 2H, N-CH2-CH2), 2.43 (dd, J = 4.8, 4.8 Hz, 4H, CH2-N—CH2). |
| 13C NMR (100 MHz, CDCl3) δ ppm: 192.92, 154.78, 139.10, 137.06, 132.53, 130.54, 128.19, 127.41, 127.15, 125.71, 125.43, 125.11, 123.80, 123.07, 123.02, 122.42, 117.89, 109.83, 107.02, 66.76, 57.51, 53.61, 44.12. | |
| MS (ESI) m/z: 401.4 [M + H+], 423.4 [M + Na+], 823.9 [2M + Na+]. | |
| HRMS (ESI) m/z: calculated for C25H24O3N2 + H+: 401.18597 [M + H+]; found: 401.18596. | |
| 11 | 1H NMR (300 MHz, CDCl3): δ 8.43 − 8.50 (m, 2H, ArH), 8.23 − 8.26 (m, 1H, ArH), 7.61 (d, J = 8.0 Hz, 1H, ArH), 7.51 − 7.55 (m, 2H, ArH), 7.48 (s, 1H, ArH), 7.34–7.39 (m, 3H, ArH), 6.73 (d, J = 8.0 Hz, 1H, ArH), 4.88 (s, 2H, O-CH2), 4.37 (q, J = 7.2 Hz, 2H, O-CH2-CH3), 4.18 (t, J = 6.5 Hz, 2H, N—CH2), 3.59 (dd, J = 4.7, 4.7 Hz, 4H, CH2-O-CH2), 2.72 (t, J = 6.5 Hz, 2H, N-CH2-CH2), 2.42 (dd, J = 4.7, 4.7 Hz, 4H, CH2-N-CH2), 1.34 (t, J = 7.2 Hz, 3H, O-CH2-CH3). |
| 13C NMR (100 MHz, CDCl3): δ 191.72, 168.62, 155.24, 138.55, 137.06, 132.72, 132.35, 127.70, 127.11, 127.03, 126.10, 125.86, 125.82, 123.68, 123.04, 122.91, 122.41, 117.89, 109.75, 103.30, 66.95, 65.77, 61.65, 57.63, 53.73, 44.27, 14.32. | |
| MS (ESI) m/z: 487.5 [M + H+], 509.5 [M + Na+], 974.0 [2M + H+], 996.0 [2M + Na+]. | |
| HRMS (ESI) m/z: calculated for C29H30O5N2 + H+: 487.22275 [M + H+]; found: 487.22285. | |
| derivative of JWH-200 | 1H NMR (300 MHz, CDCl3): δ 8.41–8.44 (m, 1H, ArH), 8.32–8.35 (m, 1H, ArH), 8.19–8.22 (m, 1H, ArH), 7.30–7.46 (m, 7H, ArH), 6.60 (d, J = 7.8 Hz, 1H, ArH), 4.66 (s, 2H, O-CH2), 4.24 (t, J = 6.8 Hz, 2H, N-CH2), 3.83 (br s, 1H, NH), 3.62 (dd, J = 4.5, 4.5 Hz, 4H, CH2-O-CH2), 2.78 (t, J = 6.8 Hz, 2H, N-CH2-CH2), 2.52 (dd, J = 4.5, 4.5 Hz, 4H, CH2-N-CH2). |
| 13C NMR (75 MHz, CDCl3): δ 191.69, 172.61, 155.58, 138.34, 136.87, 132.15, 131.70, 127.67, 127.48, 127.12, 125.92, 125.79, 125.62, 124.04, 123.10, 122.96, 122.57, 118.07, 109.84, 103.33, 66.86, 65.42, 56.44, 52.86, 42.75. | |
| MS (ESI) m/z: 459.2 [M + H+], 481.1 [M + Na+]. | |
| HRMS (ESI) m/z: calculated for C27H26O5N2 + H+: 459.19145 [M + H+]; found: 459.19109. |
2.5. 1-(Benzyloxy)naphthalene (2) [18]
Naphthalen-1-ol (1) (1442 mg, 10 mmol) was dissolved in acetonitrile (40 mL) and anhydrous potassium carbonate (2764 mg, 20 mmol) was added to the solution. After 30 min, benzyl bromide (1784 μL, 15 mmol) was gradually added. The reaction mixture was heated to reflux and stirred for 16 h. After cooling to room temperature, the mixture was diluted with ethyl acetate and washed twice with 2 M hydrochloric acid, once with water and brine. The organic layer was dried over sodium sulfate and concentrated to dryness in vacuo. The crude product was purified by column chromatography (hexane/dichloromethane 19:1) to afford 1-(benzyloxy)naphthalene (2) (2239 mg, 96%) as a colorless crystalline solid.
2.6. 1-(Benzyloxy)-4-bromonaphthalene (3) [[19], [20]]
1-(Benzyloxy)naphthalene (2) (1531 mg, 6.53 mmol) was dissolved in acetonitrile (35 mL), the solution was cooled in an ice bath and NBS (1163 mg, 6.53 mmol) was added portionwise over 20 min. The reaction mixture was stirred for 3 h at 0 °C. After warming to ambient temperature, the mixture was diluted with ethyl acetate and washed with water three times. The collected aqueous layers were extracted with ethyl acetate twice. The organic extracts were combined, dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (hexane/dichloromethane 19:1) to afford 1-(benzyloxy)-4-bromonaphthalene (3) (1885 mg, 92%) as a colorless crystalline solid.
2.7. 4-(Benzyloxy)naphthalene-1-carbonitrile (4) [[21], [22]]
1-(Benzyloxy)-4-bromonaphthalene (3) (1404 mg, 4.48 mmol) was dissolved in DMF (150 mL) and copper cyanide (605 mg, 6.72 mmol) was added to the solution. The flask was placed in heating mantle and the reaction mixture was heated to reflux for 24 h. Another portion of copper cyanide (121 mg, 1.35 mmol) was added and the reaction mixture was boiled for additional 21 h. The solvent was evaporated and the residue was suspended in water and extracted with ethyl acetate. Insoluble part was dissolved in aqueous solution of ammonia, which was then extracted with ethyl acetate two times. The organic extracts were combined, washed with 1 M hydrochloric acid and water and dried over magnesium sulfate. Ethyl acetate was evaporated and the crude product was purified by column chromatography (hexane/dichloromethane 20:1) to afford 4-(benzyloxy)naphthalene-1-carbonitrile (4) (1013 mg, 87%) as a yellowish crystalline solid.
2.8. 4-(Benzyloxy)naphthalene-1-carbaldehyde (5) [23]
4-(Benzyloxy)naphthalene-1-carbonitrile (4) (1000 mg, 3.86 mmol) was dissolved in dry tetrahydrofuran (60 mL), the solution was cooled to 0 °C in an ice bath and 1 M solution of DIBAH in hexane (7.7 mL, 7.70 mmol) was added dropwise. The reaction mixture was stirred at room temperature overnight. Excess DIBAH was decomposed by the addition of water. The aqueous phase was acidified to pH 1 with 2 M hydrochloric acid and extracted with dichloromethane twice. The organic phases were collected and dried over magnesium sulfate. The solvents were evaporated and the crude product was purified by column chromatography (hexane/ethyl acetate 9:1). 4-(Benzyloxy)naphthalene-1-carbaldehyde (5) (762 mg, 75%) was isolated as a white solid.
2.9. 4-(Benzyloxy)naphthalene-1-carboxylic acid (6) [24]
4-(Benzyloxy)naphthalene-1-carbaldehyde (5) (1544 mg, 5.89 mmol) was dissolved in ethanol (80 mL), the solution was heated to 60 °C and silver nitrate (4002 mg, 23.56 mmol) was added. Sodium hydroxide (942 mg, 23.56 mmol) was dissolved in ethanol-water mixture and the solution was gradually added to the reaction flask. After the addition was complete, the reaction mixture was stirred and heated for another 24 h. Excess silver oxide and the silver formed in the reaction were filtered off and washed with hot water. The filtrate was acidified to pH 1 with 2 M hydrochloric acid and extracted with diethyl ether twice. The organic phases were combined and dried over magnesium sulfate. The solvents were evaporated and the crude product was triturated with chloroform. Desired 4-(benzyloxy)naphthalene-1-carboxylic acid (6) (1409 mg, 86%) was isolated as a colorless crystalline solid.
2.10. 1-[2-(Morpholin-4-yl)ethyl]-1H-indole (8)
1-[2-(Morpholin-4-yl)ethyl]-1H-indole (8) was synthesized according to the procedure previously described [25]. The title compound was obtained as light yellow oil.
2.11. 3-[4-(Benzyloxy)naphthalene-1-carbonyl]-1-[2-(morpholin-4-yl)ethyl]-1H-indole (9) [26]
4-(Benzyloxy)naphthalen-1-carboxylic acid (6) (1200 mg, 4.30 mmol) was dissolved in dry dichloromethane (20 mL) and a few drops of DMF were added to the solution. Oxalyl chloride (482 μL, 5.60 mmol) was added gradually until the evolution of gas stopped. After stirring for another hour, the volatiles were evaporated in vacuo. The crude product 7 was azeotropically dried with toluene and used in the subsequent reaction without further purification. 4-(Benzyloxy)naphthalene-1-carbonyl chloride (7) (382 mg, 1.29 mmol) was dissolved in dry dichloromethane (25 mL) and the solution was cooled to −10 °C. A solution of 1-[2-(morpholin-4-yl)ethyl]-1H-indole (8) (385 mg, 1.67 mmol) in dry dichloromethane (5 mL) was added to the reaction mixture, followed by zirconium tetrachloride (450 mg, 1.93 mmol). The reaction was stirred for 1 h at −10 °C. Subsequently, the temperature was maintained at 0 °C for 3 h, before the reaction mixture was allowed to warm to room temperature. After stirring overnight, the reaction was stopped by addition of water. The aqueous phase was extracted with ethyl acetate twice. The organic extracts were combined, washed with water and dried over magnesium sulfate. The solvents were evaporated and the crude product was purified by column chromatography (hexan/aceton 3:1). 3-[4-(Benzyloxy)naphthalene-1-carbonyl]-1-[2-(morpholin-4-yl)ethyl]-1H-indole (9) (444 mg, 70%) was obtained as a white foam.
2.12. 3-(4-Hydroxynaphthalene-1-carbonyl)-1-[2-(morpholin-4-yl)ethyl]-1H-indole (10) [19]
3-[4-(Benzyloxy)naphthalene-1-carbonyl]-1-[2-(morpholin-4-yl)ethyl]-1H-indole (9) (144 mg, 0.29 mmol) was dissolved in dry ethanol (35 mL). Ammonium formate (93 mg, 1.47 mmol) was added to the solution, followed by a catalytic amount of palladium on carbon. The reaction mixture was stirred at room temperature for 1 h. The palladium catalyst was filtered off and the filtrate was concentrated in vacuo. The residue was diluted with ethyl acetate. The organic phase was washed with potassium carbonate solution and then with brine twice. The organic layer was dried over magnesium sulfate. The solvents were evaporated and the crude product was purified by column chromatography (hexan/aceton 9:4) to afford 3-(4-hydroxynaphthalene-1-carbonyl)-1-[2-(morpholin-4-yl)ethyl]-1H-indole (10) (116 mg, 99%) as a white solid.
2.13. Ethyl 2-[(4-{1-[2-(morpholin-4-yl)ethyl]-1H-indole-3-carbonyl}naphthalen-1-yl)oxy]acetate (11)
3-(4-Hydroxynaphthalene-1-carbonyl)-1-[2-(morpholin-4-yl)ethyl]-1H-indole (10) (110 mg, 0.28 mmol) was dissolved in dry acetone (30 mL), freshly annealed potassium carbonate (57 mg, 0.41 mmol) was added to the solution and the temperature was raised to 50 °C. After stirring for 30 min, ethyl 2-bromoacetate (36 μL, 0.32 mmol) was added. The reaction mixture was heated to reflux for 3 h. After cooling to room temperature, ethyl acetate was added and the organic phase was washed with water twice and once with brine. The organic layer was dried over sodium sulfate and concentrated to dryness in vacuo. The crude product was purified by column chromatography (hexane/acetone 3:1) to afford ethyl 2-[(4-{1-[2-(morpholin-4-yl)ethyl]-1H-indole-3-carbonyl}naphthalen-1-yl)oxy]acetate (11) (111 mg, 83%) as a white solid.
2.14. 2-[(4-{1-[2-(morpholin-4-yl)ethyl]-1H-indole-3-carbonyl}naphthalen-1-yl)oxy]acetic acid (derivative of JWH-200)
Ethyl 2-[(4-{1-[2-(morpholin-4-yl)ethyl]-1H-indole-3-carbonyl}naphthalen-1-yl)oxy]acetate (11) (105 mg, 0.22 mmol) was suspended in ethanol (15 mL) and 1 M solution of sodium hydroxide in water (3 mL) was added to the reaction mixture. The reaction was stirred at 50 °C for 1 h. Subsequently, ethanol was evaporated. The residual aqueous phase was acidified to pH 1 with hydrochloric acid and concentrated to dryness in vacuo. The formed sodium chloride was removed by trituration of the residue with water. The obtained crude product was purified by reverse phase chromatography (water/methanol, gradient elution 10:1 → 1:1). The title compound was isolated in the form of 2-[(4-{1-[2-(morpholin-4-yl)ethyl]-1H-indole-3-carbonyl}naphthalen-1-yl)oxy]acetic acid hydrochloride (73 mg, 68%).
2.15. Preparation of hapten-protein conjugates
The synthesized hapten was conjugated to BSA and to RSA to form an immunogen and a coating antigen, respectively. Conjugates were prepared by activated ester method, the conjugation step being carried out in reversed micellar solution [27].
Derivative of JWH-200 (1 eq) was dissolved in the exact volume of DMF to form a solution of concentration of 100 mg mL−1. DCC (1.3 eq) and NHS (1.3 eq) were added to the micro test tube, both in the form of solution in DMF (100 mg mL−1). The reaction mixture was left to stand at room temperature until the by-product N,N’-dicyclohexylurea spontaneously crystalized from the solution. After the reaction was complete (confirmed by TLC), the crystals were separated and the solution with an activated ester was directly used in subsequent reaction. A solution of BSA or RSA (0.013 eq) in bicarbonate buffer (13 μg mL−1) was mixed with a 0.3 M solution of AOT in octane to form a reversed micellar solution. The activated ester of the hapten was added to these solutions with stirring. The reaction mixture was stirred vigorously at room temperature overnight. The modified BSA or RSA was precipitated by addition of cooled acetone and the suspension was centrifuged (4 °C; 30 min; 1 500g). The supernatant was then removed and the collected precipitate was washed with cooled acetone one more time. The suspension was again centrifuged (4 °C; 20 min). After the removal of the supernatant, the crude product was air dried, dissolved in water and lyophilised to afford the conjugate in a form suitable for immunization of laboratory animals.
2.16. Immunogen (derivative of JWH-200-BSA)
The derivative of JWH-200 (10.1 mg, 20.4 μmol) was converted into the activated ester by the reaction with DCC (5.5 mg, 26.5 μmol) and NHS (3.1 mg, 26.5 μmol). Subsequent conjugation of the activated ester with BSA (18.1 mg, 0.27 μmol) afforded the immunogen (24.2 mg).
2.17. Coating antigen (derivative of JWH-200-RSA)
The derivative of JWH-200 (14.5 mg, 29.3 μmol) was converted into the activated ester by the reaction with DCC (7.8 mg, 38.1 μmol) and NHS (4.4 mg, 38.1 μmol). The conjugation of the ester with RSA (25.9 mg, 0.39 μmol) afforded the coating antigen (31.0 mg).
2.18. Antiserum against JWH-200 (Anti-JWH-200) and its specificity
Rabbits were immunized with the immunogen and an antiserum against JWH-200 was collected by using a standard procedure described previously [28]. A stock solution of the Anti-JWH-200 was prepared by dissolving 1 mg of lyophilisate in 1 mL of the PBS and stored at −20 °C.
Anti-JWH-200 specificity was investigated by cross-reactivity experiments (CR%) performed by ELISA. The calibration curves for each of the tested cannabinoids (14synthetic; 6 phytocannabinoids) were constructed. The CR% was calculated as: (IC50 of JWH-200)/IC50 of tested compound) × 100.
2.19. Gold labelled antibodies (Anti-JWH-200-Au)
The gold labelled antibodies were prepared according to the procedure described for carbon nanoparticles with minor modification [29]. To a gold colloid nanoparticles suspension (gold colloid nanoparticles 0.5 mg mL−1 of 5 mM borate buffer, pH 8.8) Anti-JWH-200 (1 mg mL−1 in PBS) were added. The mixture was stirred gently at 37 °C for 2 h. Afterwards, the suspension was centrifuged (10 °C; 60 min; 6 000g). The sediment was washed three times in a 5 mM borate buffer, pH 8.8 containing BSA (1% w/v) and NaN3 (0.02% w/v) using centrifugation (10 °C; 15 min; 13 640g). The final sediment was resuspended in 0.1 M borate buffer, pH 8.8 containing BSA (1% w/v) and NaN3 (0.02% w/v). Prepared stock suspension of the Anti-JWH-200-Au conjugate (200 μg mL−1) was stored at 4 °C in the dark.
2.20. Immunochemical methods
2.20.1. Preparation of LFIA strip and LFIA procedure
The NC membrane AE 100 was stuck to the laminated card to increase the robustness of the membrane. Subsequently, the membrane was coated with GAR at the control line and with derivative of JWH-200-RSA at the test line using the Linomat V (conc. 100 μg mL−1 in coating buffer; 0.1 μL mm−1). The conjugate pad (pre-treated with conjugate pad buffer, 5 min soaking, then drying at 37 °C for 1 h) was dispensed with the mixture of Anti-JWH-200-Au (200 μg mL−1; 1.25 μL mm−1). Than the NC membrane and the conjugate pad were dried at 37 °C overnight. On the following day, the conjugate pad, the sample pad and the absorbent pad were stuck to the laminated card with proper overlap. The membrane was cut into LFIA strips (4 mm) using a strip cutter. The strip was assembled according to the scheme shown in Fig. 2. Prepared strips were put into LFIA cassettes and were stored in sealed bags under dry conditions at laboratory temperature until used.
Fig. 2.

Construction of LFIA strip.
1–sample pad; 2–conjugate pad; 3–test line; 4–control line; 5–absorbent pad; 6–laminated card; 7–nitrocellulose membrane.
The JWH-200 standard solution (the concentration range 0–1000 ng mL−1 in the artificial saliva) or the oral fluid sample was mixed with the assay buffer (1:3) and the mixture was added into the well of the LFIA cassette (100 μL/strip).
In the diffusion flow of reactants, the JWH-200 in a sample interacted with Anti-JWH-200-Au (dried on the conjugate pad) and migrated because of capillary effects along the membrane. Samples free of JWH-200 resulted in just free Anti-JWH-200-Au captured by derivative of JWH-200-RSA dispensed on the test line and formed a red zone of maximum intensity. Otherwise, JWH-200 positive samples inhibited the interaction of the Anti-JWH-200-Au with the derivative of JWH-200-RSA resulting in decrease of red zone intensity. Therefore, the intensity of the zone (test line) inversely correlated with the JWH-200 concentration in the sample.
If the sample produced the test line appearance of the same intensity as a negative control (blank artificial saliva), it was considered to be negative (−). If the colour intensity was weaker than that of the negative control, the result was evaluated as weakly positive (±). The sample was assessed as positive (+) when the test line was absent or extremely weak. Dried strips were scanned and test line intensities converted to pixel grey volumes using TotalLab in order to obtain quantitative results.
2.20.2. ELISA procedure
ELISA was performed as an indirect competitive format. Each well of the microtiter plates was coated with the coating antigen (50 ng mL−1 in the coating buffer; 100 μL/well) and incubated at 4 °C overnight. Afterwards, the unbound antigen was removed with the wash buffer (4 times, 350 μL/well). The standards of JWH-200 (the concentration range of 0–250 ng mL−1 in the PBS) or oral fluid samples (diluted 30 times in the PBS) were added to microtiter plates (50 μL/well) followed by the corresponding solution of Anti-JWH-200 (diluted 1:40 000 in the PBS-0.1% BSA (w/v); 50 μL/well) and incubated at room temperature for 45 min. Microtiter plates were washed again (4 times, 350 μL/well) and GAR-Po (diluted 1:10 000 in the PBS; 100 μL/well) was added and incubated at room temperature for 1 h. After the washing step, the substrate solution for the enzyme was added (100 μL/well) and incubated at room temperature for 10 min. The enzyme reaction was stopped by adding the stopping solution (50 μL/well) and the absorbance was measured at 450 nm.
2.20.3. Calibration curves and interpretation of results
Sigmoid calibration standard curves were obtained by plotting the mean values of absorbance (ELISA) or pixel grey volumes (LFIA) against the logarithm of JWH-200 concentrations through a four-parameter logistic equation as we described previously [30]. The limit of detection (LOD) was defined as the concentration of an analyte corresponding to the maximum assay signal minus 3 x standard deviation (SD) in accordance with the calibration curve (the blank was calculated from 3 parallel determinations with the absence of an analyte). The IC50 corresponded to the concentration of analyte giving 50% inhibition of the asymptotic maximum. The linear working range corresponded to the analyte concentration causing the 20–80% inhibition of the maximum assay signal.
2.20.4. Oral fluid samples
Human oral fluids were obtained from five laboratory volunteers. Freshly collected oral fluids were spiked with JWH-200 in order to obtain the following concentrations: 0; 10; 50; 100; 500; 1000 ng mL−1. The samples were analysed immediately by our immunomethods and were stored at −20 °C until next analyses. Two sets of prepared samples were used to evaluate the correlation with UHPLC–MS/MS.
2.20.5. UHPLC–MS/MS analysis
For the UHPLC–MS/MS analysis Agilent 1290 Infinity UHPLC system coupled with Agilent 6460 Triple quadrupole mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) were used. Chromatographic separation was performed on Agilent Zorbax Eclipse Plus C18 column (2.1 × 50 mm; 1.8 μm). The mobile phases for gradient elution were 10 mM formic acid (A) and methanol (B). The flow rate was 0.25 mL/min and column temperature 35 °C. Gradient elution was carried out as follows: 0 min, 80:20 (A:B); 2 min, 0:100; 3.5 min, 0:100; 3.7 min, 80:20; 6 min, 80:20. Retention time of JWH 200 was 2.54 min.
The MS/MS apparatus was operating in positive mode. The applied conditions of electrospray ion source were: drying gas temperature 340 °C; drying gas flow 4 L/min; sheath gas temperature 200 °C; sheath gas flow 12 L/min; nebulizer pressure 30 psi; nozzle voltage 300 V; capillary voltage was set at 2300 V. Multiple reaction monitoring (MRM) mode was used for the detection. Three transitions of m/z were used (collision energies (eV) are given in brackets): 385.19 → 114.1 (25); 127.1 (69) and 155 (21).
Agilent Mass Hunter (Agilent Technologies, Inc.) was used for data acquisition and quantification of samples.
Simple dilution of oral fluid was used. 100 μL of oral fluid with 900 μL of 20% methanol were vortexed, centrifuged (10 min, 13 000g) and supernatant was used for the analysis.
For calibration blank oral fluid sample was spiked with different concentrations of JWH 200 and prepared the same way as described. Six final concentrations of JWH 200 ranging between 0.1 and 500 ng mL−1 were used. Peak areas of the external standard (at each concentration) were plotted against the corresponding standard concentrations using linear regression to generate standard curve.
3. Results and discussion
3.1. Hapten design and synthesis
Hapten design plays a key role in the development of immunoassays targeted on small molecules such as synthetic cannabinoids. As the structural similarity between the target compound and the designed hapten should be maximized, the hapten was derived directly from JWH-200. The most active part of a haptenic determinant is distal to the point of binding with the carrier protein, thus the linker with carboxylic functional group was placed in the position 4 of the naphthalene ring, so the most active part is the morpholine moiety characteristic for JWH-200. The aforementioned linker serves only to join the hapten to the carrier protein and it should not become an epitope, once the immunogen is applied to laboratory animals [[31], [32]].
The synthesis of the hapten was based on Friedel – Crafts acylation of N-substituted indole 8 by 4-(benzyloxy)naphthalene-1-carbonyl chloride (7) in the presence of zirconium tetrachloride as a Lewis acid [26]. The hydroxyl group was then deprotected by catalytic hydrogenolysis of benzyl protecting group using palladium on activated charcoal as a catalyst [19]. O-alkylation by ethyl 2-bromoacetate and subsequent base catalysed hydrolysis afforded the desired hapten bearing the linker with carboxylic functional group.
It is also worth noticing that an innovative synthetic route was employed for the preparation of 4-(benzyloxy)naphthalene-1-carboxylic acid (6). In the molecule of 4-(benzyloxy)-1-bromonaphtalene (3), the bromine atom was first replaced by nitrile functional group in Rosenmund – von Braun reaction [[21], [22]]. By the reduction of formed nitrile 4 with diisobutylaluminium hydride [23] and subsequent reoxidation of the intermediate aldehyde 5 with silver oxide, formed in situ from silver nitrite and sodium hydroxide [24], carboxylic functional group was introduced into the molecule. This three-step sequence afforded the desired acid 6 with higher overall yield than the published procedure based on lithiation and subsequent reaction of organolithium intermediate with carbon dioxide [19].
The hapten was prepared using a ten-step synthetic sequence from commercially available naphthalen-1-ol (1) with moderate to high yields in each step. The synthesized hapten and all the intermediates were characterized by NMR and ESI–MS (Table 1).
3.2. Preparation of hapten-protein conjugates
The synthesized hapten was coupled to BSA to form the immunogen or to RSA to form the coating antigen. The conjugate formation was confirmed by a MALDI-TOF analysis. The binding ratio of the hapten to the carrier protein was determined to be approximately 20:1 and 31:1 for the immunogen and the coating antigen, respectively. The average number of haptens bound to BSA was considered sufficient to illicit a specific immune response in immunized animals.
3.3. Optimisation of LFIA conditions and characterisation of assay
The assay was optimized using artificial saliva considering the enormous influence of the matrix (oral fluid) on the test lines appearance. The line intensities were reduced if the oral fluid samples were added compared to the intensities obtained from tests carried out in the assay buffer (data not shown). During the optimization, checkboard titration experiments were performed. Several concentrations of the derivative of JWH-200-RSA dispensed on the membrane (50–200 μg mL−1 in coating buffer; 0.1 μL mm−1) against different volume of Anti-JWH-200-Au (200 μg mL−1; 1–1.75 μL mm−1) were investigated. The amount of the immunoreagents should be kept low enough to achieve good sensitivity, but must be sufficient to provide an acceptable signal [33]. Several other factors affecting LFIA strip appearance were evaluated. Five types of NC membrane were tested (PRIMA 85, AE 99, AE 100, HiFlow Plus HF 135, HiFlow Plus HFB180). The type of membrane influenced flow time and sharpness of tested lines. Faster-flowing membranes reached endpoint more quickly but caused a loss in signal intensity and decrease of test sensitivity [[17], [34]]. As shown on Fig. 3, the best performance was observed when membrane AE100 was used. The composition of the assay buffer also influence test sensitivity [17]. In this experiment, 0.01 M PBS, pH 9.6 and 0.1 M borate buffer, pH 8.8 were tested. Additives such as BSA (0.1–2% w/v); PEG (0.1–2% v/v) and sucrose (0.1–5% w/v) and their combinations with and without surfactants Tween 20 (0.1–1% v/v) or Triton X-100 (0.1–1% v/v) were tested to further improve the LFIA sensitivity. The complete specifications of the optimized conditions are included in section Preparation of LFIA strip and LFIA procedure.
Fig. 3.

Appearance of test and control lines on different type of nitrocellulose membrane in LFIA.
CL – control line; TL – test line; 1–AE98; 2–Prima 85; 3–HF135 UB; 4–HF180; 5–AE100.
The sensitivity of the LFIA was determined from the JWH-200 calibration curve (concentration range 0–1000 ng mL−1created in the artificial saliva). The assay was carried out in triplicate under optimized conditions. The colour intensity of the test line was evaluated visually. As shown in Fig. 4, the JWH-200 concentration of 1 ng mL−1 caused a slight but visually distinguishable difference in the test line intensity compared to the negative control. Thus, 1 ng mL−1 of JWH-200 was considered to be a visual detection limit. The value of the visual LOD suggests enough sensitivity for the intended use. Published SCs concentrations in oral fluids are in the range of units to tens ng mL−1 [35]. To obtain the quantitative results, colour intensities of the test lines were converted to pixel grey volumes. The mean signal values were fitted to a sigmoid standard curve. The LOD was 0.08 ± 0.04 ng mL−1 with the linear working range of 0.3–42 ng mL−1 and the IC50 value 3.4 ± 0.6 ng mL−1.
Fig. 4.

Typical LFIA standard curve using the optimized assay protocol (mean value, n = 3).
(A) Quantitative evaluation LOD = 0.08 ± 0.04 ng mL−1; IC50 = 3.4 ± 0.6 ng mL−1; linear working range = 0.3–42 ng mL−1 (B) Visual evaluation.
To evaluate the precision of the LFIA, three standard samples with JWH-200 concentrations in the range of 10–50 ng mL−1 in human oral fluid were assayed. For the intra-assay precision study, one run of analyses (n = 3) was performed with each sample on the same day. Similarly, one run of analyses was carried out with each sample daily on three non-consecutive days for the inter-assay precision study. The intra- and inter-assay relative standard deviations (RSD) were calculated in the range 2.4–9.2% and 3.1–16.7%, respectively, indicating the acceptable precision.
3.4. ELISA characterisation and antiserum specificity
The assay was developed as an indirect competitive format using polyclonal antibody targeted at JWH-200. Checkerboard titrations were performed and suitable immunoreagent concentrations were determined when the maximum absorbance ranged from 1.2 to 1.9 and the calibration curve reached the lowest IC50 values. To enhance the sensitivity of the ELISA, several other conditions (time and temperature of incubations, composition of dilution buffers) were evaluated. The optimized conditions are listed in the section ELISA procedure. Based on the JWH-200 standard curve (concentration range 0–250 ng mL−1 in PBS), the LOD was 0.04 ± 0.02 ng mL−1 with the IC50 value of 0.42 ± 0.09 ng mL−1 and the linear working range of 0.16–1.80 ng mL−1. The parameters characterising our ELISA are comparable with the parameters published previously [9].
The influence of the human oral fluid on the assay was tested by comparing JWH-200 standard curves (concentration range 0–250 ng mL−1) obtained in the PBS and in the presence of various proportion of oral fluid (0; 10; 20; and 30-times diluted in the PBS). As can be seen in Fig. 5, undiluted oral fluid sample significantly affect obtained signal as well as IC50 values. However, 30-fold diluted oral fluid does not affect assay parameters, implying oral fluid samples could be directly applied to the immunoassay without difficult clean-up procedure (only diluted).
Fig. 5.

The comparison of ELISA curves obtained from standard prepared in assay buffer and in the presence of various proportion of oral fluid.
(○) assay buffer; (▲) oral fluid; (●) 10 x diluted oral fluid; (■) 20 x diluted oral fluid (−)30 x diluted oral fluid.
The specificity of obtained antibody was evaluated with 20 cannabinoids. Cross-reactivity values are summarized in Table 2. Substances structurally similar to the target analyte (e.g.AM-2233; AM-1220) showed medium cross-reactivity (5 %), while SCs with a different group than naphthoyl indole group interacted weakly (< 0.2 %), except the pravadoline (> 2 %).
For the precision study, the same experiments were used as for LFIA. The intra- and inter-assay RSD were calculated in the range of 1.5–8.8% and 2.0–9.7%, respectively.
3.5. Analysis of spiked oral fluid samples
The accuracy of the developed immunomethods was evaluated through recovery study with samples of human oral fluid spiked with JWH-200 at levels from 0 to 1000 ng mL−1. In the visually assessed LFIA, the negative results were obtained for non-spiked samples. The weakly positive results were obtained for samples spiked at concentrations 10 ng mL−1, while the samples spiked at level ˃50 ng mL−1of the JWH-200 were assessed as positive. Using the quantitative LFIA the recoveries ranged from 86 to 134% with RSD 2.4–16.8% and for ELISA from 82 to 131 %, with RSD 0.4–7.8 % (Table 3).
Table 3.
Results from spiked oral fluids obtained using LFIA, ELISA (n = 3).
| Saliva | Spiked concentration (ng mL−1) | LFIAa |
ELISAb |
|||||
|---|---|---|---|---|---|---|---|---|
| Visual detectionc | Mean (ng mL−1) | Recovery (%) | RSDd (%) | Mean (ng mL−1) | Recovery (%) | RSDd (%) | ||
| 1. | 0 | − − − | < LODe | – | – | < LODe | – | – |
| 10 | ± ± ± | 11.2 | 112.0 | 9.3 | 13.1 | 131.0 | 6.6 | |
| 50 | ± ± ± | 48.1 | 96.2 | 10.4 | 56.2 | 112.4 | 5.2 | |
| 100 | + + + | 94.2 | 94.2 | 9.6 | 94.6 | 94.6 | 2.3 | |
| 500 | + + + | 526.4 | 105.3 | 11.7 | 535.8 | 107.2 | 0.4 | |
| 1000 | + + + | 1274.9 | 127.5 | 4.7 | 1063.6 | 106.4 | 2.5 | |
| 2. | 0 | − − − | < LODe | – | – | < LODe | – | – |
| 10 | ± ± ± | 8.6 | 86.0 | 7.6 | 8.2 | 82.0 | 3.8 | |
| 50 | ± ± ± | 46.4 | 92.8 | 3.7 | 54.3 | 108.6 | 5.4 | |
| 100 | + + + | 89.7 | 89.7 | 8.4 | 97.7 | 97.7 | 6.4 | |
| 500 | + + + | 534.6 | 106.9 | 4.2 | 582.2 | 116.4 | 2.1 | |
| 1000 | + + + | 1206.7 | 120.7 | 2.4 | 1070.7 | 107.1 | 1.3 | |
| 3. | 0 | − − − | < LODe | – | – | < LODe | – | – |
| 10 | ± ± ± | 9.3 | 93.0 | 7.7 | 9.4 | 94.0 | 4.2 | |
| 50 | ± ± ± | 56.6 | 113.2 | 16.8 | 49.8 | 99.6 | 3.9 | |
| 100 | + + + | 92.7 | 92.7 | 9.6 | 96.2 | 96.2 | 2.5 | |
| 500 | + + + | 668.2 | 133.6 | 4.6 | 523.0 | 104.6 | 7.8 | |
| 1000 | + + + | 1278.4 | 127.8 | 12.2 | 1027.7 | 102.8 | 2.6 | |
Before the LFIA, samples were appropriately diluted with the synthetic saliva to fall into the linear working range.
Before the ELISA, samples were appropriately diluted with the assay buffer to fall into the linear working range.
Visual assessment of the test line; (–) negative result; (±) weakly positive result (the JWH-200 concentration in the range of 10–50 ng mL−1;); (+) positive result (JWH-200 concentration >100 ng mL−1).
RSD, relative standard deviation.
LOD, limit of detection.
3.6. Correlation of immunomethods with the UHPLC–MS/MS
Twelve spiked human oral fluid samples (concentration of JWH-200 from 0 to 1000 ng mL−1) were assayed three times by the newly developed ELISA and LFIA and the results were compared with those obtained using UHPLC–MS/MS (Table 4). The excellent correlations between developed immunomethods and UHPLC–MS/MS were obtained. The linear equation is y = 0.99 x (R2 ELISA = 0.99) and y = 0.79 x (R2 LFIA = 0.99). Our results demonstrate a good potential of the proposed immunomethods for using in routine analyses of JWH-200 in oral fluids.
Table 4.
Recovery obtained using the LFIA, ELISA and UHPLC–MS/MS (n = 3).
| Oral fluid | Spiked concentration (ng mL−1) | LFIAa |
ELISAb |
UHPLC–MS/MS |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (ng mL−1) | Recovery (%) | RSDc (%) | Mean (ng mL−1) | Recovery (%) | RSDc (%) | Mean (ng mL−1) | Recovery (%) | RSDc (%) | ||
| 1. | 0 | < LODd | – | – | < LODd | – | – | < LODd | – | – |
| 10 | 8.4 | 84.1 | 1.3 | 13.4 | 134.0 | 10.5 | 10.0 | 99.8 | 0.03 | |
| 50 | 46.0 | 92.0 | 6.1 | 56.5 | 113.0 | 1.3 | 50.8 | 101.6 | 0.09 | |
| 100 | 88.7 | 88.7 | 12.8 | 121.8 | 121.8 | 14.5 | 104.5 | 104.5 | 0.32 | |
| 500 | 615.8 | 123.2 | 9.7 | 468.8 | 93.8 | 2.7 | 518.2 | 103.6 | 2.02 | |
| 1000 | 1297.5 | 129.7 | 4.3 | 975.3 | 97.5 | 8.9 | 1042.8 | 104.3 | 2.56 | |
| 2. | 0 | < LODd | – | – | < LODd | – | – | < LODd | – | – |
| 10 | 8.3 | 83.5 | 3.5 | 9.4 | 94.2 | 1.3 | 94.0 | 94.2 | 0.03 | |
| 50 | 44.5 | 89.0 | 2.4 | 49.8 | 99.5 | 0.7 | 49.8 | 99.7 | 0.02 | |
| 100 | 95.1 | 95.1 | 4.9 | 96.2 | 96.2 | 0.6 | 99.1 | 99.1 | 0.11 | |
| 500 | 633.8 | 126.8 | 2.0 | 523.0 | 104.6 | 3.2 | 487.2 | 97.4 | 1.29 | |
| 1000 | 1232.4 | 123.2 | 3.6 | 1027.7 | 102.8 | 2.8 | 954.4 | 95.4 | 3.53 | |
aBefore the LFIA, samples were appropriately diluted with the artificial saliva to fall into the linear working range.
Before the ELISA, samples were appropriately diluted with the assay buffer to fall into the linear working range.
RSD, relative standard deviation.
LOD, limit of detection.
4. Conclusions
We have successfully synthesized the new hapten structurally derived from the synthetic cannabinoid JWH-200. The hapten was used to prepare the immunogen and the coating antigen. The antibodies obtained from rabbits immunized with hapten conjugated to BSA served for the development of two immunomethods for the JWH-200 detection in human oral fluids. Our developed one-step LFIA can provide a visual evidence of JWH-200 presence in oral fluids within 15 min. This assay is primarily suitable for the on-site and roadside monitoring due to the rapid qualitative results it delivers. For greater accuracy, the analyte presence should be verified by more sophisticated method such as ELISA. The ELISA proposed in this study is highly sensitive and has the excellent correlation with the UHPLC–MS/MS.
Conflict of interest
All authors declare no conflict of interest.
Acknowledgements
Financial support from specific university research (MSMT No 20-SVV/2016) and project MICR VI20172020056.
References
- 1.EMCDDA . Strategic Overview. 2016. EU drug markets report; pp. 28–29. [Google Scholar]
- 2.EMCDDA . Synthetic Cannabinoids in Europe. 2016. Perspectives on drugs. [Google Scholar]
- 3.Yeakel J.K., Logan B.K. Blood synthetic cannabinoid concentrations in cases of suspected impaired driving. J. Anal. Toxicol. 2013;37(8):547–551. doi: 10.1093/jat/bkt065. [DOI] [PubMed] [Google Scholar]
- 4.McGuinness T.M., Newell D. RISKY RECREATION synthetic cannabinoids have dangerous effects. J. Psychosoc. Nurs. Ment. Health Serv. 2012;50(8):16–18. doi: 10.3928/02793695-20120703-04. [DOI] [PubMed] [Google Scholar]
- 5.Logan B.K. Identification of synthetic cannabinoids in herbal incense blends in the United States. J. Forensic Sci. 2012;57(5):1168–1180. doi: 10.1111/j.1556-4029.2012.02207.x. [DOI] [PubMed] [Google Scholar]
- 6.Harris C.R., Brown A. Synthetic cannabinoid intoxication: a case series and review. J. Emerg. Med. 2013;44(2):360–366. doi: 10.1016/j.jemermed.2012.07.061. [DOI] [PubMed] [Google Scholar]
- 7.Uchiyama N. Identification and quantitation of two cannabimimetic phenylacetylindoles JWH-251 and JWH-250: and four cannabimimetic naphthoylindoles JWH-081, JWH-015, JWH-200, and JWH-073 as designer drugs in illegal products. Forensic Toxicol. 2010;29(1):25–37. [Google Scholar]
- 8.Wohlfarth A. Qualitative confirmation of 9 synthetic cannabinoids and 20 metabolites in human urine using LC-MS/MS and library search. Anal. Chem. 2013;85(7):3730–3738. doi: 10.1021/ac3037365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodrigues W.C. Detection of synthetic cannabinoids in oral fluid using ELISA and LC-MS-MS. J. Anal. Toxicol. 2013;37(8):526–533. doi: 10.1093/jat/bkt067. [DOI] [PubMed] [Google Scholar]
- 10.Barnes A.J. Evaluation of a homogenous enzyme immunoassay for the detection of synthetic cannabinoids in urine. Forensic Sci. Int. 2014;241:27–34. doi: 10.1016/j.forsciint.2014.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes A.J. Validation of an ELISA synthetic cannabinoids urine assay. Ther. Drug Monit. 2015;37(5):661–669. doi: 10.1097/FTD.0000000000000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arntson A. Validation of a novel immunoassay for the detection of synthetic cannabinoids and metabolites in urine specimens. J. Anal. Toxicol. 2013;37(5):284–290. doi: 10.1093/jat/bkt024. [DOI] [PubMed] [Google Scholar]
- 13.Spinelli E. Performance characteristics of an ELISA screening assay for urinary synthetic cannabinoids. Drug Test. Anal. 2015;7(6):467–474. doi: 10.1002/dta.1702. [DOI] [PubMed] [Google Scholar]
- 14.Mohr A.L. Enzyme-linked immunosorbent assay (ELISA) for the detection of use of the synthetic cannabinoid agonists UR-144 and XLR-11 in human urine. J. Anal. Toxicol. 2014;38(7):427–431. doi: 10.1093/jat/bku049. [DOI] [PubMed] [Google Scholar]
- 15.Wang K. The application of lateral flow immunoassay in point of care testing: a review. Nano Biomed. Eng. 2016;8(3):172–183. [Google Scholar]
- 16.Sajid M., Kawde A.-N., Daud M. Designs, formats and applications of lateral flow assay: a literature review. J. Saudi Chem. Soc. 2015;19(6):689–705. [Google Scholar]
- 17.Blažková M., Rauch P., Fukal L. Strip-based immunoassay for rapid detection of thiabendazole. Biosens. Bioelectron. 2010;25(9):2122–2128. doi: 10.1016/j.bios.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Velasco R., Feberero C., Sanz R. α-Lithiated aryl benzyl ethers: inhibition of [1,2]-wittig rearrangement and application to the synthesis of benzo[b]furan derivatives. Org. Lett. 2015;17(18):4416–4419. doi: 10.1021/acs.orglett.5b01964. [DOI] [PubMed] [Google Scholar]
- 19.Willis P.G. Synthesis and structure-activity relationship of a novel series of aminoalkylindoles with potential for imaging the neuronal cannabinoid receptor by positron emission tomography. J. Med. Chem. 2005;48(18):5813–5822. doi: 10.1021/jm0502743. [DOI] [PubMed] [Google Scholar]
- 20.Upender V. The synthesis and biological activity of two analogs of the anti-HIV alkaloid michellamine B. J. Heterocycl. Chem. 1996;33(4):1371–1384. [Google Scholar]
- 21.Cai L. Efficient microwave-assisted cyanation of aryl bromide. Synth. Commun. 2004;34(7):1215–1221. [Google Scholar]
- 22.Ghaffarzadeh M., Bolourtchian M., Halvagar M.M.R. High yield synthesis of aryl cyanides under microwave irradiation. J. Chem. Res. 2003;2003(12):814. [Google Scholar]
- 23.Zhang Q., Botting N.P. The synthesis of [2,3,4-13C3]glycitein. Tetrahedron. 2004;60(52):12211–12216. [Google Scholar]
- 24.Cavill G.W.K., Tetaz J.R. 696. The hydroxynaphthoate series. Part I. A synthesis of 4-hydroxynaphthoic acid and its simple esters. J. Chem. Soc. 1952;0:3634–3636. [Google Scholar]
- 25.Frost J.M. Indol-3-ylcycloalkyl ketones: effects of N1 substituted indole side chain variations on CB2 cannabinoid receptor activity. J. Med. Chem. 2009;53(1):295–315. doi: 10.1021/jm901214q. [DOI] [PubMed] [Google Scholar]
- 26.Guchhait S.K., Kashyap M., Kamble H. ZrCl4-mediated regio- and chemoselective Friedel-Crafts acylation of indole. J. Org. Chem. 2011;76(11):4753–4758. doi: 10.1021/jo200561f. [DOI] [PubMed] [Google Scholar]
- 27.Yatsimirskaya E.A. Preparation of conjugates of progesterone with bovine serum albumin in the reversed micellar medium. Steroids. 1993;58(11):547–550. doi: 10.1016/0039-128x(93)90033-j. [DOI] [PubMed] [Google Scholar]
- 28.Lapcik O. Immunoassay for biochanin a. J. Immunol. Methods. 2004;294(1–2):155–163. doi: 10.1016/j.jim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Holubova-Mickova B. Development of colloidal carbon-based immunochromatographic strip for rapid detection of carbaryl in fruit juices. Eur. Food Res. Technol. 2010;231(3):467–473. [Google Scholar]
- 30.Fojtíková L. Development of enzyme-linked immunosorbent assay for determination of boldenone in dietary supplements. Food Anal. Methods. 2016;9(11):3179–3186. [Google Scholar]
- 31.Goodrow M.H. Strategies for immunoassay hapten design, in immunoanalysis of agrochemicals. Am. Chem. Soc. 1995:119–139. [Google Scholar]
- 32.Marco M.-P., Gee S., Hammock B.D. Immunochemical techniques for environmental analysis II: Antibody production and immunoassay development. TrAC Trends Anal. Chem. 1995;14(8):415–425. [Google Scholar]
- 33.Urusov A.E. Multistage in one touch design with a universal labelling conjugate for high-sensitive lateral flow immunoassays. Biosens. Bioelectron. 2016;86:575–579. doi: 10.1016/j.bios.2016.07.027. (Supplement C) [DOI] [PubMed] [Google Scholar]
- 34.Posthuma-Trumpie G.A., Korf J., van Amerongen A. Lateral flow (immuno) assay: its strengths, weaknesses, opportunities and threats. A literature survey. Anal. Bioanal. Chem. 2009;393(2):569–582. doi: 10.1007/s00216-008-2287-2. [DOI] [PubMed] [Google Scholar]
- 35.Coulter C., Garnier M., Moore C. Synthetic cannabinoids in oral fluid. J. Anal. Toxicol. 2011;35(7):424–430. doi: 10.1093/anatox/35.7.424. [DOI] [PubMed] [Google Scholar]