

CRISPR-Cas9–mediated genome editing can be used to treat ALS in an animal model of the disease.
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal and incurable neurodegenerative disease characterized by the progressive loss of motor neurons in the spinal cord and brain. In particular, autosomal dominant mutations in the superoxide dismutase 1 (SOD1) gene are responsible for ~20% of all familial ALS cases. The clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated (Cas9) genome editing system holds the potential to treat autosomal dominant disorders by facilitating the introduction of frameshift-induced mutations that can disable mutant gene function. We demonstrate that CRISPR-Cas9 can be harnessed to disrupt mutant SOD1 expression in the G93A-SOD1 mouse model of ALS following in vivo delivery using an adeno-associated virus vector. Genome editing reduced mutant SOD1 protein by >2.5-fold in the lumbar and thoracic spinal cord, resulting in improved motor function and reduced muscle atrophy. Crucially, ALS mice treated by CRISPR-mediated genome editing had ~50% more motor neurons at end stage and displayed a ~37% delay in disease onset and a ~25% increase in survival compared to control animals. Thus, this study illustrates the potential for CRISPR-Cas9 to treat SOD1-linked forms of ALS and other central nervous system disorders caused by autosomal dominant mutations.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS; also known as Lou Gehrig’s disease) is an adult-onset neurological disorder (1) that involves the loss of motor neurons in the spinal cord, brainstem, and motor cortex. ALS leads to progressive muscle weakness and atrophy throughout the body, ultimately leading to paralysis and death within 3 to 5 years of symptom onset. There is no cure for ALS, and the only U.S. Food and Drug Administration–approved medications, riluzole and edaravone, are minimally effective, increasing survival by only 2 to 3 months in the case of riluzole (no survival data for edaravone have been reported to date) (2).
Dominant mutations in the Cu-Zn superoxide dismutase 1 (SOD1) (3) gene account for ~20% of familial forms of the disease and ~2% of all cases. The SOD1 gene encodes a metalloenzyme that converts superoxide anions into oxygen and hydrogen peroxide and is thus critical to cellular antioxidant defense. Although the mechanism behind SOD1 toxicity is not completely understood (4), transgenic animals that express mutant forms of the gene develop a progressive neurodegenerative disease that emulates the hallmarks of ALS (5, 6), including motor neuron degeneration, muscle wasting, and paralysis. Reducing mutant SOD1 expression in the spinal cord using antisense oligonucleotides (ASOs) (7, 8) or RNA interference (RNAi) (9–15) can slow disease onset and improve survival in these animal models. However, to date, ASOs have been unable to reduce mutant SOD1 protein in ALS patients (16), and both ASOs and RNAi can be hampered by incomplete knockdown, which limits their effectiveness as therapeutics.
Genome editing offers an alternative approach to treat autosomal dominant disorders, including many familial forms of ALS, via the disruption of mutant gene function. In particular, the RNA-guided Cas9 endonuclease (17) from clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated (Cas) systems has emerged as a versatile genome editing tool (18–21). Cas9 can be targeted to genomic loci to induce a DNA double-strand break (DSB) via RNA-DNA base complementarity using a single-guide RNA (sgRNA). DSBs induced by Cas9 enable the introduction of frameshift-inducing base insertions and/or deletions (indels) that can disrupt gene expression following DNA repair (22). Given these features, we reasoned that CRISPR-Cas9 could be used to treat familial ALS via genome editing following its in vivo delivery into an animal model of the disease using an adeno-associated virus (AAV) vector.
Here, we demonstrate that CRISPR-Cas9 can disrupt mutant SOD1 expression in the spinal cord of the G93A-SOD1 mouse model of ALS. Systemic administration of an AAV9 vector encoding the Cas9 nuclease with an sgRNA targeting the human SOD1G93A gene to neonatal G93A-SOD1 mice reduced mutant SOD1 protein in the spinal cord and enhanced the survival of motor neurons. This therapeutic genome editing strategy delayed disease onset, improved motor function, reduced muscle atrophy, and, critically, extended survival in ALS mice. These results thus establish that CRISPR-mediated genome editing can be used to treat familial ALS and potentially other central nervous system (CNS) disorders caused by autosomal dominant mutations.
RESULTS
Disruption of mutant SOD1 expression using CRISPR-Cas9
We used the Cas9 nuclease from Staphylococcus aureus (SaCas9) (23) to target the mutant SOD1 gene in the G93A-SOD1 mouse model of ALS (5), which carries ~25 tandem repeat copies of the hSOD1G93A transgene and recapitulates many aspects of the disease, including progressive muscle atrophy and impaired motor function. Unlike the larger Cas9 nuclease from Streptococcus pyogenes (17–21), SaCas9, along with its sgRNA and a full-length cytomegalovirus (CMV) promoter, can fit into a single AAV particle to drive expression in vivo (Fig. 1A). More than 100 mutations in SOD1 have been identified in patients with familial ALS. Because of this heterogeneity, we designed several sgRNAs that do not overlap with the G93A mutation and could be applicable to other ALS-linked SOD1 variants, including wild-type SOD1, which has been associated with sporadic ALS (24). Notably, no adverse effects were observed in a clinical trial for ALS using an ASO that targets both mutant and wild-type SOD1 (16).
Fig. 1. In vivo genome editing reduces mutant SOD1 expression in G93A-SOD1 mice.
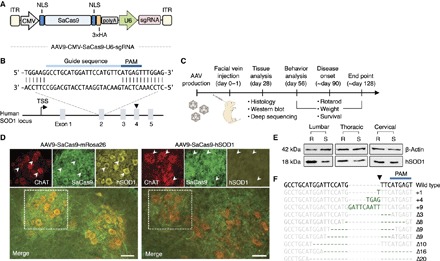
(A) AAV vector schematic. ITR, inverted terminal repeat; NLS, nuclear localization signal sequence; 3×HA, three tandem repeats of the human influenza hemagglutinin (HA) epitope tag. (B) Schematic representation of the human SOD1 locus and the sgRNA target site. The arrowhead depicts approximate position of the G93A mutation. TSS, transcriptional start site; PAM, protospacer-adjacent motif. (C) Experimental timeline for in vivo studies. (D) Immunofluorescent staining of lumbar spinal cord sections and (E) Western blot of lumbar, thoracic, and cervical spinal cord lysate 4 weeks after G93A-SOD1 mice were injected with AAV9-SaCas9-hSOD1 (S; n = 3) and AAV9-SaCas9-mRosa26 (R; n = 3) via facial vein (quantitation of Western blot results in fig. S7). Arrowheads indicate ChAT+ and SaCas9+ cells with (upward) high or (downward) low hSOD1 expression. Images were captured using identical exposure conditions. Scale bar, 50 μm. (F) Indels from whole spinal cord tissue 4 weeks after G93A-SOD1 mice were injected with AAV9-SaCas9-hSOD1 (n = 3) via facial vein. Indels are colored dark green. Wild-type sequences are colored gray. The arrowhead indicates predicted SaCas9 cleavage site (D to F). All injections were performed at P0-P1.
We evaluated the ability of SaCas9 and the designed sgRNA to disrupt the mutant SOD1 expression in mouse neuroblastoma–spinal cord–34 (NSC-34) cells stably transfected with complementary DNA (cDNA) encoding hSOD1G93A. NSC-34 cells are frequently used to study certain aspects of ALS in vitro because they share several morphological and physiological similarities with motor neurons (25). The most efficient sgRNA targeted exon 2 of the hSOD1G93A coding sequence (Fig. 1B and fig. S1) and reduced mutant protein by ~92% in SaCas9-expressing cells, which we enriched using fluorescence-activated cell sorting (FACS) following cotransfection with an enhanced green fluorescent protein (EGFP)–encoding surrogate reporter plasmid (fig. S2A). Sanger sequencing revealed the presence of indels in ~94% of hSOD1G93A transgenes analyzed from these cells (fig. S2B), confirming that mutant SOD1 expression was disrupted by SaCas9-mediated genome editing. Despite the potential for off-target (OT) disruption of the mouse SOD1 gene, we observed no significant difference (P > 0.5) in mouse SOD1 protein in NSC-34 cells expressing SaCas9 with an sgRNA targeting either the hSOD1 gene or the mouse Rosa26 locus, a genomic “safe harbor site” often used for mouse transgenesis (fig. S2A). These results indicate that SaCas9 and its sgRNA discriminate between human and mouse alleles.
In vivo genome editing in ALS mice
We next evaluated whether CRISPR-Cas9 could reduce mutant SOD1 expression in vivo following delivery to G93A-SOD1 mice using an AAV vector. AAV is a promising in vivo gene delivery vehicle found to be safe and effective in an increasing number of clinical trials, including those for hemophilia B (26), choroideremia (27), Leber’s congenital amaurosis type II (28), and lipoprotein lipase deficiency (29, 30). When administrated systemically to neonatal mice, AAV9 (31) can cross the blood-brain barrier and, within the spinal cord, transduce motor neurons, sensory fibers, and, to a lesser extent, astrocytes (32, 33). Both motor neurons (determinants of onset and early disease progression) (34) and astrocytes (secrete neurotoxic factors that can selectively kill motor neurons) (35–40) play an important role in SOD1-linked ALS. In light of this pathology, and considering a recent report highlighting the benefits of early gene therapy intervention in G93A-SOD1 mice (12), we used neonatal AAV9 delivery for this proof-of-concept study.
We injected G93A-SOD1 mice with AAV9 encoding SaCas9 and an sgRNA targeting either the hSOD1 gene (AAV9-SaCas9-hSOD1) or the mRosa26 locus (AAV9-SaCas9-mRosa26) via the facial vein at postnatal days 0 and 1 (P0-P1) (Fig. 1C and fig. S3). We used a double tyrosine mutant of AAV9 that we (41) and others (42) previously developed to enhance gene delivery to the CNS. Spinal cord sections were analyzed by immunohistochemistry for expression of (i) the motor neuron marker choline acetyltransferase (ChAT), (ii) SaCas9 via a genetically fused HA epitope, and (iii) mutant SOD1, which we detected using an antibody that preferentially recognizes human protein (fig. S4). We observed SaCas9 expression primarily in the ventral horn of the spinal cord (fig. S5) and found that ~74% of all ChAT+ cells examined throughout the anterior gray column expressed SaCas9, indicating broad motor neuron transduction. We also observed SaCas9 expression in fibers in the ventral horn, which we detected using the neurite marker β3-tubulin (fig. S6). As indicated by immunostaining for the astrocytic marker glial fibrillary acidic protein (GFAP), we observed little SaCas9 expression in gray and white matter astrocytes (fig. S7), potentially due to limited transduction by the AAV vector and/or suboptimal expression from the CMV promoter.
Compared to control animals, mutant SOD1 was reduced in transduced spinal cord cells in G93A-SOD1 mice infused with AAV9-SaCas9-hSOD1 (Fig. 1D). Western blot analysis of spinal cord lysate indicated that mutant SOD1 protein was decreased in CRISPR-treated mice by ~3-fold (P = 0.001) and ~2.5-fold (P < 0.05) in the lumbar and thoracic regions, respectively (Fig. 1E and fig. S8). Consistent with in vitro studies in NSC-34 cells (fig. S2), no significant difference (P > 0.5) in mouse SOD1 protein was observed in the spinal cord lysate from treated versus untreated animals (fig. S9). Intriguingly, despite its efficient transduction, we also observed no significant difference (P > 0.5) in mutant SOD1 protein in the cervical spinal cord of gene-edited G93A-SOD1 mice (fig. S8), potentially because of variability among treated animals.
To evaluate indel formation in vivo, we deep-sequenced hSOD1G93A transgenes amplified from dissected spinal cord tissue, which included transduced motor neurons, as well as nontransduced nerve and glial cells from the white and gray matter. These latter cell populations, which are much more numerous than motor neurons, were not expected to be gene-modified on the basis of our immunohistochemistry results. According to CRISPResso (a software pipeline that analyzes genome editing outcomes from deep sequencing data) (43), indels were present in ~0.2 and ~0.4% of sequenced hSOD1G93A transgenes from the lumbar and thoracic spinal cord of CRISPR-treated mice, respectively, corresponding to a ~7-fold (P = 0.01) and ~14-fold (P < 0.05) increase over control animals (Fig. 1F). Consistent with Western blot results showing no difference in mutant SOD1 protein in the cervical spinal cord of gene-edited mice (Fig. 1E and fig. S8), we also observed no significant increase in indel formation in cervical spinal cord samples from CRISPR-modified animals (0.05% modification frequency; P > 0.05 compared to controls).
We next investigated whether SaCas9 induced OT modifications in vivo. Using Cas-OFFinder (44), we identified 12 potential OT sites in the mouse genome that differed from the on-target SaCas9 cleavage site by up to four mismatches (fig. S10A). Deep sequencing revealed no significant increase in indel formation at each candidate OT site in CRISPR-treated mice compared to control animals (P > 0.1 for all) (fig. S10B).
Therapeutic benefits of CRISPR-mediated gene editing in a mouse model of ALS
To test whether in vivo disruption of mutant SOD1 by CRISPR-Cas9 provides therapeutic benefit, we monitored motor function, weight loss, and atrophy in G93A-SOD1 mice injected with AAV9-SaCas9-hSOD1, AAV9-SaCas9-mRosa26, or AAV9-EGFP at P0-P1. Compared to control animals, disease onset (that is, peak weight) in mice infused with AAV9-SaCas9-hSOD1 was delayed by 33 days (hSOD1, 126 ± 5.7 days; mRosa26, 93 ± 9.6; EGFP, 92 ± 8.1; uninjected, 92 ± 8.8; P < 0.0001) (Fig. 2A) and ranged from 119 to 133 days in treated animals compared to 77 to 98 days in control mice (P = 0.0001) (Fig. 2B). Mean survival also increased by 28 to 30 days in treated mice (hSOD1, 152.4 ± 7.9 days; mRosa26, 122.8 ± 6.1; EGFP, 124 ± 3.8; uninjected, 125 ± 3.5; P < 0.0001) (Fig. 2C) and ranged from 142 to 167 days in treated animals compared to 114 to 136 days in control animals (P < 0.0001) (Fig. 2D). Compared to age-matched controls, animals treated by CRISPR-Cas9 displayed improved motor function (P < 0.0001) based on rotarod performance (Fig. 2E) and maintained and/or gained weight (P < 0.0001) for 28 to 35 days beyond the expected point of disease onset (Fig. 2F). Treated mice also exhibited reduced muscular atrophy, as evidenced by a slower rate of weight loss after disease onset [hSOD1, −0.42 ± 0.015 weight (%) per day; mRosa26, −0.67 ± 0.05; EGFP, −0.63 ± 0.067; P < 0.005] (fig. S11). Notably, following the eventual onset of disease, we observed no slowing in disease progression (that is, the length of time between disease onset and end point) (fig. S12), likely due to insufficient gene editing in astrocytes, which contribute to disease progression in SOD1-linked forms of ALS (34, 35, 45–47).
Fig. 2. In vivo genome editing provides therapeutic benefit to G93A-SOD1 mice.
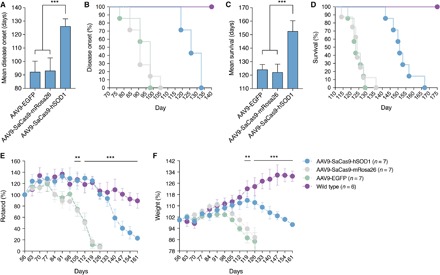
(A and B) Disease onset, (C and D) survival, (E) rotarod performance, and (F) weight of G93A-SOD1 mice injected with AAV9-SaCas9-hSOD1 (n = 7), AAV9-SaCas9-mRosa26 (n = 7), or AAV9-EGFP (n = 7) via facial vein at P0-P1. (E and F) Mean rotarod times and weights were normalized to average 56-day values for each group. Wild type (n = 6) indicates litter-matching control mice. Values are means and error bars indicate (A to C) SD and (E and F) SEM. ***P < 0.0005; **P < 0.005; (A, C) one-way or (E and F) two-way analysis of variance (ANOVA) followed by Tukey’s post hoc analysis.
Compared to control animals, immunofluorescent staining of spinal cord sections from end-stage mice revealed that animals treated by CRISPR-Cas9 had ~50% more ChAT+ neurons in the lumbar (P < 0.05) and thoracic (P < 0.001) regions of the spinal cord (Fig. 3). This suggests that CRISPR-Cas9 conferred protection to some individual motor neurons. In addition, consistent with our earlier findings (fig. S7), we observed limited SaCas9 expression in GFAP+ astrocytes in end-stage spinal cord sections from CRISPR-treated animals (fig. S13), as well as immunoreactive mutant SOD1 inclusion bodies in many of the same cells (fig. S14). Optimization of AAV-mediated gene delivery and/or SaCas9 expression in astrocytes (35, 40), microglia (34, 46, 48), and oligodendrocyte precursors (49, 50) may further enhance efficacy. Collectively, these results demonstrate that CRISPR-Cas9–mediated disruption of mutant SOD1 expression in G93A-SOD1 mice enhances the survival of spinal cord motor neurons and improves motor function and life span.
Fig. 3. G93A-SOD1 mice treated by genome editing have more ChAT+ cells at end stage compared to control mice.
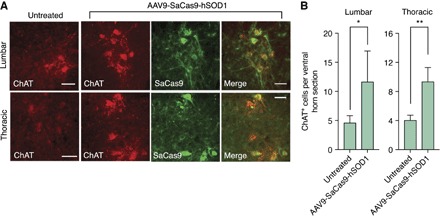
(A) Immunofluorescent staining of end-stage (top) lumbar or (bottom) thoracic spinal cord sections after G93A-SOD1 mice were injected with AAV9-SaCas9-hSOD1 or left untreated. (B) Mean number of ChAT+ neurons per end-stage (left) lumbar or (right) thoracic spinal cord hemisection after G93A-SOD1 mice were injected with AAV9-SaCas9-hSOD1 or left untreated (n = 4). Scale bars, 50 μm. Error bars indicate SD. *P < 0.05; **P < 0.001; two-tailed unpaired t test.
DISCUSSION
Genome editing technologies can be used to introduce precise genomic modifications into mammalian cells and model organisms (51) and thus hold tremendous potential for treating the genetic causes of many diseases. Here, we have shown that CRISPR-Cas9 can reduce mutant SOD1 protein in the spinal cord following systemic delivery using an AAV vector. This therapeutic genome editing strategy delayed disease onset, improved motor function, and, critically, increased survival, illustrating the utility of genome editing to treat SOD1-linked ALS and potentially other CNS disorders caused by autosomal dominant mutations, such as Huntington’s disease or the spinocerebellar ataxias. These results build on basic research studies by our laboratory (52) and others (53, 54) showing that CRISPR systems can induce gene editing in the mammalian brain and demonstrates that therapeutic genome editing can be achieved in tissues beyond the liver (55–58) and muscle (59–62) in animal models of human disease.
Both motor neurons (determinants of onset and early disease progression) (34) and astrocytes (secrete factors that selectively kill motor neurons) (35–40) play an important role in SOD1-ALS. Because of its innate capacity to cross the blood-brain barrier and transduce spinal cord motor neurons and, to a lesser extent, astrocytes, we used AAV9 to deliver our CRISPR gene editing system to G93A-SOD1 mice at P0-P1. Because neonatal delivery has proven effective for evaluating RNAi-based gene therapies in G93A-SOD1 mice (10, 12, 14), we reasoned that this strategy would enable us to assess the impact of genome editing on disease onset and progression. Notably, AAV administration to neonatal mice previously facilitated the validation of zinc-finger nuclease– and CRISPR-based strategies for correcting hemophilia B (55) and ornithine transcarbamylase deficiency (57), respectively. In addition, a phase 1 clinical trial based on AAV9-mediated delivery of the survival of motor neuron 1 (SMN1) gene for spinal muscular atrophy (63, 64) in infants is under way, underscoring the therapeutic potential of this serotype.
Immunohistochemistry analysis of spinal cord sections from CRISPR-treated mice revealed that SaCas9 expression, as well as mutant SOD1 gene disruption, was confined primarily to ChAT+ motor neurons. This finding was reinforced by end-stage histological analysis, which showed limited SaCas9 labeling in GFAP+ astrocytes and the presence of presumably toxic mutant SOD1 inclusion bodies in many of the same cells. These results collectively support the hypothesis that inefficient SOD1 disruption in astrocytes could contribute to the lack of slowing in disease progression that we observed in gene-edited mice. Advancing this first-in-class CRISPR-based gene therapy for a CNS disorder toward the clinic will therefore require optimizing gene delivery vehicles for human use (65) and enhancing Cas9 expression in motor neurons, astrocytes, and other cell types implicated in SOD1-ALS. The rapidly expanding catalog of Cas9 orthologs (several of which are smaller than SaCas9) (66, 67) may also enable the use of specialized and/or larger promoter sequences incompatible with the all-in-one AAV vector strategy used here, or additional components that can drive self-inactivation of Cas9, and thereby enhance safety.
Because of its rapid and robust phenotype, the G93A-SOD1 mouse model of ALS is among the most widely used transgenic models of the disease. This mouse carries ~25 tandem repeat copies of the hSOD1G93A transgene, and previous reports have indicated that a reduction in hSOD1G93A copy number can have a profound effect on the ALS phenotype (68). Despite the challenge that high-copy repetitive elements can pose from the perspective of genome editing, the Cas9 nuclease has facilitated the disruption of up to 62 copies of the porcine endogenous retrovirus in a kidney epithelial cell line (69), indicating its capacity for multiplexing and targeting high-copy elements. Although it is difficult to determine the number of gene-edited hSOD1G93A transgenes in transduced spinal cord cells, we showed that SaCas9 modified ~94% of hSOD1G93A alleles in transfected NSC-34 cells.
The CRISPR-based genome editing strategy used here cannot distinguish between mutant and the wild-type human SOD1, which performs important functions in cells. However, in a phase 1 clinical trial, no adverse effects were observed in ALS patients treated with an ASO targeting both mutant and wild-type SOD1 (16). If needed, an allele-specific CRISPR system configured to target disease-causing SOD1 mutations (including A4V, G37R, H46R, G85R, and G93A) or a gene knockout-and-replace therapy (70) could be used to overcome the toxicity arising from a lack of allele specificity. Because the PAM sequence recognized by SaCas9 (that is, NNGRRT) occurs less frequently than those for the more commonly used Cas9 from S. pyogenes (that is, NGG), it may be necessary to alter the PAM specificity of SaCas9 (71) to discriminate between the mutant and wild-type SOD1 alleles.
We used the Cas-OFFinder algorithm (44) to identify candidate OT cleavage sites for the SaCas9 nuclease. This software is not limited by the number of mismatches in the sgRNA sequence and allows for subtle variations in the PAM recognized by Cas9. Using deep sequencing, we observed no increase in indel formation at each candidate OT site in CRISPR-treated mice versus control animals. However, the use of unbiased genome-wide approaches that rely on the in situ capture of adapter sequences at nuclease-induced DSBs (23, 72) or cell-free digestion of genomic DNA using Cas9 ribonucleoprotein (73) could facilitate the formation of a comprehensive portrait of SaCas9 cleavage specificity. In addition, whole-exome genome sequencing of CRISPR-treated motor neurons differentiated from human-induced pluripotent stem cells derived from SOD1-ALS patients would yield critical insight into the specificity of SaCas9 in a more therapeutically relevant context. Given that the OT activity of a nuclease is proportional to its concentration and the amount of time it is exposed to the cell (74), future work elucidating the kinetics of Cas9 expression in vivo could shed light on its function and inform methods for improving its specificity.
Finally, there are several limitations to this study that should be discussed. First, we observed a ~2.5-fold reduction in mutant SOD1 protein in the lumbar and thoracic spinal cord but detected indels in only 0.2 to 0.4% of sequenced hSOD1G93A transgenes. Although the underlying reason for this difference requires further exploration, studies have demonstrated that genome editing can elicit a phenotypic effect that exceeds the measured indel frequency (23, 75). In our case, this difference could potentially be explained by CRISPR interference (76), which may function alongside SaCas9-mediated genome editing to reduce mutant SOD1 protein. Furthermore, we deep-sequenced hSOD1G93A transgenes amplified from whole spinal cord tissue, which included both transduced motor neurons and nontransduced cells from the white and gray matter, the latter of which are more numerous than motor neurons and may not express as much mutant SOD1. The Allen Spinal Cord Atlas online database indicates that, in P56 mice, SOD1 is more strongly expressed in the anterior horn of the spinal cord, where lower motor neurons reside, as well as the posterior horn. This is consistent with our own immunostaining in 28-day-old G93A-SOD1 mice (fig. S4). Thus, higher SOD1 gene expression in transduced versus nontransduced cells at the time of analysis (that is, 28 days) could possibly contribute to the observed discrepancy. Second, because of the difficulty in isolating spinal cord motor neurons from juvenile and adult mice (77), we could not measure the frequency of hSOD1G93A gene modification in transduced cells. Future work using transgenic mice expressing a GFP reporter in motor neurons (78) could facilitate analysis of indel formation or SOD1 messenger RNA following FACS enrichment. Third, we observed variable and inefficient editing in the cervical spinal cord of G93A-SOD1 mice. Further optimization of SaCas9 expression in preclinical large animal models using increased samples sizes can help resolve this point. Finally, we did not administer the AAV vector to adult animals in this proof-of-concept study. Assessing the efficacy of CRISPR-mediated disruption of mutant SOD1 in adult ALS mice both before and after disease onset will be critical in establishing the potential of this approach for clinical translation.
In conclusion, we have demonstrated that CRISPR-Cas9–mediated gene editing provides therapeutic benefit to the G93A-SOD1 mouse model of ALS. This work establishes genome editing as a possible therapy for ALS and paves the way for this technology to treat other forms of the disease, including those caused by a hexanucleotide repeat expansion in the C9orf72 gene (79, 80), which could potentially be excised by Cas9 following its coexpression with a pair of sgRNA flanking the repeat expansion.
MATERIALS AND METHODS
Study design
The objective of this study was to determine whether genome editing could be used to treat ALS. We theorized that CRISPR-Cas9–mediated disruption of the hSOD1G93A transgene in the G93A-SOD1 mouse model of ALS could slow or halt disease onset and progression, as well as provide therapeutic benefit. We used the Surveyor nuclease assay and Western blot to identify the sgRNA that could mediate disruption of hSOD1G93A in vitro. For in vivo studies, G93A-SOD1 mice were injected with the AAV vector via the facial vein at P0-P1. All treatments consisted of a single vector administration. The mice were scarified 4 weeks after injection. Mutant SOD1 protein was measured via immunohistochemistry and Western blot, and indels were evaluated using deep sequencing. For behavior studies, treatment groups were gender-balanced, and the animals were monitored daily with weight and rotarod performance measured weekly. End stage was determined as described in the “Behavior” section. No blinding was used to perform measurements. The expected effect and SD for each experiment were informed from published literature. From this information, sample sizes were determined by power calculations using α = 0.05 and β = 0.80. All animals were included in the statistical analysis, and the sample size reflects the number of independent biological replicates. Statistical methods are described in the “Statistical analysis” section.
Plasmid construction
The human Cu-Zn SOD1 gene (Entrez Gene ID, 6647) was searched for SaCas9 cleavage sites (23) using the motif 5′-G-(N)21–24-NNGRRT-3′ (where N is A, T, C, or G and R is A or G). A “G” nucleotide was used at the 5′ end of the sgRNA to ensure efficient expression from the U6 promoter. Oligonucleotides encoding sgRNA-targeting sequences were custom-synthesized (Elim Biopharm) and phosphorylated by T4 polynucleotide kinase (New England Biolabs) for 30 min at 37°C. Oligonucleotides were then annealed for 5 min at 95°C, fast cooled on ice for 10 min, and ligated into the Bsa I restriction site of pAAV-CMV-SaCas9-U6-sgRNA (Addgene, #61591) (23). Correct insertion of each sgRNA was verified by DNA sequencing. The sequences of oligonucleotides used in this study are shown in table S1.
Cell culture
Human embryonic kidney (HEK) 293T cells [University of California (UC) Berkeley Tissue Culture Facility] and mouse NSC-34 cells (Cedarlane Laboratories) (25) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Corning) supplemented with 10% (v/v) fetal bovine serum (FBS; Life Technologies) and 1% (v/v) Antibiotic-Antimycotic (Anti-Anti; Life Technologies) in a humidified 5% CO2 atmosphere at 37°C. The NSC-G93A-SOD1 cell line was generated by stable transfection of NSC-34 cells with linearized pF155-pcDNA-SOD1-G93A plasmid (Addgene, #26401) (81) followed by selection with G418 (400 μg/ml; Sigma-Aldrich). Single G418-resistant clones were isolated by limiting dilution, and hSOD1G93A expression was verified by Western blot. NSC-34–G93A–SOD1 cells were maintained in DMEM with 10% (v/v) FBS, 1% (v/v) Anti-Anti, and G418 (400 μg/ml).
Surveyor nuclease assay
NSC-34–G93A–SOD1 cells were seeded onto 24-well plates at a density of 3 × 105 cells per well. At 24 hours after seeding, the cells were transfected with 800 ng of pAAV-CMV-SaCas9-U6-sgRNA using Lipofectamine 3000 (Life Technologies) according to the manufacturer’s instructions. At 72 hours after transfection, the cells were harvested, and genomic DNA was isolated using QuickExtract DNA Extraction Solution (Epicentre). The hSOD1G93A coding sequence was amplified by nested polymerase chain reaction (PCR) using the Expand High Fidelity Taq System (Roche) with the following primers: pcDNA-SOD1-Fwd and BGH-Rev (external) and CMV-Fwd and pcDNA-SOD1-Rev (internal) (table S1). Following PCR amplification, the Surveyor Mutation Detection Kit (Integrated DNA Technologies) was used according to the manufacturer’s instructions. Cleavage products were visualized by nondenaturing tris-borate EDTA–polyacrylamide gel electrophoresis (PAGE), and the frequency of gene modification was determined by measuring the ratio of cleaved to uncleaved substrate, as described by Guschin et al. (82). Band intensity was quantitated using Image Lab software (Bio-Rad).
Sanger sequencing
NSC-34–G93A–SOD1 cells were seeded onto 12-well plates at a density of 6 × 105 cells per well. At 24 hours after seeding, the cells were transfected with 1.4 μg of pAAV-CMV-SaCas9-U6-sgRNA and 200 ng of pAAV-CAG-EGFP using Lipofectamine 3000. At 72 hours after transfection, EGFP+ NSC-34–G93A–SOD1 cells were isolated by FACS (BD Bioscience Influx Sorter; UC Berkeley Flow Cytometry Core Facility), and genomic DNA was purified as described above. The hSOD1G93A transgene was then PCR-amplified using the primers hSOD1-EcoRI-Fwd and BGH-Rev (table S1) and cloned into the Eco RI and Xho I restriction sites of pcDNA 3.1 (Thermo Fisher Scientific). Individual transformed colonies were mini-prepped and sequenced using the primer CMV-Fwd.
Western blot
G93A-SOD1 mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) and transcardially perfused with saline. Dissected and homogenized spinal cord tissue, as well as cultured NSC-34–G93A–SOD1 cells, were lysed by radioimmunoprecipitation assay buffer [10 mM tris-HCl, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate (pH 8.0)], and protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Then, 15 μg of protein was electrophoresed by SDS-PAGE and electrophoretically transferred onto a nitrocellulose membrane in transfer buffer [20 mM tris-HCl, 150 mM glycine, and 20% (v/v) methanol] for 1 hour at 160 V. Membranes were blocked with 5% (v/v) Blotting-Grade Blocker (Bio-Rad) in tris-buffered saline (TBS) [20 mM tris-HCl, 150 mM NaCl, and 0.1% (pH 7.5)] with 0.05% Tween 20 (TBST) and incubated overnight with primary antibodies in blocking solution. The following primary antibodies were used: rabbit anti-hSOD1 (1:2000; Cell Signaling Technology, 2770S), rabbit anti-m/hSOD1 (1:1000; Thermo Fisher Scientific, PA5-27240), rabbit anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:5000; Abcam, EPR16891), and rabbit anti–β-actin (1:1000; Cell Signaling Technology, 4970S). The membranes were washed three times with TBST and incubated with goat anti-rabbit secondary antibody horseradish peroxidase conjugate (1:5000, Thermo Fisher Scientific, 65-6120) in blocking solution for 1 hour at room temperature. After three washes with TBST, the blot was developed using SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific) and visualized by automated chemiluminescence using the Gel Doc XR Imaging System (Bio-Rad). Band intensity was quantitated using Image Lab software (Bio-Rad). Total mutant SOD1 protein was normalized to β-actin or GAPDH control protein in each lane.
AAV vector production
AAV vector was produced, as previously described (83). Briefly, HEK293T cells were seeded onto 15-cm plates at a density of 3 × 107 cells per plate in a serum-containing medium. At 24 hours after seeding or once cells were 90% confluent, the cells were transfected with 15 μg of pAAV-CMV-SaCas9-U6-sgRNA or pAAV-CMV-EGFP, 15 μg of pAAV9-2YF (41) [encoding the double tyrosine–to–phenylalanine mutant of the AAV9 capsid (42)], and 15 μg of pHelper using 145 μl of polyethylamine (1 μg/μl). At 48 hours after transfection, the cells were harvested by manual dissociation using a cell scraper and centrifuged at 4000g for 5 min at room temperature. The medium was removed, and the cells were resuspended in 2 ml of lysis buffer [50 mM tris-HCl and 150 mM NaCl (pH 8.0)] per 15-cm plate. The cells were freeze-thawed three times using a dry ice–ethanol bath and a 37°C water bath. Cell lysate was then incubated with 10 units of Benzonase (Sigma-Aldrich) per milliliter of cell lysate for 30 min at 37°C and centrifuged at 10,000g for 10 min at room temperature. Supernatant was then overlaid onto an iodixanol density gradient, and virus was purified by ultracentrifugation (84). AAV was washed three times with 15 ml of phosphate-buffered saline (PBS) with 0.001% Tween 20 using an Ultra-15 Centrifugal Filter Unit (Amicon) at 4000g and concentrated to ~150 μl. Virus was stored at 4°C, and the viral genomic titer was determined by quantitative real-time PCR using SYBR Green (Sigma-Aldrich) with the primers qPCR-CMV-Fwd and qPCR-CMV-Rev (table S1).
Injections
All animal procedures were approved by the Office of Laboratory Animal Care at the University of California at Berkeley and conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Eight-week-old male G93A-SOD1 mice (5) [B6SJL-Tg(SOD1*G93A)1Gur/J; The Jackson Laboratory, stock #002726] were bred with female B6SJLF1/J mice (The Jackson Laboratory, stock #100012). At 20 to 22 days after initiating breeding, P0-P1 pups were intravenously injected via the facial vein with ~4 × 1011 vector genomes of AAV9-CMV-SaCas9-U6-sgRNA or AAV9-CMV-EGFP in 40 μl of PBS with 0.001% Tween 20. Before injection, the animals were genotyped for the presence of the hSOD1G93A transgene by PCR using genomic DNA purified from a tail clip with the primers hSOD1-Tg-Fwd and hSOD1-Tg-Rev (table S1).
Behavior
Eight weeks after injections, treated and control G93A-SOD1 mice were weighed weekly and monitored for changes in body mass three times a week. Treatment groups were gender-balanced (AAV9-EGFP, male = 4 and female = 3; AAV9-SaCas9-mRosa26, male = 3 and female = 4; and AAV9-SaCas9-hSOD1, male = 4 and female = 3), and measurements were not performed in a blinded manner. Disease onset was retrospectively defined as the age at which animals reached peak weight. Motor coordination was measured once a week using a Rotamex-5 rotarod (Columbus Instruments International). The animals were placed onto the apparatus, and the latency to fall (measured in seconds) was recorded for each mouse. Each session consisted of three trials on a rotarod programmed to accelerate from 4 to 40 rpm in 180 s. All data were normalized to both starting body weight and starting rotarod time and determined at 8 weeks. Disease end stage was determined as the point when the animals could no longer turn themselves over within 10 s of being placed on their back or after full paralysis of the hindlimbs. The mice were provided with wet mashed food in their cages at the first sign of hindlimb paralysis and were henceforth monitored daily.
Immunofluorescent staining
Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. Spinal cords were postfixed in 4% paraformaldehyde overnight at 4°C and stored in 30% sucrose. The spinal cords were then harvested and cut into 40-μm coronal sections using a Microtome HM 500 cryostat. The sections were transferred to a 96-well plate and stored in cryoprotectant at −20°C. The sections were washed three times with PBS, incubated with blocking solution [PBS with 10% (v/v) donkey serum (Sigma-Aldrich) and 1% Triton X-100] for 2 hours at room temperature and stained with primary antibodies in blocking solution for 72 hours at 4°C. The stained sections were then washed three times with PBS and incubated with secondary antibodies for 2 hours at room temperature followed by a 10-min incubation with 4′,6-diamidino-2-phenylindole nuclear stain (Thermo Fisher Scientific). The sections were washed three times with PBS, mounted onto slides using VECTASHIELD Hard Set Antifade Mounting Medium (Vector Laboratories), and visualized using a Zeiss Axio Scan.Z1 and a Zeiss LSM 880 NLO Axio Examiner with optical parametric oscillator (OPO) (UC Berkeley Molecular Imaging Center). Image analysis was performed using ImageJ software (http://imagej.nih.gov/ij/).
The following primary antibodies were used for spinal cord sections: rabbit anti-hSOD1 (1:200; Cell Signaling Technology, 2770S), goat anti-ChAT (1:50; EMD Millipore; AB144P), mouse anti-HA (1:500; Abcam, ab18181), goat anti-HA (1:250; GenScript, A00168), mouse anti–β3-tubulin (1:1000; Sigma-Aldrich, T8578), chicken anti-GFAP (1:1000; Abcam, ab4674), and mouse anti-GFP (1:100, Abcam, ab1218). The following secondary antibodies were used: donkey anti-rabbit Cy3 (Jackson ImmunoResearch, 711-165-152), donkey anti-goat Alexa Fluor 647 (Thermo Fisher Scientific, A-21447), donkey anti-goat Alexa Fluor 488 (Thermo Fisher Scientific, A-11055), donkey anti-mouse Alexa Fluor 555 (Thermo Fisher Scientific, A-31570), donkey anti-chicken Alexa Fluor 647 (Jackson ImmunoResearch, 703-605-155), and donkey anti-mouse Alexa Fluor 488 (Jackson ImmunoResearch, 715-545-150).
Deep sequencing
Candidate OT cleavage sites were identified using Cas-OFFinder (44). Consistent with past reports (57, 59–61), the mouse reference genome (mm10) was searched for sequences with up to four total mismatches (up to two nucleotide mismatches and up to two DNA or sgRNA bulges) from the on-target site in the hSOD1G93A transgene. The 12 OT sites with the highest degree of sequence similarity to the 3′ end of the sgRNA were chosen for analysis. Genomic DNA was purified from whole spinal cord tissue from G93A-SOD1 mice injected with AAV9-SaCas9-hSOD1 or AAV9-EGFP using the DNeasy Blood & Tissue Kit (Qiagen). Each candidate OT site was amplified by PCR (table S2) using Phusion High-Fidelity DNA polymerase (New England Biolabs). Single-read Illumina flow cell–binding sequences and target site–specific barcodes (table S3) were incorporated during a second round of PCR. PCR products were then gel-purified using the PureLink Quick Gel Extraction and PCR Purification Combo Kit (Thermo Fisher Scientific). Barcoded amplicons were pooled together and sequenced using the MiSeq System (Illumina) with TruSeq SBS Kit v3-HS (Illumina) (QB3 Vincent J. Coates Genomics Sequencing Laboratory). Multiplexed samples were deconvoluted on the basis of unique barcodes, and adapter sequences were trimmed from the reads. Indel quantitation was performed using CRISPResso (43). Sequences were filtered for >99% confidence (phred33 ≥ 20) per read. Sequence alignment was performed on filtered reads using EMBOSS Needle using the default CRISPResso settings. Indels were measured within a 5–base pair window surrounding the predicted SaCas9 cleavage site to minimize false-positive classification.
Statistical analysis
Statistical analysis was performed using Prism 7 (GraphPad Software). Mutant SOD1 protein was compared using two-tailed paired t test. Disease onset and survival were compared using one-way ANOVA followed by Tukey’s post hoc analysis. Rotarod times and weight loss were compared using two-way ANOVA followed by Tukey’s post hoc analysis. Kaplan-Meier plots were analyzed using the log-rank test. Motor neuron survival and deep sequencing data were compared using two-tailed unpaired t test. All analyses were considered statistically significant at P < 0.05.
Supplementary Material
Acknowledgments
We thank A. Dillin for providing the rotarod, J. G. Flannery for providing the space for injections and behavioral studies, G. M. C. Rodrigues and M. M. Adil for the helpful discussion and technical assistance, and S. J. Sirk for the critical reading of the manuscript. Funding: T.G. was supported by a Ruth L. Kirschstein National Research Service Award (NRSA) (F32GM113446). D.S.O. was supported by an NSF Graduate Research Fellowship and a UC Berkeley Dissertation-Year Fellowship. L.C.B. was supported by a Ruth L. Kirschstein NRSA (F32EY023891). D.V.S. was supported by the NIH (R01EY022975) and a gift from D. Chan. Author contributions: T.G. and D.V.S. conceived the study; T.G. designed the experiments, designed the constructs, performed the molecular biology and cell culture experiments, and analyzed data; T.G. and F.K.E. packaged AAV vectors and performed behavior studies; L.C.B. and T.G. genotyped animals and L.C.B. performed injections; D.S.O. performed sectioning and immunofluorescent staining; P.L. performed deep sequencing; and T.G. and D.V.S. wrote the manuscript with contributions from all authors. Competing interests: D.V.S. is an inventor on patents related to AAV vectors and cofounder of 4D Molecular Therapeutics, a company focused on the clinical development of gene therapies for recessive diseases using engineered AAV variants. D.V.S. is also a member of the board of directors of uniQure, a company focused on clinical AAV gene therapy. D.S.O. is now an employee of Sangamo Therapeutics, and P.L. is now an employee of IGNITE Immunotherapy. L.C.B. has served as a consultant for 4D Molecular Therapeutics. T.G. and F.K.E. declare they have no competing interests. Data and materials availability: All materials are available from commercial sources, and all relevant data are reported in the study. Requests for information or materials should be addressed to D.V.S. (schaffer@berkeley.edu) or T.G. (gaj@illinois.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/eaar3952/DC1
fig. S1. Designing sgRNA to target the human SOD1 gene.
fig. S2. CRISPR-Cas9 reduced mutant SOD1 expression in NSC-34–G93A–SOD1 cells by genome editing.
fig. S3. Quality control of AAV vectors.
fig. S4. Mutant SOD1 expression in the spinal cord of untreated G93A-SOD1 mice.
fig. S5. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice leads to SaCas9 expression in ChAT+ cells in the spinal cord.
fig. S6. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice leads to SaCas9 expression in β3-tubulin+ fibers in the spinal cord.
fig. S7. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice leads to limited SaCas9 expression in GFAP+ astrocytes in the spinal cord.
fig. S8. CRISPR-Cas9–mediated genome editing reduced mutant SOD1 protein in G93A-SOD1 mice.
fig. S9. Genome editing did not affect mouse SOD1 protein in G93A-SOD1 mice.
fig. S10. Background modification at candidate OT sites in CRISPR-treated G93A-SOD1 mice.
fig. S11. G93A-SOD1 mice treated with AAV9-SaCas9-hSOD1 lose weight at a slower rate after disease onset compared to control mice.
fig. S12. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice did not delay the rate of disease progression.
fig. S13. G93A-SOD1 mice injected with AAV9-SaCas9-SaCas9 had limited SaCas9 expression in GFAP+ astrocytes at end stage.
fig. S14. Mutant SOD1 inclusion bodies were visible in end-stage spinal cord sections from CRISPR-treated G93A-SOD1 mice.
table S1. Oligonucleotides used in this study.
table S2. External primers for MiSeq analysis.
table S3. Internal primers for MiSeq analysis.
REFERENCES AND NOTES
- 1.Rowland L. P., Shneider N. A., Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Bensimon G., Lacomblez L., Meininger V.; The ALS/Riluzole Study Group , A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 330, 585–591 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Rosen D. R., Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 364, 362 (1993). [DOI] [PubMed] [Google Scholar]
- 4.Ilieva H., Polymenidou M., Cleveland D. W., Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H.-X., Chen W., Zhai P., Sufit R. L., Siddique T., Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Bruijn L. I., Becher M. W., Lee M. K., Anderson K. L., Jenkins N. A., Copeland N. G., Sisodia S. S., Rothstein J. D., Borchelt D. R., Price D. L., Cleveland D. W., ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Smith R. A., Miller T. M., Yamanaka K., Monia B. P., Condon T. P., Hung G., Lobsiger C. S., Ward C. M., McAlonis-Downes M., Wei H., Wancewicz E. V., Frank Bennett C., Cleveland D. W., Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Invest. 116, 2290–2296 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nizzardo M., Simone C., Rizzo F., Ulzi G., Ramirez A., Rizzuti M., Bordoni A., Bucchia M., Gatti S., Bresolin N., Comi G. P., Corti S., Morpholino-mediated SOD1 reduction ameliorates an amyotrophic lateral sclerosis disease phenotype. Sci. Rep. 6, 21301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raoul C., Abbas-Terki T., Bensadoun J.-C., Guillot S., Haase G., Szulc J., Henderson C. E., Aebischer P., Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat. Med. 11, 423–428 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Foust K. D., Salazar D. L., Likhite S., Ferraiuolo L., Ditsworth D., Ilieva H., Meyer K., Schmelzer L., Braun L., Cleveland D. W., Kaspar B. K., Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. 21, 2148–2159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller T. M., Kaspar B. K., Kops G. J., Yamanaka K., Christian L. J., Gage F. H., Cleveland D. W., Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann. Neurol. 57, 773–776 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dirren E., Aebischer J., Rochat C., Towne C., Schneider B. L., Aebischer P., SOD1 silencing in motoneurons or glia rescues neuromuscular function in ALS mice. Ann. Clin. Transl. Neurol. 2, 167–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borel F., Gernoux G., Cardozo B., Metterville J. P., Toro Cabreja G. C., Song L., Su Q., Gao G. P., Elmallah M. K., Brown R. H. Jr., Mueller C., Therapeutic rAAVrh10 mediated SOD1 silencing in adult SOD1G93A mice and nonhuman primates. Hum. Gene Ther. 27, 19–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoica L., Todeasa S. H., Toro Cabrera G., Salameh J. S., ElMallah M. K., Mueller C., Brown R. H. Jr., Sena-Esteves M., Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann. Neurol. 79, 687–700 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomsen G. M., Gowing G., Latter J., Chen M., Vit J.-P., Staggenborg K., Avalos P., Alkaslasi M., Ferraiuolo L., Likhite S., Kaspar B. K., Svendsen C. N., Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J. Neurosci. 34, 15587–15600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller T. M., Pestronk A., David W., Rothstein J., Simpson E., Appel S. H., Andres P. L., Mahoney K., Allred P., Alexander K., Ostrow L. W., Schoenfeld D., Macklin E. A., Norris D. A., Manousakis G., Crisp M., Smith R., Bennett C. F., Bishop K. M., Cudkowicz M. E., An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 12, 435–442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J. A., Charpentier E., A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mali P., Yang L., Esvelt K. M., Aach J., Guell M., DiCarlo J. E., Norville J. E., Church G. M., RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J., RNA-programmed genome editing in human cells. Elife 2, e00471 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho S. W., Kim S., Kim J. M., Kim J.-S., Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Santiago Y., Chan E., Liu P. Q., Orlando S., Zhang L., Urnov F. D., Holmes M. C., Guschin D., Waite A., Miller J. C., Rebar E. J., Gregory P. D., Klug A., Collingwood T. N., Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc. Natl. Acad. Sci. U.S.A. 105, 5809–5814 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ran F. A., Cong L., Yan W. X., Scott D. A., Gootenberg J. S., Kriz A. J., Zetsche B., Shalem O., Wu X., Makarova K. S., Koonin E. V., Sharp P. A., Zhang F., In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosco D. A., Morfini G., Karabacak N. M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B. A., Lemay N., McKenna-Yasek D., Frosch M. P., Agar J. N., Julien J.-P., Brady S. T., Brown R. H. Jr., Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cashman N. R., Durham H. D., Blusztajn J. K., Oda K., Tabira T., Shaw I. T., Dahrouge S., Antel J. P., Neuroblastoma × spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 194, 209–221 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Nathwani A. C., Reiss U. M., Tuddenham E. G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D., Riddell A., Pie J., Rangarajan S., Bevan D., Recht M., Shen Y.-M., Halka K. G., Basner-Tschakarjan E., Mingozzi F., High K. A., Allay J., Kay M. A., Ng C. Y., Zhou J., Cancio M., Morton C. L., Gray J. T., Srivastava D., Nienhuis A. W., Davidoff A. M., Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 371, 1994–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacLaren R. E., Groppe M., Barnard A. R., Cottriall C. L., Tolmachova T., Seymour L., Clark K. R., During M. J., Cremers F. P., Black G. C., Lotery A. J., Downes S. M., Webster A. R., Seabra M. C., Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet 383, 1129–1137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bainbridge J. W., Smith A. J., Barker S. S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G. E., Stockman A., Tyler N., Petersen-Jones S., Bhattacharya S. S., Thrasher A. J., Fitzke F. W., Carter B. J., Rubin G. S., Moore A. T., Ali R. R., Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 358, 2231–2239 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Stroes E. S., Nierman M. C., Meulenberg J. J., Franssen R., Twisk J., Henny C. P., Maas M. M., Zwinderman A. H., Ross C., Aronica E., High K. A., Levi M. M., Hayden M. R., Kastelein J. J., Kuivenhoven J. A., Intramuscular administration of AAV1-lipoprotein lipaseS447X lowers triglycerides in lipoprotein lipase–deficient patients. Arterioscler. Thromb. Vasc. Biol. 28, 2303–2304 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Carpentier A. C., Frisch F., Labbé S. M., Gagnon R., de Wal J., Greentree S., Petry H., Twisk J., Brisson D., Gaudet D., Effect of alipogene tiparvovec (AAV1-LPLS447X) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J. Clin. Endocrinol. Metab. 97, 1635–1644 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Gao G., Vandenberghe L. H., Alvira M. R., Lu Y., Calcedo R., Zhou X., Wilson J. M., Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78, 6381–6388 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foust K. D., Nurre E., Montgomery C. L., Hernandez A., Chan C. M., Kaspar B. K., Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duque S., Joussemet B., Riviere C., Marais T., Dubreil L., Douar A.-M., Fyfe J., Moullier P., Colle M.-A., Barkats M., Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol. Ther. 17, 1187–1196 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boillée S., Yamanaka K., Lobsiger C. S., Copeland N. G., Jenkins N. A., Kassiotis G., Kollias G., Cleveland D. W., Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Yamanaka K., Chun S. J., Boillee S., Fujimori-Tonou N., Yamashita H., Gutmann D. H., Takahashi R., Misawa H., Cleveland D. W., Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haidet-Phillips A. M., Hester M. E., Miranda C. J., Meyer K., Braun L., Frakes A., Song S., Likhite S., Murtha M. J., Foust K. D., Rao M., Eagle A., Kammesheidt A., Christensen A., Mendell J. R., Burghes A. H. M., Kaspar B. K., Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchetto M. C. N., Muotri A. R., Mu Y., Smith A. M., Cezar G. G., Gage F. H., Non-cell-autonomous effect of human SOD1G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657 (2008). [DOI] [PubMed] [Google Scholar]
- 38.Re D. B., Le Verche V., Yu C., Amoroso M. W., Politi K. A., Phani S., Ikiz B., Hoffmann L., Koolen M., Nagata T., Papadimitriou D., Nagy P., Mitsumoto H., Kariya S., Wichterle H., Henderson C. E., Przedborski S., Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song S., Miranda C. J., Braun L., Meyer K., Frakes A. E., Ferraiuolo L., Likhite S., Bevan A. K., Foust K. D., McConnell M. J., Walker C. M., Kaspar B. K., Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat. Med. 22, 397–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagai M., Re D. B., Nagata T., Chalazonitis A., Jessell T. M., Wichterle H., Przedborski S., Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalkara D., Byrne L. C., Lee T., Hoffmann N. V., Schaffer D. V., Flannery J. G., Enhanced gene delivery to the neonatal retina through systemic administration of tyrosine-mutated AAV9. Gene Ther. 19, 176–181 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Petrs-Silva H., Dinculescu A., Li Q., Min S.-H., Chiodo V., Pang J.-J., Zhong L., Zolotukhin S., Srivastava A., Lewin A. S., Hauswirth W. W., High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol. Ther. 17, 463–471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinello L., Canver M. C., Hoban M. D., Orkin S. H., Kohn D. B., Bauer D. E., Yuan G.-C., Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 34, 695–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bae S., Park J., Kim J.-S., Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clement A. M., Nguyen M. D., Roberts E. A., Garcia M. L., Boillée S., Rule M., McMahon A. P., Doucette W., Siwek D., Ferrante R. J., Brown R. H. Jr., Julien J.-P., Goldstein L. S. B., Cleveland D. W., Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Beers D. R., Henkel J. S., Xiao Q., Zhao W., Wang J., Yen A. A., Siklos L., McKercher S. R., Appel S. H., Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 103, 16021–16026 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Giorgio F. P., Carrasco M. A., Siao M. C., Maniatis T., Eggan K., Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frakes A. E., Ferraiuolo L., Haidet-Phillips A. M., Schmelzer L., Braun L., Miranda C. J., Ladner K. J., Bevan A. K., Foust K. D., Godbout J. P., Popovich P. G., Guttridge D. C., Kaspar B. K., Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 81, 1009–1023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang S. H., Li Y., Fukaya M., Lorenzini I., Cleveland D. W., Ostrow L. W., Rothstein J. D., Bergles D. E., Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 16, 571–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferraiuolo L., Meyer K., Sherwood T. W., Vick J., Likhite S., Frakes A., Miranda C. J., Braun L., Heath P. R., Pineda R., Beattie C. E., Shaw P. J., Askwith C. C., McTigue D., Kaspar B. K., Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 113, E6496–E6505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaj T., Sirk S. J., Shui S.-L., Liu J., Genome-editing technologies: Principles and applications. Cold Spring Harb. Perspect. Biol. 8, a023754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tervo D. G. R., Hwang B.-Y., Viswanathan S., Gaj T., Lavzin M., Ritola K. D., Lindo S., Michael S., Kuleshova E., Ojala D., Huang C.-C., Gerfen C. R., Schiller J., Dudman J. T., Hantman A. W., Looger L. L., Schaffer D. V., Karpova A. Y., A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F., In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 33, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zetsche B., Heidenreich M., Mohanraju P., Fedorova I., Kneppers J., DeGennaro E. M., Winblad N., Choudhury S. R., Abudayyeh O. O., Gootenberg J. S., Wu W. Y., Scott D. A., Severinov K., van der Oost J., Zhang F., Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 35, 31–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Haurigot V., Doyon Y., Li T., Wong S. Y., Bhagwat A. S., Malani N., Anguela X. M., Sharma R., Ivanciu L., Murphy S. L., Finn J. D., Khazi F. R., Zhou S., Paschon D. E., Rebar E. J., Bushman F. D., Gregory P. D., Holmes M. C., High K. A., In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475, 217–221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anguela X. M., Sharma R., Doyon Y., Miller J. C., Li H., Haurigot V., Rohde M. E., Wong S. Y., Davidson R. J., Zhou S., Gregory P. D., Holmes M. C., High K. A., Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 122, 3283–3287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K., Batshaw M. L., Wilson J. M., A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 34, 334–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yin H., Song C.-Q., Dorkin J. R., Zhu L. J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A., Gupta A., Bolukbasi M. F., Walsh S., Bogorad R. L., Gao G., Weng Z., Dong Y., Koteliansky V., Wolfe S. A., Langer R., Xue W., Anderson D. G., Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nelson C. E., Hakim C. H., Ousterout D. G., Thakore P. I., Moreb E. A., Castellanos Rivera R. M., Madhavan S., Pan X., Ran F. A., Yan W. X., Asokan A., Zhang F., Duan D., Gersbach C. A., In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Long C., Amoasii L., Mireault A. A., McAnally J. R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J. M., Bassel-Duby R., Olson E. N., Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351, 400–403 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabebordbar M., Zhu K., Cheng J. K. W., Chew W. L., Widrick J. J., Yan W. X., Maesner C., Wu E. Y., Xiao R., Ran F. A., Cong L., Zhang F., Vandenberghe L. H., Church G. M., Wagers A. J., In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bengtsson N. E., Hall J. K., Odom G. L., Phelps M. P., Andrus C. R., Hawkins R. D., Hauschka S. D., Chamberlain J. R., Chamberlain J. S., Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 8, 14454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Foust K. D., Wang X., McGovern V. L., Braun L., Bevan A. K., Haidet A. M., Le T. T., Morales P. R., Rich M. M., Burghes A. H. M., Kaspar B. K., Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271–274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 64.Valori C. F., Ning K., Wyles M., Mead R. J., Grierson A. J., Shaw P. J., Azzouz M., Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2, 35ra42 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Kotterman M. A., Schaffer D. V., Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 15, 445–451 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burstein D., Harrington L. B., Strutt S. C., Probst A. J., Anantharaman K., Thomas B. C., Doudna J. A., Banfield J. F., New CRISPR–Cas systems from uncultivated microbes. Nature 542, 237–241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim E., Koo T., Park S. W., Kim D., Kim K., Cho H.-Y., Song D. W., Lee K. J., Jung M. H., Kim S., Kim J. H., Kim J. H., Kim J.-S., In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 8, 14500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gurney M. E., The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J. Neurol. Sci. 152 (Suppl. 1), s67–s73 (1997). [DOI] [PubMed] [Google Scholar]
- 69.Yang L., Güell M., Niu D., George H., Lesha E., Grishin D., Aach J., Shrock E., Xu W., Poci J., Cortazio R., Wilkinson R. A., Fishman J. A., Church G., Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science 350, 1101–1104 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Chadderton N., Millington-Ward S., Palfi A., O’Reilly M., Tuohy G., Humphries M. M., Li T., Humphries P., Kenna P. F., Farrar G. J., Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol. Ther. 17, 593–599 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kleinstiver B. P., Prew M. S., Tsai S. Q., Topkar V. V., Nguyen N. T., Zheng Z., Gonzales A. P. W., Li Z., Peterson R. T., Yeh J.-R. J., Aryee M. J., Joung J. K., Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsai S. Q., Zheng Z., Nguyen N. T., Liebers M., Topkar V. V., Thapar V., Wyvekens N., Khayter C., Iafrate A. J., Le L. P., Aryee M. J., Joung J. K., GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim D., Bae S., Park J., Kim E., Kim S., Yu H. R., Hwang J., Kim J.-I., Kim J.-S., Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243 (2015). [DOI] [PubMed] [Google Scholar]
- 74.Hendel A., Fine E. J., Bao G., Porteus M. H., Quantifying on- and off-target genome editing. Trends Biotechnol. 33, 132–140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Long C., McAnally J. R., Shelton J. M., Mireault A. A., Bassel-Duby R., Olson E. N., Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 345, 1184–1188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Qi L. S., Larson M. H., Gilbert L. A., Doudna J. A., Weissman J. S., Arkin A. P., Lim W. A., Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gingras M., Gagnon V., Minotti S., Durham H. D., Berthod F., Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J. Neurosci. Methods 163, 111–118 (2007). [DOI] [PubMed] [Google Scholar]
- 78.Wichterle H., Lieberam I., Porter J. A., Jessell T. M., Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385–397 (2002). [DOI] [PubMed] [Google Scholar]
- 79.DeJesus-Hernandez M., Mackenzie I. R., Boeve B. F., Boxer A. L., Baker M., Rutherford N. J., Nicholson A. M., Finch N. A., Flynn H., Adamson J., Kouri N., Wojtas A., Sengdy P., Hsiung G.-Y. R., Karydas A., Seeley W. W., Josephs K. A., Coppola G., Geschwind D. H., Wszolek Z. K., Feldman H., Knopman D. S., Petersen R. C., Miller B. L., Dickson D. W., Boylan K. B., Graff-Radford N. R., Rademakers R., Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Renton A. E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J. R., Schymick J. C., Laaksovirta H., van Swieten J. C., Myllykangas L., Kalimo H., Paetau A., Abramzon Y., Remes A. M., Kaganovich A., Scholz S. W., Duckworth J., Ding J., Harmer D. W., Hernandez D. G., Johnson J. O., Mok K., Ryten M., Trabzuni D., Guerreiro R. J., Orrell R. W., Neal J., Murray A., Pearson J., Jansen I. E., Sondervan D., Seelaar H., Blake D., Young K., Halliwell N., Callister J. B., Toulson G., Richardson A., Gerhard A., Snowden J., Mann D., Neary D., Nalls M. A., Peuralinna T., Jansson L., Isoviita V. M., Kaivorinne A. L., Hölttä-Vuori M., Ikonen E., Sulkava R., Benatar M., Wuu J., Chiò A., Restagno G., Borghero G., Sabatelli M.; The ITALSGEN Consortium, Heckerman D., Rogaeva E., Zinman L., Rothstein J. D., Sendtner M., Drepper C., Eichler E. E., Alkan C., Abdullaev Z., Pack S. D., Dutra A., Pak E., Hardy J., Singleton A., Williams N. M., Heutink P., Pickering-Brown S., Morris H. R., Tienari P. J., Traynor B. J., A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stevens J. C., Chia R., Hendriks W. T., Bros-Facer V., van Minnen J., Martin J. E., Jackson G. S., Greensmith L., Schiavo G., Fisher E. M. C., Modification of superoxide dismutase 1 (SOD1) properties by a GFP tag – implications for research into amyotrophic lateral sclerosis (ALS). PLOS ONE 5, e9541 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guschin D. Y., Waite A. J., Katibah G. E., Miller J. C., Holmes M. C., Rebar E. J., A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 649, 247–256 (2010). [DOI] [PubMed] [Google Scholar]
- 83.Gaj T., Schaffer D. V., Adeno-associated virus–mediated delivery of CRISPR–Cas systems for genome engineering in mammalian cells. Cold Spring Harb. Protoc. 2016, pdb.prot086868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zolotukhin S., Byrne B. J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R. J., Muzyczka N., Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6, 973–985 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/12/eaar3952/DC1
fig. S1. Designing sgRNA to target the human SOD1 gene.
fig. S2. CRISPR-Cas9 reduced mutant SOD1 expression in NSC-34–G93A–SOD1 cells by genome editing.
fig. S3. Quality control of AAV vectors.
fig. S4. Mutant SOD1 expression in the spinal cord of untreated G93A-SOD1 mice.
fig. S5. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice leads to SaCas9 expression in ChAT+ cells in the spinal cord.
fig. S6. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice leads to SaCas9 expression in β3-tubulin+ fibers in the spinal cord.
fig. S7. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice leads to limited SaCas9 expression in GFAP+ astrocytes in the spinal cord.
fig. S8. CRISPR-Cas9–mediated genome editing reduced mutant SOD1 protein in G93A-SOD1 mice.
fig. S9. Genome editing did not affect mouse SOD1 protein in G93A-SOD1 mice.
fig. S10. Background modification at candidate OT sites in CRISPR-treated G93A-SOD1 mice.
fig. S11. G93A-SOD1 mice treated with AAV9-SaCas9-hSOD1 lose weight at a slower rate after disease onset compared to control mice.
fig. S12. Systemic administration of AAV9-SaCas9-hSOD1 to neonatal G93A-SOD1 mice did not delay the rate of disease progression.
fig. S13. G93A-SOD1 mice injected with AAV9-SaCas9-SaCas9 had limited SaCas9 expression in GFAP+ astrocytes at end stage.
fig. S14. Mutant SOD1 inclusion bodies were visible in end-stage spinal cord sections from CRISPR-treated G93A-SOD1 mice.
table S1. Oligonucleotides used in this study.
table S2. External primers for MiSeq analysis.
table S3. Internal primers for MiSeq analysis.