

Abstract
HdeA is a periplasmic chaperone that is rapidly activated upon shifting the pH to acidic conditions. This activation is thought to involve monomerization of HdeA. There is evidence that monomerization and partial unfolding allow the chaperone to bind to proteins denatured by low pH thereby protecting them from aggregation. We analyzed the acid-induced unfolding of HdeA using NMR spectroscopy and fluorescence measurements, and obtained experimental evidence suggesting a complex mechanism in HdeA’s acid-induced unfolding pathway, as previously postulated from molecular dynamics simulations. Counterintuitively, dissociation constant measurements show a stabilization of the HdeA dimer upon exposure to mildly acidic conditions. We provide experimental evidence that protonation of Glu37, a glutamate residue embedded in a hydrophobic pocket of HdeA, is important in controlling HdeA stabilization and thus, the acid activation of this chaperone. Our data also reveal a sharp transition from folded dimer to unfolded monomer between pH 3 and 2, and suggest the existence of a low-populated, partially folded intermediate that could assist in chaperone activation or function. Overall, this study provides a detailed experimental investigation into the mechanism by which HdeA unfolds and activates.
Keywords: chaperone, protein folding, acid, NMR
Graphical Abstract
After oral ingestion, Escherichia coli must transit through the low pH environment of the stomach before it can colonize the large intestine. The E. coli periplasmic space is sensitive to low pH conditions due to the permeable nature of the outer membrane [1], which allows the free diffusion of protons present in stomach acid into the periplasmic space. Thus, upon ingestion, the periplasmic space of E. coli rapidly equilibrates with the outside environment, causing a dramatic drop in the periplasmic pH. The acidic pH rapidly denatures periplasmic proteins [2], thereby disabling periplasmic function and endangering the cell [3]. The rapid drop in pH leaves the bacteria little time to respond using transcriptional or translational mechanisms. However, E. coli can respond to rapid pH changes at the post-translational level. It does so by the acid-induced activation of the chaperone HdeA.
At neutral pH, HdeA is present as a chaperone-inactive but well-structured dimer. With the shift to low pH in the stomach, the dimer rapidly unfolds and monomerizes, and within seconds, becomes active as a chaperone [4, 5]. When E. coli moves to the small intestine, the pH of the periplasm returns to near neutral values. HdeA is then able to release its clients, refold, and re-dimerize [6]. The mechanism and structural changes that mediate HdeA’s remarkable post-translational activation, however, are not yet clearly defined.
Recent structural and computational work has begun to provide possible explanations for how HdeA could unfold. Using coarse-grained molecular dynamics (MD) simulations, Ahlstrom et al. suggested that at low pH, HdeA unfolds upon acid shift by transitioning through a low-populated dimeric intermediate state. This intermediate differs from the HdeA dimeric state in that the C terminus is largely unfolded [7]. The presence of an intermediate in the folding pathway of HdeA has thus far gone undetected using experimental approaches. In a further study, Ahlstrom et al. investigated HdeA at various pHs and suggested that the thermodynamic stability of dimeric HdeA should be maximal at pH 5 and not simply continuously decline with decreasing pH [8]. Additionally, NMR characterization of HdeA’s unfolding process suggested a gradual loss of tertiary and quaternary structure during shifts from pH 6 to 3 [9, 10]. These NMR experiments also provided partial validation of acid dissociation constant (pKa) values for certain residues within HdeA, some of which have been postulated to be involved in the pH-dependent transition of the protein [9, 10].
Since the monomerization and unfolding of HdeA that occurs upon shift to low pH is closely associated with its rapid pH-mediated activation as a chaperone [4–6, 11], we sought to understand the pH dependence of HdeA activation in detail. Using isothermal titration calorimetry (ITC) dimer dissociation experiments, we determined the self-dissociation constant (KD) of HdeA (monomer-dimer equilibrium) to be 64 μM at pH 2.3. Additionally, we determined that self-dissociation is entropy-driven at low pH (Table 1). This result is in good agreement with previously published analytical ultracentrifugation experiments, which found the KD of HdeA dimerization to be ~ 60 μM at pH 2 [5]. However, The ITC signal becomes too weak at pH > 4 for accurate KD determination, indicating that the entropy (ΔS) of monomerization becomes less favorable at higher pH. Therefore, to determine the dimerization dissociation constants between pH 4 and 7, we switched to microscale thermophoresis (MST) experiments (Table 1). As predicted from simulations [8], the tightest binding of HdeA to itself occurs at pH 5, with the binding affinity gradually decreasing as the pH is either raised or lowered from this value. These measured KD values are in excellent quantitative agreement with the predictions from MD simulations (Table 1) [8]. They also suggest a more complex mechanism than indicated by the model of a simple gradual loosening of the structure with decreasing pH [9]. This model predicts that the KD will consistently gradually increase (the binding affinity will decrease) with decreasing pH, which was not observed in our experiments. To further our understanding of these processes, we decided to probe deeper into the counterintuitive changes in HdeA dimer stability that occur between pH 5 and 7.
Table 1.
Self-dissociation constants (KD) of HdeA and HdeA E37Q, measured by aITC or bMST. Enthalpy of dimer to monomer transition of from ITC experiment at pH 2.3: 10.6 ± 0.3 kcal/mol.
| WT HdeA | ||
|---|---|---|
|
| ||
| KD (μM) | ||
| pH | Experimental | Simulated |
| 2 | - | 84 |
| 2.3 | a64 ± 8 | - |
| 4 | b1.2 ± 0.2 | 2.5 |
| 5 | b0.64 ± 0.17 | 0.7 |
| 6 | b0.75 ± 0.31 | 1.0 |
| 7 | b2.2 ± 0.6 | 1.5 |
|
| ||
| HdeA E37Q | ||
|
| ||
| KD (μM) | ||
| pH | Experimental | Simulated |
| 5 | b0.55 ± 0.1 | - |
| 7 | b0.49 ± 0.1 | - |
MD simulation values taken from Ahlstrom et al. [8].
MD simulations suggested that the stabilization of the HdeA dimer at pH 5 is caused by the protonation of Glu37 upon shift from pH 7 to pH 5 [8]. Glu37 is a well-conserved residue that is located inside a hydrophobic pocket of HdeA and forms close interactions with Trp82 in the N-terminal region of HdeA’s C-terminal alpha helix. Simulations and NMR titrations suggested that the pKa of Glu37’s side chain is significantly higher compared to the side chains of other glutamic acid residues in HdeA, and that this increased pKa value could contribute to the stabilization of HdeA in mildly acidic conditions [8]. To test the role of Glu37 in HdeA unfolding and monomerization, we measured the KD of an E37Q HdeA mutant at pH 5 and 7. We reasoned that the E37Q mutation should mimic the protonated state of the Glu37 side chain that predominately occurs at pH values below its pKa. The E37Q substitution caused only a minor change in dimer stability at pH 5 as measured by microscale thermophoresis, but increased dimer stability at pH 7 by a factor of four (Table 1). These experimental results indicate that the dimeric stabilization around pH 5 predicted by MD simulations is likely due to deprotonation of Glu37 at higher pH, which decreases the stability of the HdeA dimer.
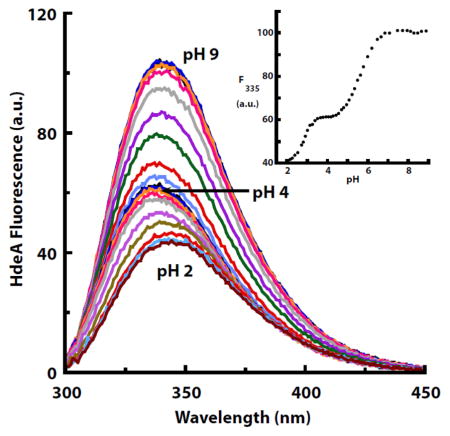
The MD simulations also suggested that HdeA uses a complex activation mechanism [8]. In contrast to a simple two state switch, activation may involve the chaperone transitioning back and forth between its dimeric-folded and monomeric-unfolded forms. To test which of these models of activation more likely applies to HdeA, we measured tryptophan fluorescence as a function of pH. These data revealed the presence of at least three states: one at low pH (<2), one at pH values around 4–5, and one at higher pH (pH > 6) (Fig. 1). Intriguingly, the intermediate state was abolished by the E37Q mutation (Fig. 1), suggesting that protonation of Glu37 may control the formation of this third state.
Fig. 1.

Tryptophan fluorescence emission spectra (280 nm excitation) of wild-type HdeA (a) and HdeA E37Q (b) as a function of pH. The insets show the fluorescence emission at 335 nm as a function of pH. HdeA contains two tryptophans: Trp82 (in close proximity to Glu37) and Trp16 (more distant from Glu37). Fit parameters for WT HdeA (a): n1 = 2.04, pKa1=2.81, n2=1.09, pKa2=5.59. Fit parameters for HdeA E37Q (b) n = 2.23, pKa = 3.14.
What is the nature of this transition at pH~6 observed in the tryptophan fluorescence titration? Conceivably, the fluorescence change we observed could reflect a change in the chemical environment of Trp82 that was directly caused by the protonation of Glu37. In this case, the fitted pKa value of 5.6 (Fig. 1) does fit with the prediction of an elevated pKa for this residue [8]. Alternatively, or additionally, the protonation of Glu37 might cause structural changes in HdeA that in turn affect the protein’s tryptophan fluorescence. Also, HdeA contains a second tryptophan (Trp16) more distant from Glu37 that could contribute to the fluorescence changes observed at pH 2. To assess in greater detail whether structural changes were occurring as a function of pH, we turned to NMR spectroscopy.
We collected 15N-1H HSQCs at various pH values ranging from 7.3 to 1.6 (Fig. 2). Upon titration from pH 7.3 to 3.1, we observed non-linear 1H and 15N chemical shift changes in fast-exchange (i.e. much faster than the chemical shift coalescence that occurs roughly in the ms timescale). This observation is in agreement with previous studies on HdeA [9] and could be induced by either a change in secondary structure or an electrostatic effect due to the protonation of neighboring acidic residues. HdeA contains 5 glutamate and 11 aspartate residues that can possibly be protonated by lowering the pH from 7.3 to 3.1, and as such, the multitude of charge neutralization could easily induce non-linear chemical shift changes [12].
Fig. 2.

Impact of pH changes on HdeA’s NMR signature. (a) Titration from pH 7.3 to 1.6, (b) from 7.3 to 3.1, (c) from 3.1 to 2.8, and (d) from 2.8 to 1.6. Color coding: from red to purple with decreasing pH, as indicated in the key at the right of each panel. The assigned spectrum of HdeA is shown in Fig. S8.
To investigate whether these changes in chemical shift are due to a change in secondary structure, we recorded additional HNCACB experiments. Observed changes in 13C chemical shifts remained modest for almost all residues except the acidic residues, which undergo protonation upon pH decrease (Fig. 3a). This result suggests that the secondary structure of HdeA remains relatively unchanged above pH 3, and that the non-linear shifts are likely almost entirely due to protonation events. Additionally, it has been previously shown by H/D exchange that protection of backbone amides in HdeA decreases at lower pH values [9]. The observation that the secondary structure remains overall unchanged suggests that the increased H/D exchange rate could be due to the presence of a low-populated unobservable intermediate; specifically, one that is possibly more flexible or partially unfolded. The existence of such a pH-dependent, low-populated intermediate has been suggested by molecular modeling [8].
Fig. 3.

NMR investigation of HdeA secondary structure upon pH variation. (a) Residue specific combined Cα and Cβ chemical shift deviations from their respective random coil values (positive values indicates α-helices, negative values indicate β-sheets) at pHs ranging from 6.9 (red) to 3.1 (purple). Vertical grey bars indicate Asp or Glu that can be protonated in this pH range. Apart from these residues, combined Cα and Cβ chemical shift deviations show only minor changes, suggesting that the previously observed increased H/D exchange upon lowering the pH is mainly due to a non-directly visible intermediate. (b) Residues disappearing (no longer observable) upon pH increase from 5.6 to 6.9 are shown in orange on the HdeA dimeric structure (left, right: two different views) (PDB ID: 1BG8) [20]. The affected residues cluster closely around Glu37 (side-chain is labeled), suggesting a key role for this residue in HdeA stabilization at mildly acidic pH.
Increasing the pH from 5.6 to 6.9 resulted in a significant number of residues showing decreased signal intensity, making them unobservable in the HNCACB experiments. Interestingly, these residues include and cluster around Glu37 (Fig. 3b), providing further evidence of the importance of this residue in the dimeric stability of HdeA at pH values in the mildly acidic range. The disappearance of these residues with the shift to pH 6.9 suggests the presence of intermediate conformational exchange at pH 7 (i.e. conformational exchange at timescales close to the chemical shift coalescence that occurs roughly in the ms), which is in line with the observation that elevating the pH above 5 destabilizes HdeA’s structure. Furthermore, this change suggests that the third state observed by fluorescence is not only due to the direct influence of Glu37 protonation, but also to a local loosening of HdeA structure, consistent with partial unfolding observed in the MD simulations [8].
Upon pH titration from pH 3 to 2, HdeA’s NMR spectrum undergoes severe changes, revealing the unfolding of HdeA on slow timescales (i.e. much slower than the chemical shift coalescence that occurs roughly in the ms timescale). Resonances corresponding to folded HdeA disappear, while signals that appear at pH 2 exhibit the low chemical shift dispersion that is expected for unfolded states. However, the absence of some resonances at pH 2 and the presence of significant line broadening at this pH suggest that HdeA is undergoing significant conformational exchange at pH 2, likely due to the sampling of residual structural elements that are still present at pH 2. No direct sign of a folded monomer could be identified, reinforcing the hypothesis of concomitant monomerization and unfolding of HdeA. These results are consistent with previous circular dichroism investigations of HdeA at pH 2.2 and molecular modeling studies [4, 7]. This apparent residual structure at pH 2 could be due to the existence of the C18-C66 disulfide bond, a linkage that has been shown to be essential for chaperone function at low pH [6].
Previous work has shown that HdeA monomerizes and unfolds under acid stress conditions, activating the protein’s chaperone activity and allowing HdeA to bind to other unfolded proteins. The results reported here provide evidence that this process occurs through a complex activation pathway, described below.
At neutral pH, HdeA is in a dimeric state in which the C-terminal region around Glu37 exhibits conformational exchange. As the pH decreases to 5, Glu37 is protonated, stabilizing this region as well as the dimer as a whole. This stabilization at pH 5 is supported by our dissociation constant measurements, fluorescence data, and NMR spectroscopy, and is consistent with previous pH-dependent MD simulations of HdeA. Continuing to decrease the pH further to ~3 does not affect the structure much. Our results, together with previous observations, suggest that HdeA undergoes several successive protonation events while retaining its dimeric, well-structured state. Alone, our data do not provide direct evidence of the presence of a low-populated intermediate. However, combined with previous H/D exchange measurements of decreased exchange protection upon lowering the pH [9], the data suggest that HdeA could simultaneously sample an ensemble of less folded states with increasing frequency. This interpretation compares well with simulated mechanisms of HdeA activation [13]. Below pH 3, the HdeA dimer unfolds and monomerizes on the ~ms to s timescale, quickly releasing a largely unfolded, active chaperone to respond to drastic environmental pH changes. The monomerization and unfolding appear to be highly correlated, and could be helped by the existence of the partially folded intermediate. Overall, our observations reveal a complex activation pathway for HdeA and confirm aspects of HdeA’s activation mechanism previously modeled using MD simulations [7, 8].
Understanding how HdeA can be so rapidly activated as a chaperone by simple pH shifts is an important component to understanding the physiology of bacteria. HdeA’s maximal dimeric stability under mildly acidic conditions could serve to prevent premature activation. Additionally, HdeA works in partnership with HdeB in acid stress protection, but HdeB is optimally active at pH 4, a value higher than the optimal pH for HdeA activity [3]. Providing HdeA with extra stability at pH 5 would be a way to efficiently differentiate its optimal pH activity from that of HdeB and thereby cover a broader pH spectrum of protection. These two chaperones appear to work synergistically in combatting acid stress [14], but how they work together is not clear. One possible mechanism could rely on HdeA containing a low population of less ordered conformations at pHs above its order-to-disorder transition, possibly allowing for some synergistic interactions in HdeA-HdeB chaperone activity.
Materials and Methods
Proteins
HdeA expression was performed using an HdeA/HdeB MG1655 knockout strain (SA152). The HdeA/HdeB knockout strain (strain SA28) was created using the pkD3 plasmid to replace the HdeA/HdeB loci with a chloramphenicol (CmR) resistance marker following the protocol described by Datsenko & Wanner [15]. Knockout primers were 5′-GAAATTGATTCGTGACGGCTCTTTCACTTTATAGTTGAGGATATTACGATGGTGTAG GCTGGAGCTGCTTC-3′ and 5′-GGTTTCATTTAAAGTAACGGTATCGCCACCTTTATATACTGTTTCTTCATGCATATGA ATATCCTCCTTAG-3′. The insertion was verified using the primers 5′-CGAAATTGATTCGTGACG – 3′ and 5′-GGAGCAGCAAGATGGCTCAAC – 3′. Subsequently, λDE3 prophage was integrated into the chromosome (strain SA152) using a λDE3 Lysogenization Kit (Novagen) following the protocol described by the vendor. HdeA expression and purification were conducted as previously described [4].
Microscale thermophoresis
Microscale thermophoresis was used to determine the dissociation constant (KD) of HdeA self-dissociation. Microscale thermophoresis was performed using a NanoTemperMonolith NT.115 instrument (NanoTemper Technologies, Munich, Germany). Wild-type (WT) HdeA and HdeA E37Q were labeled using protein labeling kit RED-NHS (Amine Reactive, dye NT-647, NanoTemper Technologies, Munich, Germany). Measurements were performed in a 100 mM citrate-phosphate buffer, adjusted to 300 mM ionic strength by addition of NaCl at the different pH values, supplemented with 0.05% Tween-20. The ionic strength of the buffer was calculated using CurTiPot 4.1.2. Samples were loaded into NT.155 standard capillaries (NanoTemper, Technologies, Munich, Germany). Measurements were performed at 25°C, 60% LED power, and 20% – 60% IR laser power (HdeA WT: 20% at pH 4.0 and 5.0, 40% at pH 6.0, and 60% at pH 7.0; HdeA E37Q: 20% at pH 5.0 and 7.0). A constant concentration of 80 nM labeled HdeA WT and 60 nM labeled HdeA E37Q, respectively, was used and increasing concentrations of unlabeled HdeA WT or HdeA E37Q were used for the titrations.
Curve fitting was performed in Kaleidagraph using equation 6, derived below. For the reaction 2 M ↔ D
| (1) |
where [M] and [D] are the concentration of monomer and dimer, respectively, and
| (2) |
where Pt is the total protein concentration. Our data are reported with respect to the monomer concentration; therefore, [D] is multiplied by 2 to maintain conservation of mass.
| (3) |
Expanding out and rearranging gives:
| (4) |
which can be directly solved, leading to equation (5) that describes [D] as a function of [Pt] in monomer:
| (5) |
ΔMST/[Pt] was included to account for the fraction bound measured by the microscale thermophoresis (MST) signal, as well as a y-intercept term (Y) to account for the signal offset:
| (6) |
Equation (6) was fit to determine the dimerization KD, ΔMST, and Y as a function of [Pt].
Isothermal calorimetry
ITC experiments were performed with a Microcal ITC200. For the ITC experiment at pH 2.3, HdeA was dialyzed into 50 mM potassium phosphate pH 2.3, 50 mM KCl. Triton X-100 was added to the dialyzed sample to a final concentration of 0.05%. The ITC experiment consisted of 20 injections at 25°C; the first injection was 0.2 μL and the remaining 19 were 2 μL. The concentration of the HdeA in the syringe was 610.5 μM. The concentration of HdeA in the cell ranged from 7.24 μM to 98.34 μM during the titration. Data were fit using dimer dissociation model in Origin.
NMR spectroscopy
The high concentrations of proteins used in NMR experiments can affect the pH of the solution. To accurately determine the pH in the NMR experiments with HdeA, we therefore used a mixture of previously characterized chemical shift pH probes. HdeA was dialyzed into buffer containing 15 mM sodium citrate, 100 mM NaCl, and 0.1 mM EDTA. 0.5 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) was used as chemical shift reference; 4 mM imidazole, 4 mM piperazine, 4 mM sodium formate [16], and 4 mM dichloroacetic acid [17] were used as pH probes. A final concentration of 0.9 mM monomer HdeA was used.
All NMR experiments were recorded on a 14.1T (600 MHz 1H Larmor frequency) Varian spectrometer equipped with a triple-resonance cryo-probe with pulse field gradients. All spectra were processed and analyzed using NMRPipe [18] and Sparky [19]. At each titration point, the pH was measured in the NMR tube using the chemical shift pH sensors listed above using a one-dimensional 1H Watergate pulse sequence.
Fluorescence pH titration
A solution of 10 μM WT HdeA or HdeA-E37Q in 1 mL 10 mM sodium citrate, 10 mM Tris, pH 9 and 150 mM NaCl was titrated with 0.5–2 μL increments of 1 M HCl at 25°C. After each addition of HCl, the pH of the solution was measured using an Accumet Excel XL15 pH meter equipped with an ORION 8220BNWP micro pH electrode. The fluorescence emission from 300 and 450 nm was recorded using a Photon International Technology fluorimeter following excitation at 280 nm. Emission spectra for each titration point were corrected for dilution from the HCl additions prior to plotting.
Fluorescence pH titrations were fit to a modified Henderson-Hasselbalch equation comprising a single pH titratable event for E37Q HdeA
or two pH titratable events for WT HdeA
where Fmin and Fmax are the minimum and maximum fluorescence values, respectively, Fint is the fluorescence of the intermediate species of the titration, n is the hill coefficient for a given pH titratable event, and pKa is the acid dissociation constant for a given pH titratable event.
Supplementary Material
Highlights.
The acid-stress chaperone HdeA undergoes activation through a complex pathway apparently involving an intermediate state.
HdeA is maximally stable at mildly acidic pH.
Acknowledgments
This work was funded by the National Institutes of Health (R01-GM102829 to J.C.A.B. and K99-GM120388 to S.H.). J.C.A.B. is a Howard Hughes Medical Investigator. The authors thank Ke Wan for his assistance in protein purification, Erik Zuiderweg for his initial NMR advice, and Vivekanandan Subramanian for maintenance of the NMR facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hancock RE. Alterations in outer membrane permeability. Annu Rev Microbiol. 1984;38:237–64. doi: 10.1146/annurev.mi.38.100184.001321. [DOI] [PubMed] [Google Scholar]
- 2.Malki A, Le HT, Milles S, Kern R, Caldas T, Abdallah J, et al. Solubilization of protein aggregates by the acid stress chaperones HdeA and HdeB. J Biol Chem. 2008;283:13679–87. doi: 10.1074/jbc.M800869200. [DOI] [PubMed] [Google Scholar]
- 3.Dahl JU, Koldewey P, Salmon L, Horowitz S, Bardwell JC, Jakob U. HdeB functions as an acid-protective chaperone in bacteria. J Biol Chem. 2015;290:65–75. doi: 10.1074/jbc.M114.612986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tapley TL, Korner JL, Barge MT, Hupfeld J, Schauerte JA, Gafni A, et al. Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc Natl Acad Sci U S A. 2009;106:5557–62. doi: 10.1073/pnas.0811811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajiwala KS, Burley SK. HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. Journal of molecular biology. 2000;295:605–12. doi: 10.1006/jmbi.1999.3347. [DOI] [PubMed] [Google Scholar]
- 6.Tapley TL, Franzmann TM, Chakraborty S, Jakob U, Bardwell JC. Protein refolding by pH-triggered chaperone binding and release. Proc Natl Acad Sci U S A. 2010;107:1071–6. doi: 10.1073/pnas.0911610107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahlstrom LS, Dickson A, Brooks CL., 3rd Binding and folding of the small bacterial chaperone HdeA. J Phys Chem B. 2013;117:13219–25. doi: 10.1021/jp403264s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahlstrom LS, Law SM, Dickson A, Brooks CL., 3rd Multiscale modeling of a conditionally disordered pH-sensing chaperone. Journal of molecular biology. 2015;427:1670–80. doi: 10.1016/j.jmb.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrison MA, Crowhurst KA. NMR-monitored titration of acid-stress bacterial chaperone HdeA reveals that Asp and Glu charge neutralization produces a loosened dimer structure in preparation for protein unfolding and chaperone activation. Protein science : a publication of the Protein Society. 2014;23:167–78. doi: 10.1002/pro.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowhurst KA. (1)(3)C, (1)(5)N and (1)H backbone and side chain chemical shift assignment of acid-stress bacterial chaperone HdeA at pH 6. Biomol NMR Assign. 2014;8:319–23. doi: 10.1007/s12104-013-9508-0. [DOI] [PubMed] [Google Scholar]
- 11.Foit L, George JS, Zhang BW, Brooks CL, 3rd, Bardwell JC. Chaperone activation by unfolding. Proc Natl Acad Sci U S A. 2013;110:E1254–62. doi: 10.1073/pnas.1222458110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakurai K, Goto Y. Principal component analysis of the pH-dependent conformational transitions of bovine beta-lactoglobulin monitored by heteronuclear NMR. Proc Natl Acad Sci U S A. 2007;104:15346–51. doi: 10.1073/pnas.0702112104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dickson A, Ahlstrom LS, Brooks CL., 3rd Coupled folding and binding with 2D Window-Exchange Umbrella Sampling. J Comput Chem. 2016;37:587–94. doi: 10.1002/jcc.24004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, He D, Yang Y, Lin S, Zhang M, Dai S, et al. Comparative proteomics reveal distinct chaperone-client interactions in supporting bacterial acid resistance. Proc Natl Acad Sci U S A. 2016;113:10872–7. doi: 10.1073/pnas.1606360113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baryshnikova OK, Williams TC, Sykes BD. Internal pH indicators for biomolecular NMR. J Biomol Nmr. 2008;41:5–7. doi: 10.1007/s10858-008-9234-6. [DOI] [PubMed] [Google Scholar]
- 17.Szakács Z, Hägele G, Tyka R. 1 H/31 P NMR pH indicator series to eliminate the glass electrode in NMR spectroscopic pK a determinations. Analytica chimica acta. 2004;522:247–58. [Google Scholar]
- 18.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol Nmr. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 19.Goddard TD, Kneller DG. Sparky 3. University of California; San Francisco, USA: 2008. [Google Scholar]
- 20.Yang F, Gustafson KR, Boyd MR, Wlodawer A. Crystal structure of Escherichia coli HdeA. Nat Struct Biol. 1998;5:763–4. doi: 10.1038/1796. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.