

Abstract
Diarylmethylamido bis(phenols) have been subjected to peptide-catalyzed, enantioselective bromination reactions. Desymmetrization of compounds in this class has been achieved such that enantioenriched products may be isolated with up to 97:3 er. Mechanistically, the observed enantioselectivity was shown to be primarily a function of differential functionalization of enantiotopic arenes, although additional studies unveiled a contribution from secondary kinetic resolution of the product (to afford the symmetrical dibromide) under the reaction conditions. Variants of the tetrapeptide catalyst were also evaluated and revealed a striking observation—enantiodivergent catalysis is observed upon changing the achiral amino acid residue in the catalyst (at the i+2 position) from an aminocyclopropane carboxamide residue (97:3 er) to an aminoisobutyramide residue (33:67 er) under a common set of conditions. An expanded set of catalysts was also evaluated, enabling structure/selectivity correlations to be considered in a mechanistic light.
Graphical Abstract
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
The development of methodologies aimed at the preparation of chiral, enantioenriched diarylmethines has emerged as an intensive activity in synthetic chemistry. Motivated by the privileged nature of this scaffold in drug discovery,1 a few general approaches have been identified to access these scaffolds with high optical enrichment,2 including asymmetric additions of arene nucleophiles to imines,3 asymmetric hydrogenation,4 and cross-coupling.5 Desymmetrizations of diarylmethine compounds are also known, such as the C–H bond functionalizations of Shi/Hartwig6 and Wang/Yu.7 Our experience with these compounds began with the challenge of desymmetrizing a simple bis(phenol), which was accomplished using a peptide-catalyzed acylation reaction.8 This study was particularly intriguing to us, given that the pro-stereogenic center is situated five bonds removed from the oxygen atom that undergoes acylation with up to 97.5:2.5 er. This study was followed by a mechanistic investigation that stimulated hypotheses about a Brønsted base mechanism for phenol activation.9
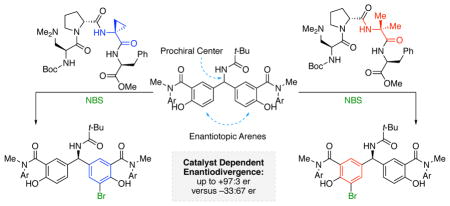
By extension, we surmised that other reactions could be subject to this type of substrate activation, and electrophilic aromatic substitution (EAS) emerged as a class of interest. Most of our studies of this process have focused on either atroposelective brominations10 or site-selective brominations of complex natural product scaffolds.11 While the details of the EAS mechanism are complicated,12 we speculated that with low pKa-phenols, we might achieve the type of desymmetrization shown in Figure 1, wherein a tertiary amine-containing peptide catalyst can differentiate the enantiotopic arenes through non-covalent interactions with the substrate. Described below are the results of selective diarylmethylami-do bis(phenol) desymmetrizations. The studies presented herein unveiled structure-selectivity correlations that show a striking degree of subtlety with respect to the effects of catalyst structure on enantioselectivity, and even the capacity to achieve enantiodivergent outcomes with catalysts that do not differ in any stereogenic element.
Figure 1.

Targeted remote desymmetrization of diarylmethylamido bis(phenols) to access drug-like, chiral molecules.
Our initial observations on the conversion of elaborated diarylmethylamido bis(phenol) compound 1 to the monobrominated product 2a and the doubly brominated product 3a (Eq 1) are described in Table 1. Most notably, when the reaction is conducted with catalyst 4 at −50 °C, with a carefully prescribed quantity of N-bromosuccinimide (NBS, 1.35 equiv), the staring material is fully consumed and the reaction delivers 2a in 66% yield with 95:5 er (Table 1, entry 1). A significant amount of the dibrominated material 3a (31% of the product mixture) is also observed. When the reaction is performed using peptide 5, in which the catalytic L-β-dimethylaminoalanine (Dmaa) is replaced with leucine, its C-isostere, the reaction becomes significantly more sluggish, such that 59% of the 1 remains and only 35% of racemic 2a is observed (Table 1, entry 2). This experiment underscores the importance of the tertiary amine moiety of Dmaa to the selective reaction pathway. Triethylamine promotes the reaction to a somewhat greater extent than peptide 5 (19% of 1 remaining; Table 1, entry 3). Under these conditions, however, product stability is an issue, and decomposition byproducts with mass/charge ratios consistent with 6a and 6b are detected in the LC/MS spectrum of the unpurified reaction mixture. These compounds may derive from a putative ipso-bromination/elimination of one aryl group. Examination of the reaction in the absence of any catalyst reveals a sluggish, but appreciable background rate that delivers 26% of racemic 2b with 74% of 1 remaining (Table 1, entry 4).
Table 1.
Peptide-catalyzed bromination of 1 to monobrominated product 2a and bisbromide 3a (Eq 1), and examination of catalysts and control reactions.

| ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Temp °C | Time h | Ratioa 1:2a:3a | er 2bb | |

|
1 | 4 | −50 | 48 | 0:69:31 (66% isolated yield) | 95:5 |
| 2 | 5 | −50 | 48 | 59:35:4 | 51.5:48.5 | |
| 3 | NEt3 | −50 | 48 | 19:43:19 | -- | |
| 4 | None | −50 | 48 | 74:26: 0 | -- | |
|
| ||||||
| 5 | 4 | −78 | 24 | 100:0:0c | -- | |
| 6d | 4 | rt | 24 | 22:64:14c | 51:49 | |
| 7e | 4 | 4 | 24 | 3:67:30c | 88:12 | |
| 8e | 4 | −10 | 5 | 1:57:42c | 92:8 | |
Product yields determined by 1H NMR analysis of the crude reaction mixture with respect to an internal standard prior to methylation.
Enantiomeric ratio determined by chiral HPLC analysis of isolated material.
Relative product ratio.
Performed with 1.0 equiv of NBS.
A solution of 4/1 was added by cannula into a vial with NBS.
Furthermore, when the 4-catalyzed reaction is attempted at −78 °C, no product is observed (Table 1, entry 5); the same reaction at ambient temperature leads to a mixture of 64% 2a, 14% 3a, and 22% 1, in which 2a (assayed as 2b) is nearly racemic (Table 1, entry 6). However, significant enantioselectivity is observed when the reaction is run at 4 °C (88:12 er; Table 1, entry 7) and also at −10 °C (92:8 er; Table 1, entry 8). The reaction is strikingly temperature dependent, with the highest selectivity noted at −50 °C. Some variation in the reaction conversion and, correspondingly the enantioselectivity, was also noted as a function of the mode NBS delivery. Various methods of addition (cannula, solid addition, etc.) were explored, as we found that it is particularly important to ensure that all of the brominating agent is cleanly delivered to the reaction solution.
Initial studies of catalyst analogs revealed a remarkable enantioselectivity dependence on a very subtle aspect of the catalyst structure. As shown in Scheme 1 and Table 2, under the optimal conditions, catalyst 4, which possesses an aminocycloprane carboxamide residue (Acpc) at the i+2 position, delivers 2a in 95:5 er (66% yield). However, α-aminoisobutyramide (Aib)-containing catalyst 7 favors the opposite absolute configuration of the product (ent-2a), albeit with a reduced er of 42:58 at 91% consumption of 1. This surprising inversion in the sense of asymmetric induction is a consequence of only 2 atomic mass units in the molecular weight of the catalyst, as the Acpc and Aib residues differ by one equivalent of molecular hydrogen. Moreover, each of the constituent amino acids of catalysts 4 and 7 are of the same stereochemical configuration, and the observed enantiodivergence stems from the perturbation of a locally achiral residue.
Scheme 1.

Table 2.
Substrate scope of the enantioselective diarylmethylamido bis(phenol) bromination as a function of i+2 substitution of the catalyst.
| Entry | Bis(phenol) | Monobromide | Ratio SM:Mono:Bisa (% Yield Mono)b | Monobromide erc | |
|---|---|---|---|---|---|

|

|
||||
| 1 | R1 = Me, R2 = Ph (1) | R1 = Me, R2 = Ph (2b) | with 4: | 0:69:31 (66) | 95:5 |
| with 7: | 9:60:31 (54) | 42:58 | |||
| 2 | R1 = Me, R2 = 4-MeOPh (8) | R1 = Me, R2 = 4-MeOPh (9) | with 4: | 0:56:44 (49) | 97:3 |
| with 7: | 3:71:26 (50) | 33:67 | |||
| 3 | R1 = Me, R2 = 4-BrPh (10) | R1 = Me, R2 = 4-BrPh (11) | with 4: | 5:59:36 (59) | 92:8 |
| with 7:d | 57:40:3 | 49:51 | |||
| 4 | R1 = i-Pr, R2 = Ph (12) | R1 = i-Pr, R2 = Ph (13) | with 4: | 1:68:31 (60) | 94:6 |
| with 7:d | 7:57:36 | 49:51 | |||
| 5 | R1 = Me, R2 = OMe (14) | R1 = Me, R2 = OMe (15) | with 4: | 7:54:39 (54) | 83:17 |
| with 7: | 10:50:40 (52) | 26:74 | |||
| 6 |
|
|
with 4: | 9:58:33 (55) | 82:18 |
| with 7: | 10:52:38 (48) | 21:79 | |||
Product ratio determined by 1H NMR analysis of the crude reaction mixture prior to methylation.
Isolated yield of indicated product (following methylation).
Enantiomeric ratio determined by chiral HPLC analysis of isolated material.
Performed with 1.3 equiv of NBS.
To the extent that enantiodivergent outcomes in organic synthesis are of fundamental interest,13 especially when parameters are varied other than the trivial swap of one enantiomer of a chiral catalyst for the other, we sought to assess the degree to which this divergence might be replicable, or even amplifiable, with other substrates. Similar to compound 1 (Table 2, entry 1), we found that p-methoxy-substituted substrate 8 shows comparable levels of enantiodivergence (Table 2, entry 2). While the examination of additional benzamide substrates 10 and 12 revealed a subtler change in enantioselectivity when going from catalyst 4 to 7, other substrates (14 and 16) provided a somewhat more dramatic enantiodivergence. For example, peptide 4 catalyzes the conversion of bis(i-propyl) ketone 16 to monobromide 17 in 82:18 er; catalyst 7 delivers the opposite enantiomer with an er of 21:79 (Table 2, entry 6).
As with many desymmetrizations,10d,14 these reactions exhibit a degree of secondary kinetic resolution as the product is partially converted to the symmetrical dibrominated compound. To establish the extent to which this secondary resolution was influencing product enantioselectivity, we performed the kinetic resolution experiment illustrated in Eq 2. When (±)-2a was subjected to the reaction conditions with catalyst 4, we observed a krel (s-factor) = 3.2. While appreciable, this observation suggests that the most significant source of enantioselection in this desymmetrization process is the peptide-enabled enantiotopic arene differentiation.
 |
(2) |
The mechanistic basis for the observed enantioselectivity and enantiodivergent behavior is not fully understood, nor easily elucidated. However, recent structural studies of similar peptide-based catalysts provide experimentally grounded basis for speculation.15 Our knowledge of the conformations of catalysts, such as 4 and 7, is based on crystallographic analysis, solution-phase NMR studies, and DFT calculations. Figure 2 shows the X-ray crystallographic structures for these peptides and their overlay, which reveals a high degree of overlap in the catalysts’ turn regions (D-Pro-Xaa), which is consistent with a type II′ β-turn motif. A greater degree of conformational variation of the i and i+3 residues is observed. Furthermore, the main chain angle of the i+2 residue, τ(i+2), is significantly wider in 4 than in 7 (116.9° vs. 111.0°, respectively) by virtue of the strain associated with the cy-clopropyl ring in 4. It seems plausible that the conformational consequences of this angle impact the selectivity of these desymmetrization reactions in a decisive way.
Figure 2.

X-ray crystal structures of enantiodivergent catalysts 4 and 7 and their overlay.
Therefore, we examined a homologous series of catalysts varying only at i+2 (4, 7, 18 – 21, Figure 3). A linear correlation between the observed enantioselectivity in the bromination of 1 and average values of τ(i+2) from our previously reported X-ray crystal structure library was revealed.10b,15a These data show that wider angles of τ(i+2) correlate with higher levels of enantioinduction (i.e., 4); the selectivity decreases steeply as τ(i+2) becomes more acute, and eventually a cross-over in selectivity is observed, as the opposite enantiomer becomes favored when the angle reaches a critical value.
Figure 3.

Identification of a linear correlation between enantioselectiv-ity and τ(i+2) in the bromination of 1. Product ratios were determined by 1H NMR analysis of the crude reaction mixture with respect to an internal standard prior to methylation. Enantiomeric excess was measured post O-methylation (2b). The average ee values are uncorrected for the extent of conversion and the associated secondary kinetic resolution (Eq 2). Additional details are provided in the Supporting Information.
Given these experimental data, we consider some speculative substrate binding and activation models for the enantiodivergence observed with peptides 4 and 7 (Figure 4). While many examples of enantioselective catalysis involve differential functionalization of the re or si face of a prochiral π-bond (Figure 4a),16 desymmetrizations such as the present case likely a substrate-rotational model for enantiomer determination (Figure 4b). For both catalysts, an activating interaction between the catalytic Dmaa residue and one enantiotopic phenol in 1 is assumed to be critical for promoting efficient bromination.10b, 17
Figure 4.

(a) Classical π-facial selectivity determinants for enantiomeric control. (b) Substrate rotation could lead to opposite enantiomers of the monobrominated products. (c) Possible reactive orientations of catalyst and substrate with subtle changes in the non-covalent interactions that may lead to enantiodivergence: (i) Catalyst 4 in type II′ β-hairpin conformation. (ii) Catalyst 7 in type II′ β-hairpin conformation. (iii) Catalyst 4 in type I′ pre-helical β-turn conformation. (iv) Catalyst 7 in type I′ pre-helical β-turn conformation.
In transition states (i) and (ii) (Figure 4c), reactive complexes are shown in which type II′ β-turns are considered for both peptides 4 and 7. For these, the i+2 amide N–H is characteristically disposed on the top face of the peptide and this moiety could act as a hydrogen bond donor to the carbonyl group in the central pivalamide of 1. Secondary attractive or repulsive interactions between the distal arene ring and the peptide could then serve to discriminate between the two enantiotopic rings, dictating the enantioselectivity of the bromination event. While the details of these secondary interactions are not presently known, perhaps the more contracted τ-angle apparent in Aib-containing catalyst 7 or the expanded τ-angle in the Acpc catalyst 4 leads to ensembles that either amplify or attenuate these interactions.
Importantly, alternative ensembles that explain the observed enantiodivergent behavior of this desymmetrization reaction can also be considered. Transition states (iii) and (iv) illustrate that peptides 4 and 7 could also react from the type I′ pre-helical β-turn conformer. Peptides possessing Acpc at the i+2 position are especially prone to nucleate the type I′ pre-helical conformer in solution and the solid-state, whereas the type II′ β-hairpin conformation could be favored with Aib.15a,18 Since these two limiting conformers orient the loop amides in opposite directions,19 the central pivalamide in 1 could then be disposed to engage such conformers in a rotationally divergent fashion. As a matter of additional complication, the mode of delivery for the electrophilic Br atom is also not precisely known. It could be delivered in a Lewis base-catalyzed manner,20 perhaps by tertiary amide carbonyl of the Dmaa residue, or even through the carbonyl of the i+3 residue, for example.21 In either modality, the depicted transition states lead to each enantiomer as shown.
In conclusion, we have discovered a unique and enantioselective desymmetrization of diarylmethylamido bis(phenols) through bromination catalyzed by tertiary amine-containing tetrapeptides. While these building blocks could be useful for further functionalization to deliver bioactive scaffolds, this study is perhaps most interesting for its stereochemical intrigue. Small changes in catalyst structure lead to enantiodivergence. Most notably, a slight change to an achiral residue distal to the catalytic moiety within catalysts that hold three stereogneic centers constant, leads to opposite product enantiomers. While mechanistic aspects of the enantiodivergence are not entirely clear at the present time, rudimentary models can be considered.
Experimental Section
General Experimental Methods
All reactions were carried out in air unless otherwise noted. Tetrahydrofuran (THF), dichloromethane (CH2Cl2), N,N-dimethylformamide (DMF) and toluene were obtained from a solvent purification system under argon. Chloroform (CHCl3) was purified by elution across basic alumina immediately prior to use in the bromination reactions. N,N-Diisopropylethylamine (i-Pr2NEt) and triethylamine (NEt3) were purified by distillation from calcium hydride (CaH2) under nitrogen. N-Bromosuccinimide (NBS) was recrystallized from hot water, dried under vacuum for 24 hours, and stored in the dark. All other reagents were obtained from commercial sources and used without further purification.
Reactions were monitored by analytical thin layer chromatography (TLC) on 60 Å silica gel F254 pre-coated plates (0.25 mm thickness) and visualized with a UV lamp (254 nm) or by staining with a solution of ceric ammonium nitrate (CAM). Preparative TLC was performed with plates pre-coated with silica gel (20 × 20 cm; 1000 μm thickness). Normal phase column chromatography was performed using 60 Å silica gel (32–63 μm). Reverse phase column chromatography was performed on a Biotage Isolera One purification system equipped with SNAP C18 columns.
For all previously uncharacterized compounds, 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained on 500 and 600 MHz Agilent spectrometers at ambient temperature, unless otherwise noted. NMR data were processed with MestReNova 10.0 using automatic phasing followed by manual phasing as necessary, Berstein polynomial fit base line correction, manual multiplet analysis, and manual integration capabilities. NMR spectral data are reported with the following convention: chemical shift (multiplicity [singlet (s), broad singlet (bs), doublet (d), triplet (t), quartet (q), heptet (hept), multiplet (m), doublet of doublets (dd), doublet of doublet of doublets (ddd), doublet of doublet of doublet of doublets (dddd), doublet of triplets (dt), and doublet of triplet of triplets (dtt)], coupling constants (Hz), integration). 1H chemical shifts were referenced to TMS (0.00 ppm), DMSO-d6 (2.50 ppm), or methanol-d4 (3.31 ppm) and 13C chemical shifts were referenced to CDCl3 (77.16 ppm). High performance liquid chromatography (HPLC) on a chiral stationary phase was performed on an Agilent 1100 series chromatograph equipped with a photodiode array detector (values reported at 230 nm) and a Chiralpak IC column (5 μm particle size, 4.5 × 250 mm). Infrared (IR) spectra were recorded on a Nicolet 6700 ATR/FT-IR spectrometer and are partially reported by convention in cm−1. Optical rotations were obtained on a Perkin-Elmer Polarimeter 341 at the sodium D-line (589 nm) using a cell of 1 dm path length. Melting point ranges (mp) were determined on an Electrothermal 1A9000 series digital melt point apparatus in sealed tubes.
Samples for high-resolution liquid chromatography-mass spectrometry (HRMS) were submitted to the Mass Spectrometry Laboratory at the University of Illinois at Urbana-Champaign. Data were acquired on a Waters Synapt G2-Si instrument equipped with an electrospray ionization (ESI) detector.
Synthesis of Diarylmethylamido Bis(phenol) Substrates
A general scheme for the substrate synthesis is shown in the Supporting Information (Scheme S1).
2-(2-(benzyloxy)-5-bromophenyl)-1,3-dioxane (S-1)
A 500 mL round bottom flask was charged with 2-(benzyloxy)-5-bromobenzaldehyde22 (1.00 equiv, 78.7 mmol, 22.9 g), p-toluenesulfonic acid monohydrate (0.05 equiv, 3.93 mmol, 748 mg), 1,3-propanediol (2.00 equiv, 157 mmol, 11.4 mL) and toluene (200 mL). The flask was fitted to a Dean–Stark apparatus and reflux condenser and heated to reflux under nitrogen for 14 hours. The reaction mixture was allowed to cool to room temperature, diluted with EtOAc and washed with brine (2x). The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. The crude product was recrystallized from hot EtOAc and hexanes to afford the desired product as a white crystalline solid (3 crops, combined yield: 59.6 mmol, 20.8 g, 76%). TLC: Rf (4:1 hexanes/EtOAc) = 0.53. IR (neat): 2965, 2853, 1489, 1379, 1237, 1098, 991, 737, 697 cm−1. 1H NMR (600 MHz, CDCl3): δ 7.77 (d, J = 2.6 Hz, 1H), 7.44 – 7.31 (m, 6H), 6.79 (d, J = 8.8 Hz, 1H), 5.88 (s, 1H), 5.09 (s, 2H), 4.25 (dddd, J = 10.5, 5.1, 1.4, 1.4 Hz, 2H), 3.98 (dddd, J = 12.7, 10.4, 2.6, 1.6 Hz, 2H), 2.24 (dtt, J = 13.5, 12.5, 5.1 Hz, 1H), 1.43 (dtt, J = 13.5, 2.6, 1.3 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 154.8, 136.8, 132.7, 130.4, 129.6, 128.7, 128.1, 127.2, 114.3, 113.7, 96.4, 70.7, 67.7, 25.9. HRMS (ESI) m/z: [M+H]+ calcd for C17H17BrO3 349.0439; found 349.0428.
4-(benzyloxy)-3-(1,3-dioxan-2-yl)benzonitrile (S-2)
An oven-dried 100 mL 3-neck round bottom flask was charged with S-1 (1.00 equiv, 33.0 mmol, 11.5 g) and DMF (55 mL) and then fitted to a reflux condenser and sparged with a balloon of argon for 15 minutes. Copper (I) cyanide (1.50 equiv, 49.5 mmol, 4.40 g) was added and the reaction mixture was sparged for another 15 minutes before the outlet was removed and the argon balloon was transferred to the top of the reflux condenser. The reaction mixture was heated to 150 °C for 20 hours at which point it was allowed to cool to room temperature and treated with saturated aqueous ammonium hydroxide (40 mL) and toluene (40 mL). After stirring for 30 minutes, the solution was transferred to a separatory funnel with toluene (100 mL) and water (100 mL) and then further diluted with additional toluene (150 mL) and saturated aqueous ammonium hydroxide (50 mL). The layers were separated and the aqueous layer was further extracted with toluene (2× 200 mL). The combined organic layers were washed with brine (2x) and then concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (5:1 to 2:1 hexanes/EtOAc), and then recrystallized from hot EtOAc and hexanes to afford the desired product as a white crystalline solid (16.8 mmol, 4.95 g, 51%). TLC: Rf (4:1 hexanes/EtOAc) = 0.29. IR (neat): 2969, 2856, 2225, 1609, 1498, 1263, 1094, 1001, 904, 738, 695 cm−1. 1H NMR (600 MHz, CDCl3): δ 7.97 (d, J = 2.1 Hz, 1H), 7.57 (dd, J = 8.6, 2.2 Hz, 1H), 7.44 – 7.33 (m, 5H), 6.96 (d, J = 8.6 Hz, 1H), 5.87 (s, 1H), 5.17 (s, 2H), 4.29 – 4.23 (m, 2H), 4.03 – 3.95 (m, 2H), 2.25 (dtt, J = 13.6, 12.5, 5.1 Hz, 1H), 1.45 (dtt, J = 13.5, 2.6, 1.3 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 158.9, 136.0, 134.5, 131.9, 128.9, 128.8, 128.4, 127.2, 119.1, 112.7, 104.5, 96.0, 70.6, 67.7, 25.8. HRMS (ESI) m/z: [M+H]+ calcd for C18H17NO3 296.1287; found 296.1282.
N-(bis(4-(benzyloxy)-3-(1,3-dioxan-2-yl)phenyl)-methyl)-pivalamide (S-3)
A flame-dried 250 mL round bottom flask was charged with S-1 (1.30 equiv, 19.8 mmol, 6.90 g), fitted with a septum, and evacuated then back-filled with nitrogen (3×). THF (100 mL) was added via cannula from an argon-purged graduated cylinder and the resulting solution was cooled to −78 °C, then slowly treated with n-BuLi (1.6 M in hexanes, 1.30 equiv, 19.8 mmol, 12.4 mL). After 45 minutes, a nitrogen-purged solution of S-2 (1.00 equiv, 15.2 mmol, 4.50 g) in THF (25 mL) was added slowly via syringe. The reaction mixture was allowed to warm to room temperature and stirred for an additional 75 minutes at which point it was quenched with MeOH (3 mL) and concentrated in vacuo. The residue was dissolved in MeOH (75 mL), cooled to 0 °C under nitrogen, and treated portionwise with NaBH4 (5.00 equiv, 76.0 mmol, 2.90 g). After 10 minutes, the ice bath was removed and the reaction mixture was stirred for 14 hours at which point it was diluted with EtOAc and water. The layers were separated and the aqueous layer was further extracted with EtOAc (2x). The combined organic layers were washed with brine and concentrated in vacuo to afford bis(4-(benzyloxy)-3-(1,3-dioxan-2-yl)phenyl)-methanamine as a pale yellow solid. The crude amine was transferred to a flame-dried 250 mL round bottom flask, dissolved in CH2Cl2 (75 mL) and NEt3 (2 equiv, 30.4 mmol, 4.20 mL) and cooled to 0 °C under nitrogen. Pivaloyl chloride (1.30 equiv, 19.8 mmol, 2.40 mL) was then added dropwise. The ice bath was removed after 10 minutes and the reaction mixture was allowed to stir at room temperature for 2 hours at which point it was quenched with saturated aqueous NaHCO3 and extracted into EtOAc. The organic layer was washed again with NaHCO3 then brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 100 g cartridge; gradient = 10% to 100% EtOAc in hexanes over 10 column volumes; flow rate = 100 mL/min; 2× runs of approximately ½ of the material) to afford the desired product as a white solid (13.8 mmol, 9.00 g, 91%). TLC: Rf (1:1 hexanes/EtOAc) = 0.32. IR (neat): 2965, 2857, 1659, 1499, 1255, 1095, 1001, 906, 730 cm−1. 1H NMR (600 MHz, CDCl3): δ 7.55 (d, J = 2.4 Hz, 2H), 7.45 – 7.41 (m, 4H), 7.41 – 7.36 (m, 4H), 7.35 – 7.30 (m, 2H), 7.04 (ddd, J = 8.5, 2.5, 0.7 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 6.18 (d, J = 7.9 Hz, 1H), 6.05 (d, J = 7.9 Hz, 1H), 5.87 (s, 2H), 5.08 (s, 4H), 4.26 – 4.19 (m, 4H), 4.01 – 3.93 (m, 4H), 2.21 (dtt, J = 13.5, 12.5, 5.1 Hz, 2H), 1.40 (dtt, J = 13.4, 2.6, 1.4 Hz, 2H), 1.23 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.1, 155.0, 137.4, 134.5, 129.4, 128.6, 127.9, 127.6, 127.2, 126.4, 112.6, 97.2, 70.5, 67.7, 67.6, 55.8, 38.9, 27.8, 26.0. HRMS (ESI) m/z: [M+H]+ calcd for C40H45NO7 652.3274; found 652.3274.
5,5′-(pivalamidomethylene)bis(2-(benzyloxy)benzoic acid) (S-4)
A solution of S-3 (1.00 equiv, 14.8 mmol, 9.00 g) in THF (37 mL) in a 250 mL round bottom flask was treated with 1 M HCl (37 mL). The reaction mixture was stirred vigorously at room temperature for 14 hours, at which point it was quenched with saturated aqueous NaHCO3 and extracted into EtOAc. The organic layer was washed with brine, dried with Na2SO4, filtered, and concentrated in vacuo to provide N-(bis(4-(benzyloxy)-3-formylphenyl)-methyl)-pivalamide as a white solid, which was carried on without further purification. The crude aldehyde was transferred to a 250 mL round bottom flask and suspended in 2-methyl-2-butene (20.0 equiv, 296 mmol, 31 mL) and t-BuOH (74 mL). A solution of NaClO2 (3.00 equiv, 44.4 mmol, 4.00 g) and NaH2PO4 (3.00 equiv, 44.4 mmol, 6.10 g) in water (15 mL) was added and the resulting reaction mixture was stirred vigorously for 24 hours. An additional portion of 2-methyl-2-butene (10 mL), NaClO2 (1.00 equiv, 14.8 mmol, 1.33 g) and NaH2PO4 (1.00 equiv, 14.8 mmol, 2.03 g) was added, and the reaction mixture was stirred for an additional 14 hours, at which point it was quenched with 1 M HCl and extracted into CH2Cl2 (3×). The combined organic layers were washed with water, dried with Na2SO4, filtered and concentrated in vacuo to afford the desired product as a pale yellow solid (13.9 mmol, 7.90 g, 94%). An analytically pure sample of the product was obtained by reverse-phase chromatography [Biotage SP4 system, KP-C18-HS column; gradient = 20% to 100% MeCN in water (0.1% formic acid buffer)], although the material was typically carried on without further purification in the synthesis of S-5, S-6, S-7, S-8 and 14. TLC: Rf (15:1 CH2Cl2/MeOH) = 0.25. IR (neat): 3276, 2968, 1723, 1611, 1496, 1226, 909, 731, 696 cm−1. 1H NMR (600 MHz, CDCl3): δ 10.73 (bs, 2H), 8.00 (d, J = 2.5 Hz, 2H), 7.46 – 7.38 (m, 12H), 7.11 (d, J = 8.6 Hz, 2H), 6.20 – 6.13 (m, 2H), 5.29 (s, 4H), 1.24 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.9, 165.0, 156.9, 135.5, 134.2, 132.3, 129.5, 129.4, 128.1, 118.6, 113.8, 72.7, 55.4, 39.0, 27.7. HRMS (ESI) m/z: [M– H]− calcd for C34H33NO7 566.2179; found 566.2184.
5,5′-(pivalamidomethylene)bis(2-(benzyloxy)-N-methyl-N-phenylbenzamide) (S-5)
An oven-dried 250 mL round bottom flask was charged with S-4 (1.00 equiv, 10.5 mmol, 6.00 g), HATU (3.00 equiv, 31.7 mmol, 12.1 g), CH2Cl2 (53 mL), and i-Pr2NEt (3.00 equiv, 31.7 mmol, 5.5 mL). The reaction mixture was stirred at room temperature under nitrogen for 10 minutes and then N-methylaniline (3.00 equiv, 31.7 mmol, 3.40 mL) was added dropwise. After stirring an additional 14 hours, the reaction mixture was diluted with EtOAc, washed with 10% aqueous citric acid (2x) and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 100 g cartridge; gradient = 30% to 100% EtOAc in hexanes over 10 column volumes, then 100% EtOAc over 7 column volumes; flow rate = 100 mL/min; 2× runs of approximately ½ of the material) to afford the desired product as a white solid (6.84 mmol, 5.10 g, 65%). TLC: Rf (2:1 EtO-Ac/hexanes) = 0.32. IR (neat): 1644, 1595, 1496, 1369, 1255, 735, 697 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.43 – 7.30 (m, 10H), 7.05 (bs, 6H), 6.89 (bs, 6H), 6.72 (bs, 2H), 6.55 (d, J = 8.5 Hz, 2H), 5.9 (d, J = 5.6 Hz, 1H), 5.78 (d, J = 6.1 Hz, 1H), 4.92 (s, 4H), 3.44 (s, 6H), 1.18 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.2, 168.8, 153.8, 143.8, 137.0, 133.8, 129.4, 128.7, 128.0, 127.5, 127.4, 127.0, 126.8, 112.5, 70.3, 54.9, 38.8, 37.2, 27.7. HRMS (ESI) m/z: [M+H]+ calcd for C48H47N3O5 746.3594; found 746.3599.
5,5′-(pivalamidomethylene)bis(2-(benzyloxy)-N-(4-meth-oxyphenyl)-N-methylbenzamide) (S-6)
An oven-dried 50 mL round bottom flask was charged with S-4 (1.00 equiv, 1.40 mmol, 800 mg), HATU (3.00 equiv, 4.20 mmol, 1.60 g), CH2Cl2 (7 mL), and i-Pr2NEt (3.00 equiv, 4.20 mmol, 730 μL). The reaction mixture was stirred at room temperature under nitrogen for 10 minutes and then 4-methoxy-N-methylaniline (3.00 equiv, 4.20 mmol, 576 mg) was added. After stirring an additional 14 hours, the reaction mixture was diluted with EtOAc, washed with 10% aqueous citric acid (2x) and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 50 g cartridge; gradient = 50% to 100% EtOAc in hexanes over 10 column volumes; flow rate = 100 mL/min) to afford the desired product as a pale purple solid (1.02 mmol, 816 mg, 73%). TLC: Rf (3:1 EtOAc/hexanes) = 0.32. IR (neat): 1640, 1512, 1376, 1247, 1035, 835, 734, 646 cm−1. 1H NMR (Major conformer, 600 MHz, CDCl3): δ 7.43 – 7.29 (m, 12H), 6.83 (d, J = 8.8 Hz, 4H), 6.73 (bs, 2H), 6.65 – 6.51 (m, 6H), 5.92 (d, J = 7.9 Hz, 1H), 5.81 (d, J = 8.0 Hz, 1H), 4.96 (s, 4H), 3.70 (s, 6H), 3.39 (s, 6H), 1.18 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.2, 169.1, 158.1, 153.6, 137.0, 136.7, 133.7, 129.3, 128.7, 128.3, 128.0, 127.7, 127.4, 127.0, 113.9, 112.5, 70.2, 55.5, 55.0, 38.8, 37.4, 27.7. HRMS (ESI) m/z: [M+H]+ calcd for C50H51N3O7 806.3805; found 806.3804.
5,5′-(pivalamidomethylene)bis(2-(benzyloxy)-N-(4-bromophenyl)-N-methylbenzamide) (S-7)
An oven-dried 50 mL round bottom flask was charged with S-4 (1.00 equiv, 1.40 mmol, 800 mg), HATU (3.00 equiv, 4.20 mmol, 1.60 g), CH2Cl2 (7 mL), and i-Pr2NEt (3.00 equiv, 4.20 mmol, 730 μL). The reaction mixture was stirred at room temperature under nitrogen for 10 minutes and then 4-bromo-N-methylaniline (3.00 equiv, 4.20 mmol, 527 μL) was added dropwise. After 24 hours, an additional portion of i-Pr2NEt (2.00 equiv, 2.80 mmol, 487 μL) and 4-bromo-N-methylaniline (2.00 equiv, 2.80 mmol, 351 μL) were added and the solution was stirred for an additional 14 hours. The reaction mixture was then diluted with EtOAc, washed with 10% aqueous citric acid (2×) and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Bio-tage SP4 system, SNAP Ultra 50 g cartridge; gradient = 30% to 100% EtOAc in hexanes over 10 column volumes; flow rate = 100 mL/min) to afford the desired product as a white solid (0.835 mmol, 755 mg, 60%). IR (neat): 1644, 1490, 1366, 1255, 1011, 832, 735 cm−1. TLC: Rf (2:1 EtO-Ac/hexanes) = 0.35. 1H NMR (Major conformer, 600 MHz, CDCl3): δ 7.42 – 7.29 (m, 12H), 7.18 (d, J = 5.3 Hz, 4H), 6.95 (bs, 2H), 6.76 (d, J = 8.1 Hz, 4H), 6.63 (bs, 2H), 5.93 (bs, 2H), 5.84 (bs, 1H), 4.94 (s, 4H), 3.40 (s, 6H), 1.19 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.4, 168.6, 153.7, 142.9, 136.7, 133.9, 131.8, 129.7, 128.8, 128.6, 128.1, 127.1, 120.2, 112.6, 70.3, 55.1, 38.9, 37.1, 27.7. HRMS (ESI) m/z: [M+H]+ calcd for C48H45Br2N3O5 902.1804; found 902.1791.
5,5′-(pivalamidomethylene)bis(2-(benzyloxy)-N-iso-propyl-N-phenylbenzamide) (S-8)
An oven-dried 50 mL round bottom flask was charged with S-4 (1.00 equiv, 1.40 mmol, 800 mg), HATU (3.00 equiv, 4.20 mmol, 1.60 g), CH2Cl2 (7 mL), and i-Pr2NEt (3.00 equiv, 4.20 mmol, 730 μL). The reaction mixture was stirred at room temperature under nitrogen for 10 minutes and then N-iso-propylaniline (3.00 equiv, 4.20 mmol, 608 μL) was added. After 24 hours, an additional portion of i-Pr2NEt (2.00 equiv, 2.80 mmol, 487 μL) and N-iso-propylaniline (2.00 equiv, 2.80 mmol, 405 μL) were added and the solution was stirred for an additional 14 hours. An additional portion of N-iso-propylaniline (1.00 equiv, 1.4 mmol, 203 μL) was added the solution was stirred for another 8 hours. The reaction mixture was then diluted with EtOAc, washed with 10% aqueous citric acid (2x) and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 50 g cartridge; gradient = 30% to 100% EtOAc in hexanes over 10 column volumes, then 100% EtOAc over 4 column volumes; flow rate = 100 mL/min) to afford the desired product as a white solid (0.75 mmol, 599 mg, 53%). TLC: Rf (1.5:1 EtO-Ac/hexanes) = 0.41. IR (neat): 1637, 1590, 1495, 1392, 1366, 1250, 1117, 733, 700 cm−1. 1H NMR (600 MHz, CDCl3): δ 7.50 – 7.30 (m, 10H), 7.12 – 7.07 (m, 6H), 6.88 (d, J = 7.4 Hz, 4H), 6.77 (bs, 2H), 6.67 – 6.58 (m, 2H), 6.55 (d, J = 8.4 Hz, 2H), 5.84 (d, J = 7.9 Hz, 1H), 5.72 (d, J = 8.0 Hz, 1H), 5.14 (qq, J = 5.8, 5.2 Hz, 2H), 4.98 (s, 4H), 1.19 (s, 9H), 1.11 (d, J = 6.7 Hz, 12H). 13C NMR (151 MHz, CDCl3): δ 177.0, 168.4, 153.3, 138.4, 137.0, 133.6, 130.6, 128.8, 128.7, 128.4, 128.2, 128.0, 127.7, 127.1, 126.9, 112.2, 70.2, 54.9, 46.6, 38.8, 27.7, 21.1. HRMS (ESI) m/z: [M+H]+ calcd for C52H55N3O5 802.4220; found 802.4224.
5,5′-(pivalamidomethylene)bis(2-hydroxy-N-methyl-N-phenylbenzamide) (1)
An oven-dried 250 mL round bottom flask was charged with S-5 (1.00 equiv, 6.84 mmol, 5.10 g), fitted with a septum, and evacuated then back-filled with nitrogen (3×). The nitrogen inlet was removed and replaced with an argon balloon, and the solution was cooled to 0 °C. A solution of BCl3 (1 M in CH2Cl2, 5.00 equiv, 34 mmol, 34 mL) was added slowly and the reaction mixture was stirred vigorously. After 60 minutes, the reaction was quenched by the dropwise addition of water, allowed to warm to room temperature, and then diluted with EtOAc and brine. The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (1:1 EtOAc/hexanes) to afford the desired product as a white solid (5.00 mmol, 2.83 g, 73%). mp: 194 – 195 °C. TLC: Rf (1:1 EtOAc/hexanes) = 0.31. IR (neat): 1633, 1589, 1495, 1368, 1280, 910, 731, 698 cm-1. 1H NMR (600 MHz, CDCl3): δ 10.78 (s, 2H), 7.25 – 7.15 (m, 6H), 6.98 (d, J = 6.9 Hz, 4H), 6.79 (d, J = 8.5 Hz, 2H), 6.49 (dd, J = 8.5, 2.2 Hz, 2H), 6.23 (d, J = 2.3 Hz, 2H), 5.22 (d, J = 7.4 Hz, 1H), 5.14 (d, J = 7.4 Hz, 1H), 3.44 (s, 6H), 1.08 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 176.9, 171.3, 159.8, 145.2, 131.3, 130.9, 129.8, 129.2, 127.3, 126.7, 118.2, 115.9, 55.2, 39.4, 38.6, 27.6. HRMS (ESI) m/z: [M+H]+ calcd for C34H35N3O5 566.2655; found 566.2645.
5,5′-(pivalamidomethylene)bis(2-hydroxy-N-(4-methoxy-phenyl)-N-methylbenzamide) (8)
A flame-dried 50 mL round bottom flask was charged with S-6 (1.00 equiv, 0.95 mmol, 775 mg), fitted with a septum, and purged with argon for 15 minutes. The outlet was then removed, CH2Cl2 was added via syringe, and the solution was cooled to −78 °C. A solution of BCl3 (1 M in CH2Cl2, 2.50 equiv, 2.4 mmol, 2.4 mL) was added slowly and the reaction mixture was stirred vigorously at −78 °C for 60 minutes, then warmed to 0 °C for an additional 2 hours. The reaction was quenched by the dropwise addition of water, allowed to warm to room temperature, and then diluted with EtOAc and brine. The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 50 g cartridge; gradient = 2% to 10% MeOH in CH2Cl2 over 10 column volumes; flow rate = 100 mL/min) to afford the desired product as a white solid (0.79 mmol, 494 mg, 82%). TLC: Rf (2:1 EtOAc/hexanes) = 0.33. IR (neat): 1633, 1511, 1442, 1386, 1248, 1034, 834, 731 cm−1. 1H NMR (600 MHz, CDCl3): δ 10.91 (s, 2H), 6.91 (d, J = 8.7 Hz, 4H), 6.80 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.8 Hz, 4H), 6.53 (dd, J = 8.5, 2.2 Hz, 2H), 6.36 (d, J = 2.3 Hz, 2H), 5.31 – 5.22 (m, 2H), 3.77 (s, 6H), 3.40 (s, 6H), 1.09 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 176.9, 171.2, 159.9, 158.5, 137.9, 131.2, 130.9, 129.0, 127.8, 118.3, 115.9, 114.9, 55.6, 55.4, 39.7, 38.6, 27.6. HRMS (ESI) m/z: [M+H]+ calcd for C36H39N3O7 626.2866; found 626.2859.
5,5′-(pivalamidomethylene)bis(N-(4-bromophenyl)-2-hydr-oxy-N-methylbenzamide) (10)
A flame-dried 50 mL round bottom flask was charged with S-7 (1.00 equiv, 0.80 mmol, 725 mg), fitted with a septum, and purged with argon for 15 minutes. The outlet was then removed, CH2Cl2 was added via syringe, and the solution was cooled to 0 °C. A solution of BCl3 (1 M in CH2Cl2, 5.00 equiv, 4.0 mmol, 4.0 mL) was added slowly and the reaction mixture was stirred vigorously. After 60 minutes, the reaction was quenched by the dropwise addition of water, allowed to warm to room temperature, and then diluted with EtOAc and brine. The organic layer was dried with Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 50 g cartridge; gradient = 50% to 100% EtOAc in hexanes over 8 column volumes; flow rate = 100 mL/min) to afford the desired product as a white solid (0.64 mmol, 462 mg, 80%). TLC: Rf (1.5:1 EtOAc/hexanes) = 0.28. IR (neat): 1635, 1583, 1488, 1367, 1278, 1012, 831, 731 cm−1. 1H NMR (600 MHz, CDCl3): δ 10.59 (s, 2H), 7.40 – 7.36 (m, 4H), 6.92 (d, J = 8.6 Hz, 2H), 6.90 – 6.86 (m, 4H), 6.58 (dd, J = 8.6, 2.3 Hz, 2H), 6.33 (d, J = 2.3 Hz, 2H), 5.37 – 5.25 (m, 2H), 3.43 (s, 6H), 1.11 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.1, 171.3, 159.9, 144.3, 133.0, 131.7, 131.1, 128.7, 128.3, 120.8, 118.9, 115.6, 55.3, 39.3, 38.7, 27.6. HRMS (ESI) m/z: [M+H]+ calcd for C34H33Br2N3O5 722.0865; found 722.0846.
5,5′-(pivalamidomethylene)bis(2-hydroxy-N-iso-propyl-N-phenylbenzamide) (12)
A flame-dried 25 mL round bottom flask was charged with S-8 (1 equiv, 0.71 mmol, 570 mg), fitted with a septum and purged with argon for 15 minutes. The outlet was then removed, CH2Cl2 was added via syringe, and the solution was cooled to 0 °C. A solution of BCl3 (1 M in CH2Cl2, 5.00 equiv, 3.6 mmol, 3.6 mL) was added slowly and the reaction mixture was stirred vigorously for 60 minutes. The reaction was quenched by the dropwise addition of water, allowed to warm to room temperature, and then diluted with EtOAc and brine. The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (Biotage SP4 system, SNAP Ultra 50 g cartridge; gradient = 40% to 100% EtOAc in hexanes over 8 column volumes; flow rate = 100 mL/min) to afford the desired product as a white solid (0.55 mmol, 341 mg, 77%). TLC: Rf (1.5:1 EtOAc/hexanes) = 0.42. IR (neat): 1632, 1588, 1494, 1399, 1351, 1226, 1116, 731, 702 cm−1. 1H NMR (600 MHz, CDCl3): δ 10.72 (s, 2H), 7.24 – 7.15 (m, 6H), 6.92 (d, J = 6.6 Hz, 4H), 6.77 (d, J = 8.5 Hz, 2H), 6.45 (dd, J = 8.5, 2.3 Hz, 2H), 6.21 (d, J = 2.2 Hz, 2H), 5.22 (d, J = 7.5 Hz, 1H), 5.11 (d, J = 7.5 Hz, 1H), 5.02 (hept, J = 6.8 Hz, 2H), 1.20 (d, J = 6.8 Hz, 6H), 1.17 (d, J = 6.8 Hz, 6H), 1.11 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 176.8, 171.0, 159.5, 140.0, 130.9, 130.8, 130.0, 129.2, 128.9, 127.9, 117.8, 117.4, 55.2, 49.0, 38.6, 27.7, 21.0, 20.8. HRMS (ESI) m/z: [M+H]+ calcd for C38H43N3O5 622.3281; found 622.3281.
5,5′-(pivalamidomethylene)bis(2-hydroxy-N-meth-oxy-N-methylbenzamide) (14)
An oven-dried 25 mL round bottom flask was charged with S-4 (1.00 equiv, 0.800 mmol, 450 mg), HATU (3.00 equiv, 2.40 mmol, 913 mg), CH2Cl2 (4 mL), and i-Pr2NEt (3.00 equiv, 2.40 mmol, 420 μL). The reaction mixture was stirred at room temperature under nitrogen for 10 minutes, and then N,O-dimethylhydr-oxylamine hydrochloride (3.00 equiv, 2.40 mmol, 234 mg) was added. After stirring an additional 14 hours, the reaction mixture was diluted with EtOAc, washed with 10% aqueous citric acid (2x) and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude reaction mixture was partially purified by flash column chromatography on silica gel (1:1 hexanes:EtOAc to EtOAc) to afford 5,5′-(pival-amidomethylene)bis(2-(benzyloxy)-N-methoxy-N-methylbenzamide) as a white solid: TLC: Rf (EtOAc) = 0.53. 1H NMR (600 MHz, CDCl3): δ 7.42 – 7.28 (m, 10H), 7.20 –7.05 (m, 4H), 6.87 (d, J = 8.6 Hz, 2H), 6.23 (s, 1H), 6.13 (d, J = 7.6 Hz, 1H), 5.08 (s, 4H), 3.89 – 2.86 (m, 12H), 1.22 (s, 9H). The partially purified material was transferred to a flame-dried 50 mL round bottom flask, dissolved in CH2Cl2 (7 mL), and cooled to 0 °C under argon. A solution of BCl3 (1 M in CH2Cl2, 5.00 equiv, 3.5 mmol, 3.5 mL) was added slowly and the reaction mixture was stirred vigorously. After 60 minutes, the reaction was quenched by the dropwise addition of water, allowed to warm to room temperature, and then diluted with EtOAc and brine. The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel (1:1 EtOAc/hexanes to EtOAc) to afford the desired product as a white solid (0.500 mmol, 236 mg, 62% over 2 steps). TLC: Rf (2:1 EtOAc/hexanes) = 0.28. IR (neat): 1632, 1486, 1366, 1256, 1200, 834, 731 cm−1. 1H NMR (600 MHz, CDCl3): δ 11.21 (s, 2H), 7.80 (d, J = 2.3 Hz, 2H), 7.23 (dd, J = 8.6, 2.3 Hz, 2H), 6.96 (d, J = 8.6 Hz, 2H), 6.08 (d, J = 7.4 Hz, 1H), 5.99 (d, J = 7.5 Hz, 1H), 3.49 (s, 6H), 3.35 (s, 6H), 1.24 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.5, 169.5, 160.6, 132.9, 131.9, 128.6, 118.5, 114.2, 61.3, 55.8, 38.9, 34.0, 27.8. HRMS (ESI) m/z: [M+H]+ calcd for C24H31N3O7 474.2240; found 474.2224.
N-(bis(4-methoxyphenyl)methyl)pivalamide (S-9)
A stirred solution of bis(4-methoxyphenyl)methanamine23 (1.00 equiv, 16.4 mmol, 4.00 g), CH2Cl2 (80 mL) and NEt3 (2.00 equiv, 32.8 mmol, 4.6 mL) in an oven-dried 250 mL round bottom flask was cooled to 0 °C under nitrogen. Pivaloyl chloride (1.20 equiv, 19.7 mmol, 2.4 mL) was added dropwise and the ice bath was then removed. After stirring at room temperature for 2 hours, the reaction mixture was quenched with saturated aqueous sodium bicarbonate, extracted into EtOAc, dried with Na2SO4, filtered, and concentrated in vacuo to afford the desired product as a tan solid (13.0 mmol, 4.85 g, 80%). TLC: Rf (2:1 hexanes/EtOAc) = 0.32. IR (neat): 1632, 1510, 1248, 1176, 1035, 829, 569 cm−1. 1H NMR (600 MHz, CDCl3): δ 7.11 (d, J = 8.5 Hz, 4H), 6.85 (d, J = 8.7 Hz, 4H), 6.11 (d, J = 7.6 Hz, 1H), 6.08 (d, J = 7.8 Hz, 1H), 3.79 (s, 6H), 1.23 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.4, 158.9, 134.3, 128.5, 114.1, 55.8, 55.4, 38.9, 27.8. HRMS (ESI) m/z: [M+H]+ calcd for C20H25NO3 328.1913; found 328.1914.
N-(bis(4-hydroxy-3-isobutyrylphenyl)methyl)-pivalamide (16)
A stirred suspension of AlCl3 (4.00 equiv, 8.6 mmol, 1.1 g) in 1,2-DCE (8.6 mL) in an oven-dried 25 mL 2-neck flask was cooled to 0 °C under nitrogen. Isobu-tyryl chloride (4.50 equiv, 9.7 mmol, 1 mL) was added dropwise, and then the ice bath was removed and the reaction mixture stirred for 20 minutes at room temperature. Over 60 minutes, S-9 (1.00 equiv, 2.15 mmol, 800 mg) was added in 4 portions. After an additional 2.5 hours, the reaction was quenched with ice, then diluted with EtOAc and saturated aqueous sodium bicarbonate. The organic layer was further washed with sodium bicarbonate and brine, dried with Na2SO4, filtered, and concentrated in vacuo. The crude reaction mixture was transferred to a flame-dried 50 mL round bottom flask, dissolved in CH2Cl2 (11 mL), and cooled to 0 °C under argon. A solution of BBr3 (1 M in CH2Cl2, 6.00 equiv, 12.9 mmol, 12.9 mL) was added slowly and the reaction mixture was stirred vigorously for 2 hours, at which point it was quenched with water and extracted into EtOAc (3×). The combined organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. The crude residue was dissolved in MeOH, further concentrated in vacuo (3×), and then purified by flash column chromatography on silica gel (5:1 to 2:1 hexanes/EtOAc) to afford the desired product as a white solid (1.08 mmol, 473 mg, 50%). TLC: Rf (2:1 hexanes/EtOAc) = 0.39. IR (neat): 1640, 1485, 1273, 1196, 1150, 995, 733 cm−1. 1H NMR (600 MHz, CDCl3): δ 12.51 (s, 2H), 7.62 (d, J = 2.2 Hz, 2H), 7.28 (dd, J = 8.7, 2.3 Hz, 2H), 7.00 (d, J = 8.6 Hz, 2H), 6.15 (d, J = 7.5 Hz, 1H), 6.05 (d, J = 7.6 Hz, 1H), 3.47 (hept, J = 6.8 Hz, 2H), 1.26 (s, 9H), 1.21 (d, J = 6.8, 6H), 1.20 (d, J = 6.8, 6H). 13C NMR (151 MHz, CDCl3): δ 210.6, 177.7, 162.8, 135.0, 131.6, 128.8, 119.6, 118.0, 55.6, 39.0, 35.2, 27.8, 19.4, 19.3. HRMS (ESI) m/z: [M+H]+ calcd for C26H33NO5 440.2437; found 440.2426.
Synthesis and Characterization of Peptide Catalysts
The solution phase synthesis of catalysts 4–20 was accomplished using the N-tert-butoxycarbonyl (Boc) protecting group strategy adapted from Miller and coworkers.10e,24 Following the final coupling, peptide catalysts were purified by reverse-phase chromatography (Biotage SP4 system, KP-C18-HS, gradient = 20% to 100% MeCN in H2O). If necessary, a second purification by reverse-phase chromatography (20% to 100% MeOH in H2O with 0.1% formic acid additive) was performed. The clean column fractions were pooled and partially concentrated in vacuo, then diluted with CH2Cl2 and washed with saturated NaHCO3 (2x) and brine (1x). The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo to afford the pure desired peptide. Boc-L-β-Dimethylaminoalanine (Boc-Dmaa-OH) was synthesized according to a literature procedure.25 All other amino acid residues and coupling reagents were purchased from commercial suppliers. Once synthesized, peptides were stored at 0 °C to prevent epimerization and other adverse side-reactivity. Yields are not optimized.
Boc-Dmaa-D-Pro-Acpc-Phe-OMe (4)
Overall yield, 31%. IR (neat): 3319, 1669, 1641, 1532, 1445, 1367, 1250, 1170, 1043, 652 cm−1. (c = 1.0, CHCl3). 1H NMR (600 MHz, CDCl3): δ 7.61 (s, 1H), 7.51 (d, J = 7.5 Hz, 1H), 7.30–7.22 (m, 3H), 7.22–7.15 (m, 2H), 5.73 (s, 1H), 4.70 (q, J = 7.2 Hz, 1H), 4.43 (t, J = 6.2 Hz, 1H), 4.31–4.25 (m, 1H), 4.03–3.95 (m, 1H), 3.61 (s, 3H), 3.56 (q, J = 8.2 Hz, 1H), 3.15–3.07 (m, 2H), 2.66 (t, J = 11.1 Hz, 1H), 2.45–2.36 (m, 1H), 2.28 (s, 6H), 2.19–2.11 (m, 2H), 2.01–1.91 (m, 2H), 1.69–1.57 (m, 1H), 1.42 (s, 9H), 1.37–1.30 (m, 1H), 1.01–0.95 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 172.2, 172.2, 171.5, 171.3, 156.7, 137.0, 129.6, 128.4, 126.7, 80.7, 61.6, 59.4, 54.7, 52.0, 50.7, 47.8, 45.7, 38.4, 34.6, 29.4, 28.4, 24.7, 17.3, 17.1. HRMS (ESI) m/z: [M+H]+ calcd for C29H43N5O7 574.3241; found 574.3235.
Boc-Leu-D-Pro-Acpc-Phe-OMe (5)
Overall yield, 89%. 1H NMR (600 MHz, CDCl3): δ 7.54 (d, J = 7.7 Hz, 1H), 7.30 – 7.15 (m, 6H), 5.26 (d, J = 6.5 Hz, 1H), 4.72 (q, J = 7.3 Hz, 1H), 4.39 (t, J = 6.2 Hz, 1H), 4.30 (dt, J = 8.8, 6.1 Hz, 1H), 4.02 – 3.94 (m, 1H), 3.60 (s, 3H), 3.53 (dt, J = 9.8, 7.5 Hz, 1H), 3.15 – 3.08 (m, 2H), 2.13 (q, J = 6.9 Hz, 2H), 2.04 – 1.93 (m, 2H), 1.75 – 1.65 (m, 1H), 1.64 – 1.47 (m, 3H), 1.43 – 1.34 (m, 1H), 1.41 (s, 9H), 0.99 – 0.93 (m, 2H), 0.98 (d, J = 4.4 Hz, 3H), 0.97 (d, J = 4.5 Hz, 3H). 13C NMR (151 MHz, CDCl3): δ 173.1, 172.3, 172.2, 171.3, 156.7, 136.9, 129.6, 128.4, 126.8, 80.6, 61.4, 54.6, 52.0, 51.1, 47.6, 40.6, 38.5, 34.6, 29.1, 28.4, 24.9, 24.8, 23.4, 22.2, 17.5, 17.3. HRMS (ESI) m/z: [M+H]+ calcd for C30H44N4O7 573.3288; found 573.3287.
Boc-Dmaa-D-Pro-Aib-Phe-OMe (7)
Overall yield, 31%. 1H NMR (600 MHz, CDCl3): δ 7.28–7.26 (m, 2H), 7.22–7.16 (m, 3H), 7.11 (d, J = 8.0 Hz, 1H), 6.90 (s, 1H), 5.71 (bs, 1H), 4.81 (app q, J = 7.1 Hz, 1H), 4.44–4.34 (m, 2H), 3.92–3.84 (m, 1H), 3.64 (s, 3H), 3.58–3.51 (m, 1H), 3.23–3.01 (m, 2H), 2.58–2.48 (m, 2H), 2.28 (s, 6H), 2.29–2.19 (m, 1H), 2.13–2.03 (m, 1H), 2.02–1.90 (m, 2H), 1.48 (s, 3H), 1.42 (s, 9H), 1.37 (s, 3H). 13C NMR (151 MHz, CDCl3): δ 174.1, 172.6, 171.3, 170.8, 155.9, 136.8, 129.5, 128.5, 126.9, 79.9, 61.1, 59.9, 57.2, 53.6, 52.2, 50.9, 47.5, 45.8, 38.2, 28.5, 28.4, 26.1, 24.9, 24.9. HRMS (ESI) m/z: [M+H]+ calcd for C29H41N5O7 576.3397; found 576.3389.
Boc-Dmaa-D-Pro-Gly-Phe-OMe (18)
Overall yield, 28%. 1H NMR (600 MHz, CDCl3): δ 7.63 (s, 1H), 7.33 – 7.14 (m, 6H), 5.74 (s, 1H), 4.76 (q, J = 6.9 Hz, 1H), 4.55 (dd, J = 8.5, 3.2 Hz, 1H), 4.33 – 4.26 (m, 1H), 4.05 – 3.94 (m, 2H), 3.73 (dd, J = 17.0, 5.5 Hz, 1H), 3.65 (s, 3H), 3.55 (dt, J = 9.7, 8.0 Hz, 1H), 3.18 – 3.04 (m, 2H), 2.67 (t, J = 11.1 Hz, 1H), 2.50 – 2.36 (m, 1H), 2.28 (s, 6H), 2.27 – 2.08 (m, 2H), 2.04 – 1.93 (m, 2H), 1.40 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 171.9, 171.7, 171.2, 169.1, 156.7, 136.6, 129.5, 128.5, 126.9, 80.7, 61.3, 59.3, 53.9, 52.2, 50.8, 47.7, 45.7, 43.4, 38.2, 29.4, 28.4, 28.4, 24.6. HRMS (ESI) m/z: [M+H]+ calcd for C27H41N5O7 + H 548.3084; found 548.3082.
Boc-Dmaa-D-Pro-Acbc-Phe-OMe (19)
Overall yield, 59%. 1H NMR (600 MHz, CDCl3): δ 7.43 (s, 1H), 7.29 – 7.14 (m, 6H), 5.67 (s, 1H), 4.73 (q, J = 7.1 Hz, 1H), 4.49 – 4.43 (m, 1H), 4.33 – 4.28 (m, 1H), 4.00 – 3.90 (m, 1H), 3.61 (s, 3H), 3.53 (q, J = 8.2 Hz, 1H), 3.09 (qd, J = 13.8, 6.9 Hz, 2H), 2.77 – 2.56 (m, 3H), 2.42 (dd, J = 12.6, 5.1 Hz, 1H), 2.27 (s, 6H), 2.27 – 1.85 (m, 8H), 1.39 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 173.2, 172.3, 171.1, 171.0, 156.2, 136.9, 129.5, 128.4, 126.8, 80.3, 60.9, 59.5, 59.3, 54.1, 52.0, 50.9, 47.5, 45.6, 38.3, 31.5, 28.8, 28.5, 28.3, 24.7, 15.9. HRMS (ESI) m/z: [M+H]+ calcd for C30H45N5O7 588.3397; found 588.3398.
Boc-Dmaa-D-Pro-Cle-Phe-OMe (20)
Overall yield, 54%. 1H NMR (600 MHz, CDCl3): δ 7.28 – 7.17 (m, 6H), 6.97 (s, 1H), 5.69 (d, J = 4.8 Hz, 1H), 4.77 (q, J = 7.2 Hz, 1H), 4.44 – 4.31 (m, 2H), 3.92 – 3.84 (m, 1H), 3.62 (s, 3H), 3.59 – 3.51 (m, 1H), 3.15 – 3.04 (m, 2H), 2.59 – 2.47 (m, 2H), 2.28 (s, 6H), 2.27 – 2.20 (m, 1H), 2.19 – 2.08 (m, 1H), 2.07 – 1.79 (m, 6H), 1.77 – 1.56 (m, 4H), 1.42 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 173.8, 172.5, 171.4, 171.0, 156.0, 136.9, 129.5, 128.4, 126.8, 80.1, 67.3, 61.1, 59.8, 53.9, 52.1, 50.9, 47.6, 45.8, 38.3, 37.5, 36.8, 28.5, 28.5, 28.4, 24.9, 24.6, 24.5. HRMS (ESI) m/z: [M+H]+ calcd for 602.3554; found 602.3561.
Boc-Dmaa-D-Pro-Achc-Phe-OMe (21)
Overall yield, 34%. 1H NMR (600 MHz, CDCl3): δ 7.28 – 7.15 (m, 6H), 6.89 (s, 1H), 5.69 (d, J = 5.6 Hz, 1H), 4.80 (q, J = 7.3 Hz, 1H), 4.50 – 4.39 (m, 2H), 3.94 – 3.85 (m, 1H), 3.62 (s, 3H), 3.58 (q, J = 8.9 Hz, 1H), 3.16 – 3.01 (m, 2H), 2.64 – 2.49 (m, 2H), 2.34 – 2.28 (m, 1H), 2.29 (s, 6H), 2.20 – 1.90 (m, 5H), 1.79 (td, J = 13.5, 3.7 Hz, 1H), 1.69 (td, J = 13.2, 3.9 Hz, 1H), 1.64 – 1.52 (m, 3H), 1.41 (s, 9H), 1.36 – 1.19 (m, 3H). 13C NMR (151 MHz, CDCl3): δ 174.3, 172.6, 171.6, 170.6, 155.8, 136.9, 129.4, 128.4, 126.8, 79.9, 61.2, 60.3, 60.1, 53.6, 52.1, 50.8, 47.6, 45.8, 38.3, 32.0, 31.5, 28.5, 28.4, 28.2, 25.3, 25.0, 21.6, 21.5. HRMS (ESI) m/z: [M+H]+ calcd for C32H49N5O7 616.3710; found 616.3716.
General Procedure for Desymmetrization Reactions
An oven-dried 4-dram vial was charged with substrate (1.00 equiv, 0.05 mmol), peptide catalyst (10 mol %, 0.005 mmol, 2.9 mg) and a stir bar. Chloroform (830 μL) was added and the reaction mixture was agitated until fully homogenous and then toluene (1.67 mL) was added. The vial was sealed with a Teflon cap and cooled to −50 °C for at least 30 minutes. NBS (1.35 equiv, 0.0675 mmol, 12.0 mg) was added in one portion, and the vial was re-capped quickly, further sealed with Teflon tape and stirred at −50 °C for 48 hours.
For reactions performed using cannula transfer: in an oven-dried 4-dram vial equipped with a stir bar, the substrate (1.00 equiv, 0.05 mmol) and peptide catalyst (10 mol %, 0.005 mmol, 2.9 mg) were dissolved in 2.0 mL of 2:1 PhCH3/CHCl3 and stirred at −50 °C for 30 minutes. The mixture was transferred via a 14 cm-long 18 gauge Teflon cannula into an oven-dried 4-dram vial equipped with a stir bar and NBS (1.35 equiv, 0.0675 mmol, 12.0 mg), which had also been stirring at −50 °C for 30 minutes. The vial that contained the peptide catalyst and substrate mixture was rinsed with 0.25 mL of 2:1 PhCH3:CHCl3 (x2). The vial containing peptide catalyst, substrate, and NBS was then sealed with a Teflon cap and stirred at −50 °C for 48 hours.
The reaction was then quenched with a solution of butyl vinyl ether (1 M in MeOH, 5.00 equiv, 0.25 mmol, 250 μL), warmed to room temperature, passed across a short plug of silica gel with EtOAc, and concentrated in vacuo. The crude reaction mixture was analyzed by 1H NMR spectroscopy with PhTMS as an internal standard in either CDCl3 (with respect to phenol O–H peaks) or methanol-d4 (with respect to diarylmethine C–H peaks). Refer to Supporting Information for full characterization of phenols.
The crude residue was transferred to an oven-dried 4-dram vial, dried in vacuo for at least 2 hours, and treated with acetone (1.0 mL), anhydrous K2CO3 (6 equiv, 0.30 mmol, 41.5 mg) and MeI (30.0 equiv, 1.5 mmol, 93 μL). The reaction vial was sealed with a Teflon cap, submerged in a pre-heated oil bath at 65 °C, and stirred until full conversion of the product mixture was observed by LCMS analysis (8–24 hours). The reaction mixture was allowed to cool to room temperature, passed through a short plug of Celite with CH2Cl2, and concentrated in vacuo. The crude residue was purified by flash column chromatography on silica gel and, as necessary, by reverse-phase C-18 chromatography to remove any remaining N-methylsuccinimide. Enantiomeric ratios were determined by chiral HPLC analysis of the purified product (unless otherwise indicated).
(R)-3-Bromo-2-methoxy-5-((4-methoxy-3-(methyl-(phenyl)carbamoyl)phenyl)(pivalamido)-methyl)-N-methyl-N-phenylbenzamide (2b, Table 2, entry 1)
The crude residue was purified by flash column chromatography on silica gel (2:1 EtOAc:hexanes) to afford the desired product as a white solid (0.033 mmol, 22.3 mg, 66%). HPLC (Chiralpak IC column, 20:80 EtOH/hexanes, 1 mL/min): RT(major) = 19.1 min, RT(minor) = 28.1 min. (c = 0.5, CH2Cl2). mp: 117 – 120 °C. TLC: Rf (2:1 EtOAc/hexanes) = 0.24. IR (neat): 1641, 1594, 1496, 1370, 1257, 1139, 911, 730, 698 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.21 – 6.90 (m, 11H), 6.78 – 6.60 (m, 3H), 6.52 (d, J = 8.1 Hz, 1H), 5.80 (d, J = 7.1 Hz, 1H), 5.66 (d, J = 6.3 Hz, 1H), 3.96 (s, 3H), 3.65 (s, 3H), 3.47 (s, 3H), 3.46 (s, 3H), 1.17 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.2, 168.6, 167.7, 154.9, 152.7, 143.8, 143.5, 138.5, 133.4, 132.8, 132.5, 129.6, 129.2, 128.7, 127.4, 127.4, 127.0, 127.0, 126.9, 126.1, 117.6, 111.1, 62.6, 55.5, 54.7, 38.8, 37.4, 37.1, 27.7. High temp experiment (single conformer): 1H NMR (500 MHz, DMSO-d6, 110 °C): δ 7.60 (d, J = 6.7 Hz, 1H), 7.38 – 6.99 (m, 13H), 6.94 (d, J = 7.2 Hz, 1H), 6.74 (d, J = 8.3 Hz, 1H), 5.93 (d, J = 8.5 Hz, 1H), 3.84 (s, 3H), 3.63 (s, 3H), 3.33 (s, 3H), 3.31 (s, 3H), 1.16 (s, 9H). HRMS (ESI) m/z: [M+H]+ calcd for C36H38BrN3O5 672.2073; found 672.2072.
5,5′-(pivalamidomethylene)bis(3-bromo-2-methoxy-N-methyl-N-phenylbenzamide) (3b)
Experimental procedure reported above for purification of 2b; additional purification by reverse-phase column chromatography (Biotage SP4 system, KP-C18-HS 12 g column; gradient = 15% to 100% MeCN in H2O over 12 column volumes, then 100% MeCN over 5 column volumes; flow rate = 12 mL/min) to afford the title compound as a white solid (0.013 mmol, 9.8 mg, 26%). TLC: Rf (2:1 EtOAc/hexanes) = 0.39. IR (neat): 1639, 1594, 1471, 1371, 1257, 997, 731, 698 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.23 – 7.11 (m, 6H), 6.97 (d, J = 7.6 Hz, 4H), 6.90 (s, 2H), 6.57 (s, 2H), 5.72 (d, J = 7.6 Hz, 1H), 5.56 (d, J = 7.8 Hz, 1H), 3.96 (s, 6H), 3.47 (s, 6H), 1.17 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.2, 167.5, 153.1, 143.3, 137.5, 133.6, 132.6, 129.3, 127.6, 126.9, 126.2, 118.0, 62.6, 54.4, 37.3, 29.9, 27.7. HRMS (ESI) m/z: [M+H]+ calcd for C36H37Br2N3O5 750.1168; found 750.1178.
(R)-3-bromo-2-methoxy-5-((4-methoxy-3-((4-methoxy-phenyl)(methyl)carbamoyl)phenyl)-(pivalamido)methyl)-N-(4-methoxyphenyl)-N-methylbenzamide (9, Table 2, entry 2)
The crude residue was purified by flash column chromatography on silica gel (2:1 EtOAc/hexanes to EtOAc; 2x) to afford the desired product as a white solid (0.025 mmol, 18.0 mg, 49%). HPLC: (Chiralpak IC column, 20:80 EtOH/hexanes, 1 mL/min): RT(major) = 25.5 min, RT(minor) = 36.1 min. (c = 0.5, CH2Cl2). TLC: Rf (3:1 EtOAc/hexanes) = 0.25. IR (neat): 1639, 1512, 1374, 1248, 1036, 836, 730 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.06 (s, 1H), 6.95 – 6.82 (m, 5H), 6.72 – 6.63 (m, 5H), 6.59 – 6.48 (m, 2H), 5.83 (d, J = 7.7 Hz, 1H), 5.72 (d, J = 7.5 Hz, 1H), 3.95 (s, 3H), 3.71 (s, 3H), 3.71 (s, 3H), 3.68 (s, 3H), 3.42 (s, 3H), 3.41 (s, 3H), 1.17 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.3, 168.9, 167.9, 158.4, 158.2, 154.6, 152.5, 138.5, 136.7, 136.3, 133.7, 132.6, 132.5, 129.4, 128.1, 127.4, 127.2, 126.1, 117.6, 114.4, 113.9, 111.1, 62.6, 55.5, 54.7, 38.8, 37.6, 37.4, 27.7. HRMS (ESI) m/z: [M+H]+ calcd for C38H42BrN3O7 732.2284; found 732.2293.
(R)-3-bromo-N-(4-bromophenyl)-5-((3-((4-bromo-phenyl)(methyl)carbamoyl)-4-methoxyphenyl)-(pival-amido)methyl)-2-methoxy-N-methylbenzamide (11, Table 2, Entry 3)
The crude residue was purified by preparative thin layer chromatography on silica gel (2:1 EtO-Ac/hexanes; 3×), followed by reverse-phase column chromatography (Biotage SP4 system, KP-C18-HS 30 g column; gradient = 30% to 100% MeCN in H2O over 10 column volumes, then 100% MeCN over 5 column volumes; flow rate = 25 mL/min) to afford the desired product as a white solid (0.030 mmol, 24.5 mg, 53%). HPLC (Chiralpak IC column, 10:90 EtOH/hexanes, 1 mL/min): RT(major) = 37.2 min, RT(minor) = 50.4 min. (c = 0.5, CH2Cl2). TLC: Rf (2:1 EtOAc/hexanes) = 0.23. IR (neat): 1643, 1489, 1367, 1258, 1011, 832, 731 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.31 – 7.23 (m, 4H), 7.14 (s, 1H), 6.96 – 6.83 (m, 5H), 6.71 – 6.47 (m, 3H), 5.83 (s, 1H), 5.75 (s, 1H), 3.95 (s, 3H), 3.66 (s, 3H), 3.44 (s, 3H), 3.42 (s, 3H), 1.18 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.4, 168.5, 167.5, 154.7, 152.8, 142.9, 142.6, 138.6, 133.4, 132.9, 132.7, 132.4, 131.9, 129.8, 128.6, 128.5, 127.3, 126.8, 126.2, 120.8, 120.5, 117.8, 111.3, 62.7, 55.6, 54.8, 38.9, 37.3, 37.2, 27.7. HRMS (ESI) m/z: [M+H]+ calcd for C36H36N3Br3O5 828.0283; found 828.0279.
(R)-3-bromo-N-isopropyl-5-((3-(isopropyl(phenyl)-carb-amoyl)-4-methoxyphenyl)(pivalamido)methyl)-2-methoxy-N-phenylbenzamide (13, Table 2, entry 4)
The crude residue was purified by flash column chromatography on silica gel (1.5:1 EtOAc/hexanes) followed by reverse-phase column chromatography (Biotage SP4 system, KP-C18-HS 30 g column; gradient = 30% to 100% MeCN in H2O over 10 column volumes, then 100% MeCN over 5 column volumes; flow rate = 25 mL/min) to afford the desired product as a white solid (0.030 mmol, 21.8 mg, 60%). HPLC (Chiralpak IC column, 10:90 EtOH/hexanes, 1 mL/min): RT(major) = 22.9 min, RT(minor) = 29.1 min. (c = 0.5, CH2Cl2). TLC: Rf (1.5:1 EtOAc/hexanes) = 0.32. IR (neat): 1634, 1495, 1471, 1347, 1251, 1117, 730, 701 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.18 – 7.07 (m, 6H), 7.03 – 6.79 (m, 5H), 6.64 (bs, 2H), 6.55 (bs, 1H), 6.50 (d, J = 8.5 Hz, 1H), 5.76 (d, J = 7.7 Hz, 1H), 5.65 (d, J = 7.4 Hz, 1H), 5.16 (h, J = 6.8 Hz, 1H), 5.10 (h, J = 6.8 Hz, 1H), 3.94 (s, 3H), 3.73 (s, 3H), 1.18 (s, 9H), 1.19 – 1.14 (m, 12H). 13C NMR (151 MHz, CDCl3): δ 177.0, 168.2, 167.4, 154.4, 151.9, 138.4, 138.4, 138.0, 134.3, 132.3, 132.1, 130.3, 128.9, 128.6, 128.2, 128.1, 128.1, 127.9, 127.0, 125.7, 117.3, 110.9, 62.5, 55.5, 54.6, 46.9, 46.6, 38.8, 27.7, 21.3, 21.2, 21.1, 21.0. HRMS (ESI) m/z: [M+H]+ calcd for C40H46BrN3O5 728.2699; found 728.2701.
(R)-3-bromo-N,2-dimethoxy-5-((4-methoxy-3-(methoxy-(methyl)carbamoyl)phenyl)(pivalamido)-methyl)-N-methylbenzamide (15, Table 2, entry 5)
The crude residue was purified by preparative thin layer chromatography (EtOAc) to afford the desired product as a white semi-solid (15.6 mg, 0.027 mmol, 54%). HPLC (Chiralpak IC column, 20:80 EtOH/hexanes, 1 mL/min): RT(major) = 28.7 min, RT(minor) = 42.8 min. (c = 0.5, CH2Cl2). TLC: Rf (EtOAc) =0.27. IR (neat): 1647, 1501, 1473, 1259, 1184, 997, 732 cm−1. Major conformer: 1H NMR (600 MHz, CDCl3): δ 7.41 (s, 1H), 7.19 (d, J = 7.7 Hz, 1H), 7.10 – 7.00 (m, 2H), 6.90 (d, J = 8.6 Hz, 1H), 6.12 (d, J = 7.5 Hz, 1H), 6.05 (d, J = 7.5 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.41 (bs, 3H), 3.32 (bs, 3H), 1.22 (s, 9H). 1H NMR (500 MHz, DMSO-d6, 110 °C): δ 7.89 (d, J = 8.5 Hz, 1H), 7.56 (s, 1H), 7.31 (d, J = 8.6 Hz, 1H), 7.21 (s, 2H), 7.17 (s, 1H), 7.05 (d, J = 8.6 Hz, 1H), 6.16 (d, J = 8.5 Hz, 1H), 3.82 – 3.76 (m, 6H), 3.53 – 3.45 (m, 6H), 3.21 (s, 3H), 3.16 (s, 3H), 1.17 (s, 9H). 13C NMR (126 MHz, DMSO-d6, 110 °C): δ 176.2, 154.3, 151.3, 139.4, 133.2, 132.2, 130.5, 128.9, 128.4, 127.5, 125.9, 125.5, 125.2, 115.2, 111.4, 60.8, 60.2, 60.0, 55.5, 53.7, 37.6, 32.5, 27.5, 26.7. HRMS (ESI) m/z: [M+H]+ calcd for C26H34BrN3O7 580.1658; found 580.1662.
(R)-N-((3-bromo-5-isobutyryl-4-methoxyphenyl)(3-iso-butyryl-4-methoxyphenyl)methyl)pivalamide (17, Table 2, entry 6)
The crude residue was purified by flash column chromatography on silica gel (2:1 hexanes:EtOAc) to afford the desired product as a white semi-solid (15.0 mg, 0.027 mmol, 55%). HPLC (Chiralpak IC column, 10:90 EtOH/hexanes): RT(minor) = 10.1 min, RT(major) = 11.2 min. (c = 0.5, CH2Cl2). TLC: Rf (2:1 hexanes/EtOAc) = 0.25. IR (neat): 1640, 1485, 1363, 1273, 1196, 995, 733 cm−1. 1H NMR (600 MHz, CDCl3): δ 7.46 (d, J = 2.1 Hz, 1H), 7.35 (d, J = 2.4 Hz, 1H), 7.22 (dd, J = 8.6, 2.4 Hz, 1H), 7.15 (d, J = 2.1 Hz, 1H), 6.92 (d, J = 8.6 Hz, 1H), 6.11 (d, J = 7.5 Hz, 1H), 6.06 (d, J = 7.5 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.44 (hept, J = 6.9 Hz, 1H), 3.30 (hept, J = 6.9 Hz, 1H), 1.24 (s, 9H), 1.12, (d, J = 6.8 Hz, 6H), 1.12 (d, J = 6.9 Hz, 6H). 13C NMR (151 MHz, CDCl3): δ 207.6, 207.6, 177.7, 157.5, 154.1, 139.2, 135.9, 134.6, 132.9, 132.0, 129.3, 128.9, 126.8, 118.5, 112.1, 62.8, 55.9, 55.2, 40.3, 40.2, 38.9, 27.7, 18.7, 18.6, 18.6. HRMS (ESI) m/z: [M+H]+ calcd for C28H36BrN3O5 546.1855; found 546.1865.
Kinetic Resolution Studies
Experimental details for kinetic resolutions (Eq 2)
Performed according to general procedure for desymmetrization reactions with 0.45 equiv NBS (0.023 mmol, 4.0 mg) to control reaction conversion. Conversion values were determined by 1H NMR analysis of the crude bromination reaction mixtures in methanol-d4. The enantiomeric excess of 2b was used to calculate krel values by the method of Kagan26 as determined by chiral HPLC analysis of the crude reaction mixture following methylation (IC column, Chiralpak IC column, 20:80 EtOH/hexanes, 1 mL/min): RT(major) = 19.1 min, RT(minor) = 28.1 min). Additional details are provided in the Supporting Information.
(±)-3-bromo-2-hydroxy-5-((4-hydroxy-3-(methyl-(phenyl)carbamoyl)phenyl)(pivalamido)methyl)-N-methyl-N-phenylbenzamide [(±)-2a]
An oven-dried 250 mL round bottom flask was charged with 5,5′-(pivalamidomethylene)bis(2-hydroxy-N-methyl-N-phenyl-benzamide) (1), CHCl3 (22 mL) and toluene (43 mL). NBS (1.3 equiv, 1.7 mmol, 305 mg) was added in 3 portions over 5 minutes. The reaction mixture was stirred at room temperature under nitrogen in the dark for 16 hours, at which point it was quenched with a solution of butyl vinyl ether (1 M in MeOH, 2 equiv, 2.6 mmol, 260 μL) and concentrated in vacuo. The crude residue was purified by reverse-phase chromatography (Biotage SP4 system, KP-C18-HS 120 g cartridge; gradient = 10% to 100% MeCN in H2O over 14 column volumes; flow rate = 50 mL/min) to afford the desired product as a white solid (0.45 mmol, 289 mg, 34%). TLC: Rf (1:1 EtOAc/hexanes) = 0.29. IR (neat): 1633, 1581, 1495, 1278, 1192, 909, 732, 699 cm−1. 1H NMR (600 MHz, CDCl3): δ 11.43 (s, 1H), 10.90 (s, 1H), 7.33 – 7.16 (m, 6H), 7.01 – 6.96 (m, 4H), 6.81 (d, J = 8.5 Hz, 1H), 6.79 (dd, J = 2.1, 0.6 Hz, 1H), 6.49 (dd, J = 8.6, 2.2 Hz, 1H), 6.25 (dd, J = 2.1, 0.6 Hz, 1H), 6.15 (d, J = 2.3 Hz, 1H), 5.17 (d, J = 7.2 Hz, 1H), 5.09 (d, J = 7.3 Hz, 1H), 3.45 (s, 3H), 3.44 (s, 3H), 1.09 (s, 9H). 1H NMR (500 MHz, Methanol-d4): δ 7.55 (d, J = 8.4 Hz, 1H, –NHPiv, partially exchanges), 7.30 – 7.03 (m, 11H), 6.71 – 6.63 (m, 3H), 6.61 (d, J = 8.3 Hz, 1H), 5.69 – 5.64 (m, 1H), 3.43 (s, 3H), 3.42 (s, 3H), 1.16 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.0, 171.1, 170.6, 160.2, 156.4, 145.1, 144.8, 133.9, 132.0, 131.4, 130.3, 129.9, 129.8, 129.4, 128.4, 127.6, 127.6, 126.7, 126.7, 118.4, 117.2, 115.9, 111.9, 54.9, 39.6, 39.3, 38.7, 27.6. HRMS (ESI) m/z: [M+H]+ calcd for C34H34BrN3O5 644.1760; found 644.1760.
5,5′-(pivalamidomethylene)bis(3-bromo-2-hydroxy-N-methyl-N-phenylbenzamide) (3a)
Experimental procedure reported above for (±)-2a. TLC: Rf (1:1 hexanes/EtOAc) = 0.38. IR (neat): 1631, 1581, 1495, 1454, 1349, 1276, 1191, 1135, 731, 700 cm−1. 1H NMR (600 MHz, CDCl3) δ 11.59 (s, 2H), 7.33 – 7.20 (m, 6H), 6.98 (d, J = 6.9 Hz, 4H), 6.80 (d, J = 2.1 Hz, 2H), 6.16 (d, J = 2.1 Hz, 2H), 5.15 (d, J = 7.2 Hz, 1H), 5.03 (d, J = 7.2 Hz, 1H), 3.45 (s, 6H), 1.10 (s, 9H). 1H NMR (500 MHz, Methanol-d4): δ 7.60 (d, J = 8.2 Hz, 1H, –NHPiv, partially exchanges), 7.29 – 7.16 (m, 6H), 7.10 (d, J = 7.5 Hz, 4H), 7.05 (d, J = 1.3 Hz, 2H), 6.59 (s, 2H), 5.58 (bs, 1H), 3.43 (s, 6H), 1.15 (s, 9H). 13C NMR (151 MHz, CDCl3): δ 177.0, 170.3, 156.8, 144.7, 134.0, 131.3, 129.9, 128.6, 127.9, 126.7, 117.1, 112.1, 54.5, 39.5, 38.7, 27.6. HRMS (ESI) m/z: [M+H]+ calcd for C34H33Br2N3O5 722.0865; found 722.0860.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of General Medical Science of the National Institutes of Health (GM-068649) for research support. A.J.M acknowledges the NSF Graduate Research Fellowship Program for early support. E.A.S. acknowledges the research support of the NIH Molecular Biophysics Predoctoral Training Grant (T32 GM008283). We would also like to thank Dr. Brandon Mercado for solving the X-ray crystal structures of 1 and 2a.
Footnotes
Notes
The authors declare no competing financial interests. Crystallographic data are deposited with the Cambridge Crystallographic Data Center under the accession number CCDC 1573490 (1), 1570899 (2a), 1510509 (4), CCDC 1510523 (7).
Associated Content
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Scheme for the substrate synthesis (Scheme S1); 1H and 13C NMR spectra of all compounds; HPLC traces of 2b, 9, 11, 13, 15, and 17; additional data for the τ-angle and kinetic resolution studies; and X-ray crystallographic data for compounds 1 and 2a.
References
- 1.(a) Ameen D, Snape TJ. Med Chem Comm. 2013;4:893. [Google Scholar]; (b) Naito R, Yonetoku Y, Okamoto Y, Toyoshima A, Ikeda K, Takeuchi M. J Med Chem. 2005;48:6597. doi: 10.1021/jm050099q. [DOI] [PubMed] [Google Scholar]; (c) Plobeck N, Delorme D, Wei ZY, Yang H, Zhou F, Schwarz P, Gawell L, Gagnon H, Pelcman B, Schmidt R, Yue SY, Walpole C, Brown W, Zhou E, Labarre M, Payza K, St-Onge S, Kamassah A, Morin PE, Projean D, Ducharme J, Roberts E. J Med Chem. 2000;43:3878. doi: 10.1021/jm000228x. [DOI] [PubMed] [Google Scholar]; (d) Calderon SN, Rice KC, Rothman RB, Porreca F, Flippen-Anderson JL, Kayakiri H, Xu H, Becketts K, Smith LE, Bilsky EJ, Davis P, Horvath R. J Med Chem. 1997;40:695. doi: 10.1021/jm960319n. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt F, Stemmler RT, Rudolph J, Bolm C. Chem Soc Rev. 2006;35:454. doi: 10.1039/b600091f. [DOI] [PubMed] [Google Scholar]
- 3.Selected examples: Wangweerawong A, Bergman RG, Ellman JA. J Am Chem Soc. 2014;136:8520. doi: 10.1021/ja5033452.Chen CC, Gopula B, Syu JF, Pan JH, Kuo TS, Wu PY, Henschke JP, Wu HL. J Org Chem. 2014;79:8077. doi: 10.1021/jo5012653.Shao C, Yu HJ, Wu NY, Feng CG, Lin GQ. Org Lett. 2010;12:3820. doi: 10.1021/ol101531r.Wang ZQ, Feng CG, Xu MH, Lin GQ. J Am Chem Soc. 2007;129:5336. doi: 10.1021/ja0710914.Jagt RB, Toullec PY, Geerdink D, de Vries JG, Feringa BL, Minnaard AJ. Angew Chem Int Ed. 2006;45:2789. doi: 10.1002/anie.200504309.Duan HF, Jia YX, Wang LX, Zhou QL. Org Lett. 2006;8:2567. doi: 10.1021/ol060755w.Tokunaga N, Otomaru Y, Okamoto K, Ueyama K, Shintani R, Hayashi T. J Am Chem Soc. 2004;126:13584. doi: 10.1021/ja044790e.Kuriyama M, Soeta T, Hao X, Chen Q, Tomioka K. J Am Chem Soc. 2004;126:8128. doi: 10.1021/ja0475398.Hayashi T, Kawai M, Tokunaga N. Angew Chem Int Ed. 2004;43:6125. doi: 10.1002/anie.200461338.
- 4.Selected examples: Touge T, Nara H, Fujiwhara M, Kayaki Y, Ikariya T. J Am Chem Soc. 2016;138:10084. doi: 10.1021/jacs.6b05738.Hou G, Tao R, Sun Y, Zhang X, Gosselin F. J Am Chem Soc. 2010;132:2124. doi: 10.1021/ja909583s.
- 5.Selected examples: Yamamoto E, Hilton MJ, Orlandi M, Saini V, Toste FD, Sigman MS. J Am Chem Soc. 2016;138:15877. doi: 10.1021/jacs.6b11367.Tabuchi S, Hirano K, Miura M. Angew Chem Int Ed. 2016;55:6973. doi: 10.1002/anie.201602075.Guduguntla S, Hornillos V, Tessier R, Fañanás-Mastral M, Feringa BL. Org Lett. 2016;18:252. doi: 10.1021/acs.orglett.5b03396.Friis SD, Pirnot MT, Buchwald SL. J Am Chem Soc. 2016;138:8372. doi: 10.1021/jacs.6b04566.Xu B, Li ML, Zuo XD, Zhu SF, Zhou QL. J Am Chem Soc. 2015;137:8700. doi: 10.1021/jacs.5b05086.Kim B, Chinn AJ, Fandrick DR, Senanayake CH, Singer RA, Miller SJ. J Am Chem Soc. 2016;138:7939. doi: 10.1021/jacs.6b03444.
- 6.(a) Su B, Zhou TG, Xu PL, Shi ZJ, Hartwig JF. Angew Chem Int Ed. 2017;56:7205. doi: 10.1002/anie.201702628. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Su B, Zhou TG, Li XW, Shao XR, Xu PL, Wu WL, Hartwig JF, Shi ZJ. Angew Chem Int Ed. 2017;56:1092. doi: 10.1002/anie.201609939. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lee T, Wilson TW, Berg R, Ryberg P, Hartwig JF. J Am Chem Soc. 2015;137:6742. doi: 10.1021/jacs.5b03091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Laforteza BN, Chan KS, Yu JQ. Angew Chem Int Ed. 2015;54:11143. doi: 10.1002/anie.201505204. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chu L, Wang XC, Moore CE, Rheingold AL, Yu JQ. J Am Chem Soc. 2013;135:16344. doi: 10.1021/ja408864c. [DOI] [PubMed] [Google Scholar]; (c) Cheng XF, Li Y, Su YM, Yin F, Wang JY, Sheng J, Vora HU, Wang XS, Yu JQ. J Am Chem Soc. 2013;135:1236. doi: 10.1021/ja311259x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2006;128:16454. doi: 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]
- 9.Lewis CA, Gustafson JL, Chiu A, Balsells J, Pollard D, Murry J, Reamer RA, Hansen KB, Miller SJ. J Am Chem Soc. 2008;130:16358. doi: 10.1021/ja807120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Miyaji R, Asano K, Matsubara S. J Am Chem Soc. 2015;137:6766. doi: 10.1021/jacs.5b04151. [DOI] [PubMed] [Google Scholar]; (b) Diener ME, Metrano AJ, Kusano S, Miller SJ. J Am Chem Soc. 2015;137:12369. doi: 10.1021/jacs.5b07726. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barrett KT, Metrano AJ, Rablen PR, Miller SJ. Nature. 2014;509:71. doi: 10.1038/nature13189. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mori K, Ichikawa Y, Kobayashi M, Shibata Y, Yamanaka M, Akiyama T. J Am Chem Soc. 2013;135:3964. doi: 10.1021/ja311902f. [DOI] [PubMed] [Google Scholar]; (e) Barrett KT, Miller SJ. J Am Chem Soc. 2013;135:2963. doi: 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Garand E, Kamrath MZ, Jordan PA, Wolk AB, Leavitt CM, McCoy AB, Miller SJ, Johnson MA. Science. 2012;335:694. doi: 10.1126/science.1214948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gustafson JL, Lim D, Barrett KT, Miller SJ. Angew Chem Int Ed. 2011;50:5125. doi: 10.1002/anie.201101147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gustafson JL, Lim D, Miller SJ. Science. 2010;328:1251. doi: 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Pathak TP, Miller SJ. J Am Chem Soc. 2013;135:8415. doi: 10.1021/ja4038998. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pathak TP, Miller SJ. J Am Chem Soc. 2012;134:6120. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galabov B, Nalbantova D, Schleyer P, Schaefer HF., 3rd Acc Chem Res. 2016;49:1191. doi: 10.1021/acs.accounts.6b00120. [DOI] [PubMed] [Google Scholar]
- 13.Sibi MP, Liu M. Curr Org Chem. 2001;5:719. [Google Scholar]
- 14.Schreiber SL, Schreiber TS, Smith DB. J Am Chem Soc. 1987;109:1525. for a relevant recent example, see also: ref 10d. [Google Scholar]
- 15.(a) Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Hurtley AE, Miller SJ. J Am Chem Soc. 2017;139:492. doi: 10.1021/jacs.6b11348. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Miller SJ. Chem Commun. 2016;52:4816. doi: 10.1039/c6cc01428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For example: Denmark SE, Fu J. Chem Rev. 2003;103:2763–2794. doi: 10.1021/cr020050h.Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069–1094. doi: 10.1021/cr980414z.Wong OA, Shi Y. Chem Rev. 2008;108:3958–3987. doi: 10.1021/cr068367v.Eliel EL, Wilen S, Mander LN. Stereochemistry of Organic Compounds. Wiley Interscience; New York, NY: 1994. Corey EJ, Kürti L. Enantioselective Chemical Synthesis: Methods Logic and Practice. Direct Book Publishing; Dallas, TX: 2010.
- 17.(a) Gustafson JL, Lim D, Miller SJ. Science. 2010;328:1251–1255. doi: 10.1126/science.1188403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Garand E, Kamrath MZ, Jordan PA, Work AB, McCoy AB, Miller SJ, Johnson MA. Science. 2012;335:694–698. doi: 10.1126/science.1214948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barrett KT, Miller SJ. J Am Chem Soc. 2013;135:2963–2966. doi: 10.1021/ja400082x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selected prior structural studies on C-α,α disubstituted glycine-containing peptides: Crisma M, De Borggraeve WM, Peggion C, Formaggio F, Royo S, Jimenez AI, Cativiela C, Toniolo C. Chemistry. 2005;12:251. doi: 10.1002/chem.200500865.Toniolo C, Crisma M, Formaggio F, Peggion C. Biopolymers. 2001;60:396. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. and references therein.
- 19.(a) Venkatraman J, Shankaramma SC, Balaram P. Chem Rev. 2001;101:3131. doi: 10.1021/cr000053z. [DOI] [PubMed] [Google Scholar]; (b) Gunasekaran K, Gomathi L, Ramakrishnan C, Chandrasekhar J, Balaram P. J Mol Biol. 1998;284:1505. doi: 10.1006/jmbi.1998.2154. [DOI] [PubMed] [Google Scholar]; (c) Wilmot CM, Thornton JM. J Mol Biol. 1998;203:221. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]; (d) Haque TS, Little JC, Gellman SH. J Am Chem Soc. 1996;118:6975. [Google Scholar]; (e) Haque TS, Little JC, Gellman SH. J Am Chem Soc. 1994;116:4105. [Google Scholar]; (f) Hutchinson EG, Thornton JM. Protein Science. 1994;3:2207. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denmark SE, Beutner GL. Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 21.(a) Denmark SE, Burk MT. Proc Nat Acad Sci USA. 2010;107:20655–20660. doi: 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maddox SM, Nalbandian CJ, Smith DE, Gustafson JL. Org Lett. 2015;17:1042–1045. doi: 10.1021/acs.orglett.5b00186. [DOI] [PubMed] [Google Scholar]
- 22.Gagnepain J, Castet F, Quideau S. Angew Chem Int Ed. 2007;46:1533. doi: 10.1002/anie.200604610. [DOI] [PubMed] [Google Scholar]
- 23.Buckingham F, Kirjavainen AK, Forsback S, Krzyczmonik A, Keller T, Newington IM, Glaser M, Luthra SK, Solin O, Gouverneur V. Angew Chem Int Ed. 2015;54:13366. doi: 10.1002/anie.201506035. [DOI] [PubMed] [Google Scholar]
- 24.General review: El-Faham A, Albericio F. Chem Rev. 2011;111:6557. doi: 10.1021/cr100048w.. For additional experimental details, see also: ref 10e.
- 25.Zhang LH, Kauffman GS, Presti JA, Yin JJ. J Org Chem. 1997;62:6918. [Google Scholar]
- 26.(a) Kagan HB, Fiaud JC. In: In Topics in Stereochemistry. Eliel EL, Fiaud JC, editors. Vol. 18. Wiley; New York: 1988. p. 24. [Google Scholar]; (b) Keith JM, Larrow JF, Jacobsen EN. Adv Synth Catal. 2001;343:5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.