

Summary
Given the limited efficacy of clinical approaches that rely on ex vivo generated dendritic cells (DC), it is imperative to design strategies that harness specialized DC subsets in situ. This requires delineating the expression of surface markers by DC subsets among individuals and tissues. Here, we performed a multiparametric phenotypic characterization and unbiased analysis of human DC subsets in blood, tonsil, spleen and skin. We uncovered previously unreported phenotypic heterogeneity of human cDC2 among individuals, including variable expression of functional receptors such as CD172a. We found marked differences in DC subsets localized in blood and lymphoid tissues versus skin, and a striking absence of the newly discovered Axl+ DC in the skin. Finally, we evaluated the capacity of anti-receptor monoclonal antibodies to deliver vaccine components to skin DC subsets. These results offer a promising path for developing DC subset-specific immunotherapies that cannot be provided by transcriptomic analysis alone.
eTOC Blurb
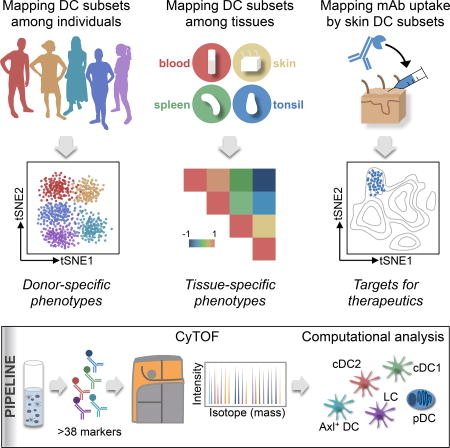
Dendritic cells (DC) are potent initiators of immune responses; however, human DC subsets have yet to be successfully harnessed for immunotherapies. By combining CyTOF and unbiased analysis, Alcantara-Hernandez et al. profile the heterogeneity of human DC subsets among individuals and tissues, providing comprehensive insights for the development of DC-based therapeutics.
Introduction
Dendritic cells (DC) are unique in their capacity to initiate and modulate distinct types of immune responses, providing immunity against a wide range of pathogens as well as tolerance to self and innocuous environmental antigens (Banchereau and Steinman, 1998). The capacity to induce different types of immune responses has been attributed, at least in part, to the presence of functionally specialized DC subsets (Merad et al., 2013). However, human DC subsets localized in tissues have not yet been harnessed for immunotherapies (Palucka and Banchereau, 2013). This is in part due to our limited understanding of DC subsets, especially the phenotypic heterogeneity among individuals and tissues. Mapping protein targets in humans is ultimately essential for the development of DC subset-based immunotherapeutics.
DC subsets have been classified based on transcriptome analysis and ontogeny (Heidkamp et al., 2016; Merad et al., 2013; Villani et al., 2017). The DC network is composed of two major DC subsets, i.e., classical DC (cDC) expressing the transcription factor (TF) ZBTB46 and plasmacytoid DC (pDC) expressing the TF TCF4 (Cisse et al., 2008; Meredith et al., 2012; Satpathy et al., 2012). cDC can be further divided into cDC1 and cDC2 with distinct lineage-specific TFs (Hildner et al., 2008; Schlitzer et al., 2013). pDC are generally grouped together despite the fact that some reports suggest the existence of phenotypically and functionally distinct pDC subsets (Matsui et al., 2009; Zhang et al., 2017). Altogether, these analyses have constituted powerful tools to dissect the diversity of the DC network and identify correlations among mouse and human DC subsets, as well as tissues (Guilliams et al., 2016; Reynolds and Haniffa, 2015). However, RNA expression does not necessarily predict protein expression (Luber et al., 2010). For example, the mouse C-type lectin Langerin is expressed homogeneously at the RNA level in cDC1; however, at the protein level it is differentially expressed among tissues (Idoyaga et al., 2013) and mouse lines (Cheong et al., 2007). This dichotomy between RNA and protein expression is likely to also emerge in genetically diverse humans exposed to distinct environmental stimuli. Consequently, transcriptome analysis alone may not be sufficient to recognize important DC subset phenotypic differences. Additional factors precluding a more accurate representation of human DC phenotypic diversity include limited tissue sources, small sample sizes, and commonly employed biased flow cytometry analysis of a limited set of surface receptors (<12) (Granot et al., 2017; Haniffa et al., 2012). Therefore, the need remains for a high-dimensional single-cell characterization and unbiased analysis of DC subsets across tissues and individuals at the protein level.
Until now, human DC-based therapeutic strategies have mainly relied on ex vivo generated DC differentiated from CD14+ blood monocytes in the presence of granulocyte monocyte colony stimulating factor (GM-CSF) and interleukin-4 (IL-4) (Sallusto and Lanzavecchia, 1994). Unfortunately, monocyte-derived DC (moDC) have demonstrated limited efficacy in clinical trials (Palucka and Banchereau, 2013). This has been attributed to several obstacles, including the limited capacity of these cells to migrate to T cell-rich areas (Adema et al., 2005). Accordingly, identifying alternative therapeutic strategies that harness human DC subsets localized in tissues will likely yield improved treatments. In situ DC subsets can be targeted by anti-receptor monoclonal antibodies (mAb) carrying antigens and adjuvants. This approach has shown robust results in murine models, increasing the potency of vaccines by at least 100-fold (Trumpfheller et al., 2011). Several targeting receptors have been identified in mice, including DEC205 (Trumpfheller et al., 2011), Langerin (Idoyaga et al., 2011), CLEC9A (Idoyaga et al., 2011; Sancho et al., 2008) and XCR1 (Fossum et al., 2015). Similarly, several receptors have been proposed in humans and tested in vitro in blood DC or moDC (Sehgal et al., 2014). However, to bring this strategy to the clinic, it is imperative to understand: 1) the surface expression of these receptors in DC subsets localized at sites of vaccination (e.g., skin); and 2) variation in expression among individuals.
With the goal of examining the surface phenotype of DC subsets in humans, we used mass cytometry (CyTOF) and unbiased analysis. We observed more interindividual phenotypic heterogeneity of cDC2 than expected, and found evidence that the phenotype of skin DC subsets differ significantly from that of their blood and lymphoid-organ homologs. We characterized a range of phenotypes exhibited by Axl+ DC and their tissue distribution, which aligned with pDC and not cDC. Our study represents an easily accessible data resource on DC subsets’ phenotypic differences among individuals and tissues that can be exploited for the development of therapeutics. To exemplify this, we correlated phenotype with the efficiency of mAb against surface receptors to deliver vaccine components to distinct skin DC subsets. This dataset reveals unique information that has not been recognized using either transcriptome or low-dimensional flow cytometry analysis, and highlights strategies to harness DC subsets for immunotherapeutics.
Results
CyTOF analysis shows unappreciated surface phenotypic diversity within human blood DC subsets
To characterize the surface phenotype of myeloid cells in a highly parametric unsupervised fashion, we designed a CyTOF panel of 38 heavy metal-conjugated mAb (Table S1). This panel included markers for the exclusion of Lin+ cells, basophils and innate lymphocytes (ILC). It also integrated a broad range of identity markers that allowed the segregation of previously described human DC subsets, such as BDCA1, BDCA2 and BDCA3, hematopoietic receptors, pattern recognition receptors, co-stimulatory and co-inhibitory molecules, and chemokine receptors. Parallel analysis by CyTOF and flow cytometry showed similar frequencies of previously described DC subsets, corroborating our CyTOF approach (Figure S1A and S1B).
We then used our CyTOF panel to analyze peripheral blood mononuclear cells (PBMC) from 5 healthy donors (Figure 1A). To detect cell subsets we used X-shift clustering, which selects the optimal k-nearest neighbor (kNN) value to separate phenotypically distinct clusters of cells without over-fragmentation (Samusik et al., 2016). We then visualized X-shift-detected clusters through a t-Distribution Stochastic Neighbor Embedding (viSNE), which uses all pairwise distances to place single cells in a 2-dimensional scatter plot (Amir et al., 2013). The viSNE plot generated from the merged data represents a map of all myeloid cells present in PBMC of these 5 donors (Figure 1B). Monocytes were the main cells identified and were split into 3 clusters that mapped to well-established subsets: (a) CD14+CD16− classical monocytes, (b) non-classical CD14−CD16+ monocytes, and (c) intermediate CD14+CD16+ monocytes, representing ~80%, ~10%, and ~2% of Lin− PBMC, respectively (Figure 1C). We summarized the median intensity of the markers used in our CyTOF panel for each subset in a heatmap (Figure 1D). CD14+ monocytes (both CD16+ and CD16−) had high expression of CD163 and CD11b in congruence with their inflammatory phenotype. CD16+ monocytes (CD14+ and CD14−) had high expression of CX3CR1 and CD115, the phenotypic hallmark of patrolling monocytes (Cros et al., 2010). Importantly, these 3 subsets of monocytes were phenotypically homogeneous among the individuals analyzed (Figure S1C).
Figure 1. Unsupervised CyTOF analysis of myeloid cells in human blood.

(A) Schematic of experimental design and analysis of 5 donors in 4 independent exp. using CyTOF panel 1. (B) X-shift delineated clusters (a, b and c) were manually colored. (C) Biaxial plots of CD14 and CD16 expression for clusters identified in B. Shown is frequency +/− SD of Lin events. (D) Heatmap of marker median intensity in clusters detected in B. (E) As in B, but X-shift clusters were delineated after CD14+ and CD16+ exclusion. (F) Heatmap of marker median intensity in clusters identified in E. (G) DMT representing clusters identified in E. Shown are marker cutoff values that define each sub-branch. Please see Figure S1 for CyTOF validation and PCC of donors’ monocytes.
Given the predominance of monocytes in PBMC samples, X-shift analysis was unable to detect the diversity of minor myeloid cell populations such as DC subsets. Consequently, to explore the complexity of the DC network, we excluded CD14+ and CD16+ cells by biaxial gating (Figure S1B) as previously described (Breton et al., 2015). Ten distinct clusters of cells were detected by this method, and the median intensity of each marker was quantified and summarized in a heatmap (Figure 1E and 1F). Cluster 7 corresponded to basophils (HLA-DR−CD11b+CD123+). We identified two clusters (9 and 10) with variable expression of CD34, HLA-DR, CD117 and CCR7, which corresponded to hematopoietic stem cells and early DC progenitors (Villani et al., 2017). We also found cells with variable (negative to low) expression of HLA-DR that represented ILC (Simoni et al., 2017) and other cells that could not be deduced from their surface expression profile (combined in cluster 8).
We recognized cluster 1 as pDC (HLA-DR+CD123+BDCA2+CD11c−) and cluster 6 as cDC1 (BDCA3hiHLA-DR+CD11c+CLEC9A+). cDC1 had high expression of CD135, as previously described (Breton et al., 2015), and PD-L1, which can inhibit T cell function (Keir et al., 2008). We identified 3 distinct clusters (3, 4, and 5) as cDC2 based on their high expression of CD11c, HLA-DR and BDCA1. Finally, we observed a cluster of cells (2) that localized near pDC but expressed CD11c, probably corresponding to the newly identified Axl+ DC (Villani et al., 2017).
To identify the key markers that separate each X-shift-derived cluster, we used a divisive marker tree (DMT) (Samusik et al., 2016) (Figure 1G). DMT analysis showed that circulating cDC1 diverged from a single node separated by the expression of CLEC9A, a previously reported cDC1-specific marker (Huysamen et al., 2008). Circulating cDC2, on the other hand, derived from the BDCA1 node and separated into 3 distinct clusters based on the expression of CD163 (the scavenger receptor of hemoglobin-haptoglobin-complexes) and CD172a (the ligand of CD47). Finally, pDC and cluster 2 derived from the same HLA-DRhi node and bifurcated based on the expression of CD11c. Taken together, these data show that high-dimensional unsupervised CyTOF analysis allows delineating formerly described DC subsets, as well as displaying previously uncharacterized heterogeneity in the DC network.
Cluster 2 corresponds to Axl+ DC and is undetectable in human skin
Our CyTOF analysis clearly identified a minor cluster of cells (cluster 2) localized near pDC in the viSNE map and unsupervised hierarchical clustering (Figure 1E–G). We first confirmed our X-shift results using principal component analysis (PCA), where cluster 2 again localized between pDC and cDC2 (Figure 2A). Clustering and Pearson correlation coefficients (PCC) based on surface phenotype showed that cluster 2 was present in every donor and grouped separately from pDC and cDC2 (Figure S2A). Biaxial gating of CyTOF data and flow cytometry validation showed that cluster 2 fell within the pDC gate (HLA-DR+CD123+BDCA2+) but expressed higher CD34, CD11c, HLA-DR and BDCA3, and slightly lower CD123 and BDCA2 (Figure S2B–E). We then examined the distribution of cluster 2 in lymphoid and non-lymphoid tissues (Figure 2B). Unbiased analysis clearly detected cluster 2 in tonsil and spleen but not in healthy skin, where pDC were also absent as previously reported (Gregorio et al., 2010). We conclude that cluster 2 follows the same tissue distribution as pDC.
Figure 2. Tissue distribution and characterization of Axl+ DC.

(A) PCA of cDC1, pDC, cDC2 (clusters 3–5) and cluster2 identified in Fig. 1E. (B) viSNE contour map of merged data from all organs and individuals was overlaid with pDC and cluster 2. (C) viSNE of PBMC analyzed with CyTOF Panel 2 (n=1). (D) Upper: Biaxial plots of CyTOF data from C. Black dots represent gated Axl+ cells. Lower: Flow cytometry gating strategy (1 representative of 4 donors in 2 independent exp.). (E) gMFI is shown for populations gated as in D (n=4, 1 of 2 independent exp.). (F) MLR for sorted populations from D (n=2, 2 independent exp.). (G) MLR of “pure” Axl pDC freshly-isolated or after 2d culture with CD40L+IL-3 (2 representative donors). (H) As in G, but expressed as fold change +/− SD relative to average of freshly-isolated pDC (n=7, 4 independent exp.) (I) gMFI of markers and percentage CD11c+ in “pure” Axl pDC cultured with CD40L+IL-3 for 0, 2, or 6d (n=6–7, 4–5 independent exp.). Please see Figure S2 for additional phenotypic profiling of Axl+ DC by flow cytometry and CyTOF, and Figure S3 for sort strategy and purity.
pDC subpopulations have been previously segregated based on the expression of CD2, CD5, CD81 and Axl (Matsui et al., 2009; Zhang et al., 2017). Recently, a distinct DC subset that clustered near pDC was identified by scRNA-seq based on the expression of Axl and Siglec6 (Villani et al., 2017). These cells acquired some cDC markers after culture with stromal cells and hematopoietins, suggesting they may be DC precursors (See et al., 2017). We asked if cluster 2 represented Axl+ DC by designing a new CyTOF panel that incorporated these markers (Table S1, panel 2). Unbiased viSNE analysis with this new panel again segregated a discrete population of cells bridging pDC and cDC2 (Figure 2C). Cluster 2 from our first CyTOF panel mapped within this population of cells, but only made up a fraction of them (arrow, light blue, upper left corner). This bridging population ranged from pDC-like to cDC2-like, expressing high-to-low CD123 and BDCA2, and low-to-high CD11c, BDCA3, CD5, CD2, BDCA1, CD33 and CX3CR1 (Figure 2C and S2F). This population also expressed CD81 and the unique markers Axl and Siglec 6. We conclude that cluster 2 falls within the recently described Axl+ DC, which is a considerably diverse population of cells.
We then explored the diversity observed in Axl+ DC. Biaxial gating of CyTOF data showed that Axl+ DC (black dots) fell within traditional pDC and cDC2 gates (Figure 2D). We validated our observations by flow cytometry and identified 3 populations of Axl+ cells representing different parts of the spectrum: CD11c−Axl+ (purple) and CD11c+Axl+ (light blue) that fell within the pDC gate, and BDCA1+Axl+ (orange) that fell within the cDC2 gate. Our original cluster 2 corresponded to the CD11c+Axl+ population (light blue). Analysis of the surface markers on these 3 populations together with pDC and cDC2 corroborated a transition between pDC and cDC2 phenotypes (Figure 2E). This transition was accompanied by lower TCF4 expression, a TF known to maintain pDC phenotype and function (Reizis et al., 2011).
The discovery that Axl+ DC are contaminating the traditional pDC gate has brought into question some attributes of pDC (See et al., 2017; Villani et al., 2017), in particular, their capacity to present antigen and become cDC-like when activated. Using our flow cytometry purification strategy (Figure S3A), we re-evaluated this issue. Freshly-isolated Axl− pDC (“pure”) were less potent at initiating allogeneic reaction compared to cDC2 as previously described (See et al., 2017; Villani et al., 2017). However, unlike these previous reports, we found that the antigen-presentation capability of Axl+ DC varied across the population, correlating with their similarity to pDC or cDC2 (Figure 2F).
We then asked if “pure” pDC could increase their antigen presentation capabilities and become cDC-like cells upon activation with CD40L and IL-3 (Grouard et al., 1997). “Pure” pDC activated for 2 days were more potent at promoting allogeneic T cell proliferation than freshly-isolated pDC (Figure 2G–H). Furthermore, activated “pure” pDC upregulated HLA-DR, CD80 and CD11c, suggesting their conversion to a cDC-like phenotype (Figure 2I). This conversion was accompanied by rapid downregulation of TCF4 (Figure 2I and S2G), a TF that plays a role in enforcing the immature phenotype of pDC (Ghosh et al., 2010; Reizis et al., 2011). Altogether, our results argue against the recent statements that pDC are unable to become cDC-like cells through phenotypic and functional activation.
Clusters of blood cDC2 are differentially represented among individuals
Our unbiased X-shift analysis on merged data from 5 individuals defined 3 clusters of circulating cDC2 cells (3, 4 and 5) (Figure 1E–G). We next asked whether these cDC2 clusters were present in every donor. Thus, a viSNE contour map of the merged data (Figure 3A, upper left) was overlaid with each donor’s cells and colored by clusters. We found that cDC1 from all 5 individuals localized to the same position of the viSNE map, suggesting homogeneity in this subset. Similarly, pDC and Axl+ DC were present in all donors analyzed and occupied similar regions of the viSNE map, confirming co-existence of these distinct DC subsets in the blood of healthy donors. In contrast, individual viSNE plots showed interindividual variation in the representation of the 3 cDC2 clusters. To confirm these results, we calculated the normalized frequency of each cluster across donors (Figure 3B). Although frequencies varied slightly, cDC1, pDC and Axl+ DC were present in every donor. On the contrary, the frequency of cDC2 clusters varied dramatically between individuals. Donors 3 and 5 had a predominance of cluster 4, whereas donors 1, 2 and 4 had a predominance of clusters 3 and 5. These results suggest that the observed diversity in cDC2 may be explained by different representation of each cluster among individuals.
Figure 3. Circulating cDC2 are highly heterogeneous among healthy individuals.

(A) viSNE contour map of merged data from Figure 1E was overlaid with each donor’s plot (D1–5). Axl+ DC refers to cluster 2 (CD11c+Axl+). (B) Heatmap of normalized frequency of DC clusters in each donor (D1–5). (C) PCC calculated between donors. All cDC2 clusters analyzed had >5 cells per donor. (D) Heatmap of marker variance across donors and clusters for each population. (E) Violin plots of highly variable markers in cDC2 of each donor (D1–5). Please see Figure S4 for phenotypic profiling at the single cell level.
To further dissect donor-donor variability, we analyzed the phenotypic profiles of each DC subset. We calculated PCC between donors based on cellular phenotype and represented them in a heatmap (Figure 3C). In accordance with their localization in the viSNE map, we found that cDC1, pDC and Axl+ DC had high PCC, indicating that their phenotype was homogeneous among individuals. We found lower correlations within cDC2, suggesting that their phenotype varied between individuals, even within the same cluster. The differences between individuals and clusters could be explained by variance in the expression of CD172a, CD32, BDCA1, and CD163 (Figure 3D). Indeed, cDC2 clusters displayed a range of marker expression across individuals as visualized by violin plots (Figure 3E and Figure S4). Altogether, our results suggest that the observed cDC2 heterogeneity is indicative of interindividual variation rather than conserved subpopulations of cDC2. Thus, genetic variation and environmental stimuli may greatly influence the phenotype of circulating human cDC2.
Human skin DC subsets are distinct from blood and lymphoid tissues
We decided to extend our unbiased analysis to evaluate the relationship between DC subsets from blood, lymphoid organs (tonsils and spleen), and a peripheral tissue, the skin. We chose skin given its use as a vaccination site in humans. We detected significant differences in the frequency of the major DC subsets between organs (Figure 4A). Tonsils harbored the highest frequency of pDC, whereas spleens harbored the greatest frequency of Axl+ DC. cDC1 and cDC2 were found in every organ analyzed, though at variable frequencies in each tissue. The highest frequency of cDC1 was in the spleen (21%), whereas cDC2 were the predominant DC subset in the skin (93%). As expected, we identified a subset of DC corresponding to Langerhans cells (LC) only present in the skin.
Figure 4. Skin DC subsets are different from blood and lymphoid tissues.

(A) Frequency of DC subsets in each tissue (n=3–7, 1–5 independent exp.). Numbers represent relative contribution of each subset to total DC pool. Axl+ DC refers to cluster 2 (CD11c+Axl+). (B) viSNE contour map of all organs and individuals was overlaid with each organ (columns). DC subsets were manually annotated (rows). Colors represent expression of BDCA3 and BDCA1 (rows). Analysis was performed with CyTOF panel 1. (C) Clustered heatmap of PCC based on phenotype for tissues analyzed in A. (D) Expression of markers in cDC1 and cDC2 from tissues analyzed in A. Statistics for skin vs. other organs in black; skin vs. blood in blue and brackets.
To analyze DC subsets among organs, we generated a viSNE contour plot of the merged data (Figure 4B, upper corner). This viSNE plot represents a map of DC subsets in all 4 organs and 18 individuals analyzed. We then overlaid this map with cells from each tissue colored by expression of BDCA3 and BDCA1 for the visualization of cDC1 and cDC2, respectively (Figure 4B). We found that cDC1 and cDC2 from blood and lymphoid organs mapped to similar places on the viSNE map (Figure 4B), suggesting that these subsets have a conserved phenotype across these tissues as previously described for blood and normal spleen (Mittag et al., 2011). In contrast, skin cDC1 and cDC2 subsets localized to different regions of the viSNE map, suggesting that skin cDC had a different phenotype. We confirmed these results by calculating PCC displayed in a clustered heatmap (Figure 4C). As expected by ontogeny studies, each DC subset clustered together and had high correlation coefficients independent of the tissue analyzed. Nevertheless, skin cDC1 and skin cDC2 had lower correlations with their blood or lymphoid-organ counterparts, suggesting that skin-derived signals likely influence the phenotype of cDC subsets. We then investigated which markers were differentially expressed in skin cDC (Figure 4D). Compared to blood and lymphoid-organ cDC1, skin cDC1 expressed significantly more CD1a, CD83, HLA-DR and BDCA1. Furthermore, skin cDC1 expressed less CD11c and CD135 than blood cDC1. CLEC9A expression, which is a blood cDC1 discriminatory marker (Villani et al., 2017), was expressed much less in tonsil and undetectable in skin cDC1. Similarly, skin cDC2 were distinguished from blood and lymphoid tissue cDC2 by expression of several markers including CD11b, CD123, CD206, CD1a and CD83. Altogether, our CyTOF analysis indicates that the skin imprints a distinct phenotype on cDC subsets.
Human skin DC subsets exhibit interindividual diversity
Although skin cDC subsets differed from blood and lymphoid-organ cDC, it was unclear if they were homogeneous among individuals. Thus, we investigated skin cDC by visualizing individual viSNE plots and analyzing variance across donors (Figure 5 and S5). Unlike blood, we were unable to detect 3 clusters of cDC2. We observed donor-specific patterns in skin cDC2, with little overlap in the viSNE maps, again indicating phenotypic heterogeneity among individuals. On the other hand, there was much less interindividual heterogeneity in skin cDC1 and LC. These observations were confirmed by calculating PCC based on cellular phenotype (Figure S5A–C).
Figure 5. Skin DC subsets are highly heterogeneous between individuals.

(A) viSNE contour map of merged skin data (n=7, 5 independent exp.) was overlaid with a plot for each donor (D1–7). DC subsets were manually annotated. (B) Heatmap of marker variance across donors (n=7) in skin DC subsets. (C) Violin plots of highly variable surface markers in skin DC. Each dot represents a donor. Please see Figure S5 for PCC of skin DC subsets.
We then asked which markers displayed variance between individuals (Figure 5D and 5E). We found that variance in cDC2 was based on expression of CD1a, CD32 and CD172a, receptors involved in antigen uptake and presentation. We also observed interindividual variability in the expression of CD83 and ICOSL, which are co-stimulatory and co-inhibitory molecules, respectively. LC and cDC1 showed variance in fewer markers, e.g., Langerin. From these results, we concluded that the skin harbors DC subsets that are heterogeneous among individuals, especially cDC2. This interindividual variation can be explained by the different expression of molecules involved in antigen uptake, presentation, and T cell co-stimulation and inhibition.
Ex vivo generated monocyte-derived DC do not represent blood or tissue DC subsets
DC generated from monocytes (moDC) have shown limited therapeutic efficacy (Palucka and Banchereau, 2013). This phenomenon has been attributed to the lack of similarities between moDC and tissue DC subsets. Importantly, the in vivo equivalent of moDC has only been examined so far using a few parameters by flow cytometry and more recently by global transcriptome analysis of gene expression (Balan et al., 2014). Given the high-dimensional single-cell nature of our approach that allows the unbiased identification of DC subsets based on protein expression, we inquired as to the in vivo equivalents of moDC. Monocytes cultured for 5 days in the presence of GM-CSF and IL-4 gave rise to a homogeneous population of cells expressing DC-SIGN, one of the hallmark surface markers of moDC (Figure 6A).
Figure 6. moDC differ from in situ DC subsets.

(A) FACS analysis of in vitro generated moDC (1 representative of 2 independent exp.). (B) PCA of moDC and myeloid subsets from tissues and donors analyzed in Figure 4. Ellipse marks 95% confidence region for each cell type. (C) viSNE contour map generated from merged blood, skin, and moDC (n=2–7, 2–5 independent exp.) data was overlaid with each sample type (columns), colored by marker expression (rows). Please see Figure S6 for PCA variance.
We then analyzed moDC by CyTOF and compared them with DC subsets from blood, lymphoid tissues and skin. PCA showed that moDC were distinct from any in vivo populations, located far from blood monocytes, as well as blood and tissue DC subsets (Figure 6B). This separation of moDC was explained by their differential expression of many cellular markers including CD172a, CD163, and CD115 (Figure S6). Of note, skin DC subsets (cDC1, cDC2, and LC) were clearly segregated from blood and lymphoid organ DC subsets, confirming our previous results (Figure 4).
We then visualized moDC together with blood and skin DC subsets on a viSNE map (Figure 6C). Even though moDC express prototypic DC markers such as CD11c and HLA-DR, these cells did not overlap with any DC subset. We conclude that as a population of cells generated ex vivo, moDC are not representative of any DC subsets present in healthy individuals in vivo.
Skin explants serve as a tool to evaluate DC receptors for therapeutic targets
DC express several C-type lectins that act as antigen uptake receptors. These receptors have been exploited as targets for antigen delivery using mAb as carriers. So far, these anti-receptor mAb have been administered subcutaneously (s.c.) or intradermally (i.d.) in humans, targeting mainly skin DC subsets. However, the expression of these receptors has mostly been analyzed in moDC or blood DC. Given our observed DC variation among donors and tissues, we evaluated the expression of uptake receptors in skin DC subsets. We used viSNE (Figure S7A) and violin (Figure 7A) plots to depict the expression of each receptor. Surprisingly, we found that the expression of most of these receptors varied slightly among donors, but was not conserved between tissues. DEC205 was detected in most myeloid cells analyzed, with higher expression in blood than skin. Dectin1 and CD206 were highly expressed by skin macrophages, cDC1 and cDC2; however, blood cDC2 almost completely lacked expression of either of these receptors. Similarly, CLEC9A was present in blood cDC1 but completely absent in skin cDC1. As expected, Langerin was robustly expressed on LC. XCR1 expression was very low and restricted to a fraction of cDC1 cells in blood and skin. Our results show that skin DC subsets have a specific pattern of C-type lectins that is not shared with blood DC as previously assumed.
Figure 7. Uptake of anti-receptor mAb by skin DC subsets.

(A) Violin plots of uptake receptors in blood and skin myeloid subsets analyzed using CyTOF panel 3. Each dot represents a donor (n=4–7, 6 independent exp.). (B) Schematic of the mAb inoculation (upper panel). Individual viSNE display expression (Stained) or capture (Inoculated) of mAb (1 representative of 3 independent exp.). (C) Uptake of A647-labeled anti-Clec9A–or control mAb in C57BL/6 mice 24h after s.c. inoculation (1 representative of 2 exp.). Please see Figure S7 for blood vs. skin comparison and flow cytometry validation of mAb capturing.
We next designed a strategy to evaluate the uptake of anti-receptor mAb by CyTOF using skin explants (Figure 7B). We found that metal-conjugated mAb injected i.d. were efficiently up-taken by skin DC subsets, and the capture pattern recapitulated the expression of the surface receptor in each DC subset (“Stained” vs. “Inoculated”; Figure 7B). We further corroborated these results by flow cytometry and confirmed receptor specificity using control mAb without receptor affinity (Figure S7B). As expected, anti-Langerin mAb were captured by LC, whereas anti-DEC205, -Dectin1 and -CD206 were captured by cDC1, cDC2, and macrophages. Finally, anti-XCR1 mAb were only captured by a small number of skin cDC1, whereas anti-CLEC9A mAb were not captured by any DC subsets, consistent with the lack of CLEC9A expression in the skin.
We and others have reported in mice that mAb against CLEC9A are efficiently captured by splenic CD8+ cDC1 (Idoyaga et al., 2011), but CLEC9A targeting has not been reported to our knowledge in CD103+ skin migratory cDC1. Thus, we revised anti-CLEC9A mAb targeting (Figure 7C). Confirming our human findings, we found that anti-CLEC9A mAb were efficiently captured by CD8+ lymphoid-resident cDC1, but not by CD103+ skin migratory cDC1. These results validate that CLEC9A is not expressed by skin cDC1 in both humans and mice.
Altogether, we have demonstrated that human skin explants combined with CyTOF provide a reliable model to study anti-receptor mAb targeting to distinct DC subsets in situ. Furthermore, we show that care should be taken when extrapolating surface receptor expression results from blood to skin DC subsets, from RNA to protein expression, or from mouse to human. Importantly, although uptake receptors are differentially expressed between tissues, we found only slight heterogeneity among individuals, supporting the use of these receptors as targets for in vivo DC subset-based therapies administered i.d.
Discussion
An active area of research in DC immunobiology has been the classification and alignment of DC subsets among species and tissues, for which transcriptome analysis and ontogeny have been powerful assets (Merad et al., 2013; See et al., 2017; Villani et al., 2017). However, it remains unclear whether DC subsets that share the same ontogeny have conserved phenotypic characteristics when exposed to distinct external cues. To address this question, we characterized phenotypic adaptations of human DC subsets through comprehensive protein profiling at a single-cell resolution. Our dataset reveals striking differences in the phenotype of human DC subsets among individuals and tissues that, to our knowledge, have not been reported with transcriptome or flow cytometry analysis. Furthermore, our study unmasks diversity in the human DC network critical for the development of new therapeutic strategies.
There is no doubt that ontogeny plays an important role in determining DC lineages; however, we found that donor-specific cues are also involved in fine-tuning each subset’s phenotype. We observed that blood and skin cDC2 were the most heterogeneous DC among individuals. In blood, cDC2 could be grouped in 3 distinct clusters based on CD172a and CD163 expression; however, these clusters were differentially represented in each donor. Furthermore, circulating cDC2 did not correlate with skin cDC2, arguing against the presence of subsets in healthy humans. A recent study identified two cDC2 subpopulations based on CD163 and CD32 expression (Villani et al., 2017); however, only one individual was analyzed. It is not our main aim here to take a position on whether human cDC2 should be further divided based on their ontogeny and function, but rather to bring to light the limitations in the approaches used until now to analyze these cells. These limitations include the number of parameters used by flow cytometry (<12), the few a priori-determined parameters used to isolate DC subsets for transcriptome analysis and the number of donors and tissues analyzed. Our analysis of several healthy donors and tissues argues against the existence of cDC2 subpopulations and favors the hypothesis that cDC2 exist in a continuum of cellular phenotypes among donors. This scenario may be different during inflammation, when cDC2 could stem from both DC precursors and monocytes (Menezes et al., 2016). Nevertheless, interindividual heterogeneity in cDC2 may have important functional consequences. Indeed, the capacity of cDC2 to cross-present antigen is still unclear (Palucka and Banchereau, 2013), but may be modulated by phenotypically and functionally distinct cDC2 present in some individuals, but not all.
Our CyTOF analysis allowed us to independently identify a cluster of cells localized at the intersection of pDC and cDC2, demonstrating that this analysis is sufficiently powered to distinguish small populations. Further assessment revealed that this cluster coincides with previously described pDC subsets (Matsui et al., 2009; Zhang et al., 2017) and the recently identified Axl+ DC (Villani et al., 2017). The origin of Axl+ DC is still a matter of debate. Villani et al describe these cells as a distinct subset based on their function and limited proliferation capacity. In contrast, See et al argue that these cells are circulating DC progenitors. We found that healthy skin lacks Axl+ DC, a tissue known to also lack pDC (Gregorio et al., 2010), but not DC progenitors (Merad et al., 2013). On the other hand, lymphoid organs harbor both pDC and Axl+ DC. These observations position Axl+ DC more closely to pDC and argue against these cells being DC progenitors. Further studies will be required to dissect the origin, function and plasticity of this newly-identified population in human health and disease.
We found that Axl+ DC were not a homogeneous population. Their phenotype ranged from pDC- to cDC-like and correlated with their capacity to present antigen. We also found variation in TCF4 expression within this population, with cells more similar to pDC expressing more TCF4. TCF4 is required to maintain a pDC phenotype (Reizis et al., 2011), and its loss causes acquisition of a cDC-like phenotype (Ghosh et al., 2010), which can explain the continuum of phenotypes observed among Axl+ DC. We also showed that the capacity of pDC to promote the proliferation of allogeneic T cells is not solely attributable to Axl+ contamination; rather, “pure” pDC require activation with CD40L and IL-3 to become potent antigen-presenting cells, which is associated with TCF4 downregulation and acquisition of a cDC-like phenotype. These results agree with a model in which TCF4 represses the cDC developmental program in human pDC as previously suggested (Reizis et al., 2011). Considering their TCF4 expression and transitional pDC to cDC phenotype, it is possible that circulating TCF4lowAxl+ DC represent in situ plasticity of the pDC lineage.
We analyzed skin DC because of the broad adoption of this tissue for human immunization. We opted to analyze adult skin rather than newborn foreskin given that adults are the target population for DC-based vaccines. We found CyTOF to be uniquely suited to analyze this tissue. First, the number of parameters allowed by CyTOF facilitated the use of unbiased analysis methods. Second, minor populations were not excluded based on pre-determined conceptions about phenotype. Finally, skin tissue auto-fluorescence that confounds flow cytometry analysis (Haniffa et al., 2012) is absent from CyTOF. Our analysis reveals that skin cDC1 and cDC2 do not overlap with their blood or lymphoid-organ homologs, suggesting that the phenotype of human DC subsets is in part dictated by environmental cues. There is evidence in mice that tissue signals can fine-tune the phenotype and function of myeloid cells (Lavin et al., 2015). The high genetic and environmental diversity inherent in human populations may have a more critical role in determining human DC subsets’ phenotype and possibly function. Understanding the functional specialization of DC subsets imprinted in the skin is imperative for harnessing these cells effectively.
Our cross-sectional sampling of de-identified human skin did not allow us to dissect the sources of observed heterogeneity in cDC2, which could include age, gender and anatomical location of the samples. We cannot eliminate the possibility of pathology; yet, various observations argued against heterogeneity based merely on immune status. First, our skin samples had no visible abnormalities. Second, while cDC2 phenotype was heterogeneous, skin LC and cDC1 phenotypes were fairly conserved among individuals. This observation was in line with the heterogeneity observed in blood cDC2, samples obtained from young healthy donors. Finally, we did not find pDC in the skin, cells reported to be present in the skin during pathological states (Gregorio et al., 2010). A potential caveat of tissue cell suspension analysis is the need of enzymatic digestion, which could cleave surface receptors. We showed that the expression of several uptake receptors was equivalent during staining of cell suspensions or mAb inoculation in undigested skin explants, which suggests that our digestion procedure supports little-to-no cleavage of surface markers. Further studies will aim to understand the source of interindividual heterogeneity and investigate its association with health and disease.
Although moDC have been FDA-approved for cancer therapeutics, this strategy has shown limited efficacy (Palucka and Banchereau, 2013). This failure has been attributed to several factors including uncharacterized differences between moDC and in situ DC. Indeed, our CyTOF analysis showed that moDC do not overlap with any DC subset localized in human tissues. An alternative to moDC is the targeting of in situ DC subsets using anti-receptor mAb as carriers for vaccine components. Our study highlights previously unappreciated variation in the expression of surface receptors between blood and skin DC subsets. For instance, CLEC9A has been suggested as a target for human cDC1 (Tullett et al., 2016); yet, we found CLEC9A to be absent in skin cDC1. This observation agrees with the previously observed lack of CLEC9A staining in human migratory DC (Segura et al., 2012), and argues that CLEC9A expression in skin cDC1 may only be at the RNA level (Haniffa et al., 2012). Of note, cDC1 are known to reduce expression of CLEC9A during development (Schraml et al., 2013), which could explain its lack of expression in the skin. Alternatively, different isoforms of CLEC9A that cannot be detected with the same mAb may be present in each tissue (Hanč et al., 2016).
Our CyTOF analysis detected Langerin expression only in LC. Recently, Langerin was reported in another subpopulation of skin cDC2 (Bigley et al., 2015). This discrepancy may represent sensitivity differences between flow cytometry and CyTOF, a transitional state of LC found in only some donors or variation in body sites analyzed. Nonetheless, the specificity of Langerin expression could be exploited therapeutically. Unlike mouse LC, human LC are reported to cross-present antigen and prime CD8+ T cells (Klechevsky et al., 2008). In future studies, we will test whether this can be achieved by antigen targeting using anti-Langerin mAb.
In humans, humoral and T cell responses can be induced by antigen delivery using mAbs that are captured by both cDC1 and cDC2, i.e., anti-CD206 and -DEC205 (Dhodapkar et al., 2014; Morse et al., 2011). These observations re-open the long-standing debate of antigen delivery to multiple vs. specific DC subsets as a vaccine strategy for immunization. Mouse studies have shown that the priming of CD8+ and CD4+ T cells is similar whether anti-receptor mAb deliver antigen only to cDC1 or to multiple other DC subsets at the same time (Idoyaga et al., 2011). On the other hand, the use of anti-receptor mAb as a therapeutic strategy for autoimmunity may require the delivery of antigen to distinct tolerogenic DC subsets (Idoyaga et al., 2013).
Our study opens an important portal into human DC biology and therapeutics. The next steps should aim to understand the environmental and genetic cues that determine differences in human DC subset phenotype and function among individuals and tissues. Understanding the foundations of human DC diversity and the consequences of this heterogeneity for the induction of adaptive immune responses may hold the key to developing personalized immunotherapies.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Juliana Idoyaga (jidoyaga@stanford.edu).
Experimental Model and Subject Details
Human Specimens
All donors provided informed consent in accordance with IRB protocols approved by Stanford University Administrative Panel on Human Subjects in Medical Research. De-identified blood and buffy coats (from whole blood or Leukocyte Reduction System (LRS) chambers) were obtained from healthy (without acute diseases) 20–40 years old adults following the guidelines of the Research and Laboratory Environmental Health and Safety program of Stanford University or from the Stanford Blood Center. Skin (10 donors), spleen (3 donors) and tonsils (3 donors) were from de-identified adult donors who provided informed consent and were subject to surgery at Stanford Hospital. Skin obtained from patients undergoing cosmetic surgery was not heavily scarred, irradiated or infected, and had no visible abnormalities such as masses, ulcers, discoloration or abrasions. Tonsils were obtained from adults with obstructive sleep apnea. Spleens obtained from the Stanford Tissue Bank, classified by a pathologist as normal, were from distal pancreatectomy from patients not subjected to any chemotherapy before organ extraction. Blood and all tissues were processed within 2h of organ procurement. In agreement with IRB protocols approved for this study, all human samples were de-identified, without demographic information such as gender or sex of donors. Given the de-identified nature of the samples, associations between phenotype and demographic information could not be performed.
Mice
Female C57BL/6 (B6) mice were purchased from The Jackson Laboratory. Animals were maintained under specific pathogen-free conditions and used at 6–8 week of age in accordance with the Stanford University Administrative Panel on Laboratory Animal Care and overseen by the Institutional Animal Care and Use Committee.
Method Details
Preparation of PBMC and monocyte-derived DC
Blood was collected using EDTA-coated tubes (BD Biosciences). PBMC were isolated by density gradient centrifugation using Ficoll-Paque PLUS (GE Healthcare). Cells were washed with PBS, counted and cryopreserved in liquid nitrogen at a concentration of 4–8×106 cells per mL in freezing medium (90%FBS-10%DMSO) until assay time. For differentiation of moDC, CD14+ monocytes were separated from PBMC using anti-CD14 magnetic beads and LS columns (Miltenyi Biotec) following manufacturer’s protocol. This resulted in >95% CD14+ monocytes as determined by FACS analysis (not shown). Purified CD14+ monocytes were incubated in a six-well culture plate (1×106 cells/mL) in RPMI 1640 medium containing 5% heat-inactivated human serum (Gem Cell), 2mM L-glutamine (Corning), 100 IU Penicillin (Corning), 100 mg/mL Streptomycin (Corning), 50 ng/mL GM-CSF (Peprotech) and 25 ng/mL IL-4 (R&D or Peprotech). Fresh medium was added at day 2 and 4 of culture. moDC were analyzed at day 5, when >95% of the cells expressed CD11c and HLA-DR.
Preparation of human tissue cell suspensions
Skin was cleaned by removing subcutaneous tissue and cut into 1×1cm pieces with a scalpel. Skin was digested overnight (12–16h) with 1 mg/mL Collagenase IV (Worthington) and 0.05 mg/mL DNAse I (Roche) in RPMI containing 10% FBS, 2mM L-glutamine, 100 IU Penicillin and 100 mg/mL Streptomycin. Incubation was done at 37°C with continuous shaking. Digestion was stopped by adding 5 mM EDTA (Corning). Cell suspension was washed with RPMI containing 10% FBS (Gibco) and filtered through 100 µm cell strainers. CD45+ cells were counted using CountBright Absolute Counting beads (ThermoFisher Scientific) and cryopreserved at a concentration of 2×106 CD45+ cells per /mL in freezing medium. Tonsil was cut in small pieces (0.2×0.2 cm) and minced with castro scissors. Pieces were transferred to a 100 µm strainer, pushed with a syringe plunger and rinsed with RPMI containing 10% FBS. 3 grams of spleen were perfused with 10 mL of digestion medium consisting of RPMI containing 10% FBS, 2µM L-glutamine (Corning), 100 IU Penicillin, 100 µg/mL Streptomycin (Corning), 0.8 mg/mL Collagenase IV and 0.05 mg/mL DNAse I. After perfusion, spleen was cut in small pieces and incubated in digestion medium for 30 min at 37°C with continuous shaking. Digestion was stopped by adding 5 mM EDTA. Cell suspensions were filtered through 100 µm cell strainers, centrifuged and enriched in PBMC by density gradient centrifugation using Ficoll-Paque PLUS.
Tissue leukocyte enrichment
Skin cell suspensions were thawed and incubated with 50 µg/mL human gamma-globulin (ThermoFisher Scientific) to block nonspecific binding. Cell suspensions were incubated with purified mAb against TE-7 (EMD Millipore), CD140a (clone αR1, BD Biosciences), DARC (R&D Systems) and desmoglein-3 (clone 5G11, ThermoFisher Scientific), following incubation with anti-mouse magnetic Dynabeads (Thermo Scientific) at a concentration of 10 magnetic beads per target cell, and following manufacturer’s instructions. Tonsil and spleen cell suspensions were thawed and incubated with 50 µg/mL human gamma-globulin (ThermoFisher Scientific) to block nonspecific binding. Cell suspensions were incubated with purified mAb against CD3 (OKT3) and CD20 (2H7), followed by anti-mouse magnetic beads (Dynabeads, ThermoScientific) at a concentration of 4 beads per target cell.
Cell sorting
Buffy coats and LRS chamber were obtained from healthy adult donors (<40 years old). PBMC were isolated with Ficoll-Paque PLUS density gradient centrifugation as described above. Next, myeloid cells were negatively enriched using mAb against CD3 (OKT3), CD19 (HIB19), CD14 (HCD14), CD335 (9E2), and CD66b (G10F5) followed by anti-mouse magnetic beads (Dynabeads, ThermoScientific) at a concentration of 2–4 beads per target cell. Cells were sorted to >99% purity (see Figure S4) using a FACSAria Fusion cell sorter (BD Biosciences) in the Stanford Shared FACS Facility. For purification of Axl− pDC (“pure pDC”), PBMC were first enriched via negative selection using pDC Isolation Kit II (Miltenyi Biotec) following manufacturer’s instructions and further sorted to 99% purity based on the expression of BDCA4 and lack of Axl.
Stimulation of Purified pDCs
“Pure” pDCs were cultured in 200µL complete R10 media containing 10% FBS, 2mM L-glutamine (Corning), 100 IU Penicillin, 100 µg/mL Streptomycin (Corning), 25mM HEPES (Corning), 1mM Sodium Pyruvate (Corning), 100 µM MEM Nonessential Amino Acids (Corning) and 55µM 2-Mercaptoethanol (Gibco) in 96 well U-bottom plates. Cells were stimulated with 10ng/mL IL-3 (R&D) plus recombinant human 200ng/mL CD40L (R&D).
Mixed Leukocyte Reaction
PBMC obtained by gradient centrifugation with Ficoll-Paque Plus were washed twice with PBS. A maximum of 107 cells were resuspended in 1.7nM CFSE (Sigma), incubated at 37°C in a water bath for 10 min and washed with MACS solution (2% BSA from Sigma plus 2nM EDTA in PBS). After CFSE labeling, T cells were obtained using Pan T Cell Isolation Kit (MACS Miltenyi Biotec), according to manufacturer’s instructions. Dendritic cells were sorted following the gating strategy shown in Figure S3. For each subset, 5,000 dendritic cells were plated with 100,000 CFSE labeled T cells (1:20 ratio). Percentage and number of proliferated T cells were analyzed at day 5. In some cases, “pure” (Axl−) pDCs were cultured for 2–6 days with CD40L+IL3, after which, cells were re-counted and plated with allogeneic T cells (1:20 ratio) in the presence of CD40L+ IL3. Same T cell donor was used to perform the experiments with freshly-isolated vs. activated pDC. Results expressed as frequency of CFSElo T cells, total number of CFSElo T cells, or fold change relative to the average frequency of CFSElo T cells cultured with freshly-isolated pDC.
Staining cell suspensions for CyTOF and FACS
Metal-conjugated mAb were obtained from Fluidigm or labeled using MaxPar X8 labeling kit (Fluidigm) according to manufacturer’s instructions (see Table S1). Anti-DEC205 (3G9) and the corresponding isotype control were generously provided by Celldex Therapeutics. MAb for FACS were purchased from Biolegend, R&D and eBiosciences (see Table S2). For FACS analysis, cryopreserved samples were thawed and washed twice with media containing benzonase 25×108 U/mL (Pierce MAb) followed by incubation with human gamma-globulin to block nonspecific binding for 15 minutes on ice. Cells were incubated with antibody mixes in human FACS buffer (2mM EDTA, 2% Donor equine serum in PBS) for 20 minutes on ice. Cells were acquired on a 5-laser LSRFortessa X20 (BD Biosciences). Control samples included unstained and single-fluorochrome-stained compensation beads (BD Biosciences) for accurate compensation, and fluorescence minus one (FMO) for DC markers. FACS data were analyzed using FlowJo software (Tree Star, Inc.). Histograms despite the median fluorescence intensity or MFI. For mass cytometry analysis, thawed samples were stained with 1 mL of 0.25 µM cisplatin (Fluidigm) for 5 min at room temperature to exclude dead cells. Cells were then washed with CyFACS buffer (BSA 1%, EDTA 2mM in PBS) and stained with heavy-metal-labeled mAb cocktail for 30 min on ice. Cells were washed twice with CyFACS then fixed with 2% fresh paraformaldehyde (SIGMA) in PBS overnight. After fixation, cells were permeabilized with Perm/wash buffer (eBioscience) and stained with intracellular mAb for 30 minutes in ice. After washing, cells were incubated with Iridium intercalator 125nM (Fluidigm) for 20 minutes in PBS. Cells were washed with water, filtered and acquired in a CyTOF2 (Fluidigm).
Targeting in situ assays
Skin was cleaned from subcutaneous tissue and cut in 2 cm × 2 cm pieces. 0.3µg of monoclonal antibodies (mAb) against surface receptors (or the corresponding control antibody without receptor affinity) were injected intradermally (i.d.) in two different sites (25 µL per site). Skin was incubated at 37°C for 6h in complete RPMI medium. After incubation, skin was processed, digested and stained as described above.
In vivo CLEC9A Targeting
Anti-CLEC9A (10B4) and control Ig (GL117) mAb were labeled with Alexa 647 per the manufacturer’s instructions (Life Technologies) as previously described (Idoyaga et al., 2011). C57BL/6 mice were inoculated with 10 µg of Alexa 647-labeled mAb s.c. in the footpad and euthanized 24h later. Skin draining lymph nodes were harvested and single cells suspensions were obtained by enzymatic digestion for 25 min at 37°C in Hanks’ buffer (Life Technologies) containing 400 units/mL Collagenase D (Roche) and 50 µg/mL DNasel (Roche). A total of 5 mM EDTA (Life Technologies) was added for the last 5 min. Cell suspensions were washed, incubated for 10 min at 4°C with 2.4G2 mAb to block Fc receptors, and stained with mAb against surface molecules for 20 min at 4°C. Stained cells were acquired on a LSRFortessa ×20, and data were analyzed with FlowJo Software. For gating strategy, please see (Idoyaga et al., 2013).
Quantification and Statistical Analysis
CyTOF2 files were exported as .fsc and normalized using the Nolan Lab’s normalizer (https://github.com/nolanlab/bead-normalization). Events were gated in FlowJo to identify leukocytes (CD45+) that were lineage negative (CD3− CD19− CD335− and CD66b− to exclude T, B, NK cells and granulocytes, respectively) and not monocytes (CD16− CD14−) in blood and lymphoid organs. For X-shift clustering analysis, .fcs were imported and transformed in Vortex (Samusik et al., 2016) using 5,000 events for each donor. All CyTOF data was transformed using hyperbolic arcsin (asinh x/5). Clustering analysis was performed using Euclidean distances in X-shift and the number of neighbors for density estimate (K) value was selected based on the elbow point cluster number in each analysis. Events expressing HLA-DR−/lo in blood were merged into a single cluster. For visualization, .fcs files from individual donors were exported from Vortex and processed using the CYT (SightOf) implementation for Matlab (Amir et al., 2013). For viSNE maps in Figures 4 and 6, all donors were merged and 15,000 events from each tissue were used to build the map. viSNE contour plots were used as a reference map. Events from individual donors or organs were then plotted and overlaid into the reference viSNE map. Values corresponding to the median intensity of expression for all CyTOF data were obtained from Vortex and used for further statistical analysis. For statistical analysis of MLR comparing different DC populations, a one-way ANOVA plus Tukey’s multiple comparison post-test correction was performed. For analysis of MLR before and after “pure” pDC activation, a paired student’s t-test was run. To compare the median intensities between organs, an unpaired student’s t-test was performed. All statistical tests run with GraphPad Prism 6. Statistical significance was annotated as follows: *p<0.05, **p<0.01, ***p<0.001. Principal Component Analysis (PCA) was performed and visualized in R using prcomp and the rgl package. Pearson correlation coefficient (PCC) heatmaps were calculated in R and visualized with the corrplot package. Violin plots and expression heatmaps were drawn with the ggplot2 package.
Supplementary Material
Highlights.
CyTOF reveals interindividual heterogeneity among DC subsets, especially cDC2.
Human skin harbors cDC with a unique phenotype and lacks Axl+ DC.
Axl+ DC display phenotypic and functional diversity, and pDC exhibit plasticity.
Receptor profiling identifies targets for antigen delivery to skin DC subsets.
Acknowledgments
M.A-H is recipient of a CONACyT (Mexico) and Stanford Medicine Dean’s Office fellowships. R.L. is the recipient of an NSF GRFP fellowship. This work was supported by NIH grant 1DP2AR069953 (to JI), the Baxter and Freidenrich Foundations (to JI) and U19AI057229 (to MMD). Further support comes from NIH P30 CA124435 utilizing the Shared FACS Facility and Tissue Bank. Cell sorting was performed on instruments funded through S10RR025518 and S10OD016318. We thank the blood and tissue donors for their participation. We also thank the surgical pathology department, the surgeons and nurses, Drs. Dekker and Capasso, Research Nurse Swope and Janet Bueno for tonsil, spleen and skin sample acquisition. We also thank Drs. Maecker, Leipold, Dermadi and Fragiadakis for helpful discussion about CyTOF and analysis. We thank the Idoyaga Lab members for technical support, and Drs. Gottfried, Carette, and Fernandez Vidal for discussions and critical reading of the manuscript. E.G.E. is a scientific cofounder, shareholder and member of the board of directors of Medeor Therapeutics and Bolt Biopharmaceutics. T.K. is an employee and shareholder of Celldex Therapeutics. G.P.N. is a consultant for Fluidigm and the inventor of CyTOF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors Contributions
Conceptualization, M.A-H., R.L. and J.I.; Methodology, M.A-H., R.L., G.N. and J.I.; Investigation, M.A-H. and R.L.; Formal Analysis, M.A-H. and R.L.; Resources, L.W., P.M., E.G.E. T.K., M.M.D. and G.N.; Writing, M.A-H., R.L. and J.I.; Visualization, M.A-H. and R.L.; Funding Acquisition, J.I.; Supervision, J.I.; Project administration, J.I.
References
- Adema GJ, de Vries IJM, Punt CJ, Figdor CG. Migration of dendritic cell based cancer vaccines: in vivo veritas? Current Opinion in Immunology. 2005;17:170–174. doi: 10.1016/j.coi.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Amir E-AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balan S, Ollion V, Colletti N, Chelbi R, Montanana-Sanchis F, Liu H, Manh T-PV, Sanchez C, Savoret J, Perrot I, et al. Human XCR1+ Dendritic Cells Derived In Vitro from CD34+ Progenitors Closely Resemble Blood Dendritic Cells, Including Their Adjuvant Responsiveness, Contrary to Monocyte-Derived Dendritic Cells. J Immunol. 2014;193:1622–1635. doi: 10.4049/jimmunol.1401243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Bigley V, McGovern N, Milne P, Dickinson R, Pagan S, Cookson S, Haniffa M, Collin M. Langerin-expressing dendritic cells in human tissues are related to CD1c+ dendritic cells and distinct from Langerhans cells and CD141high XCR1+ dendritic cells. Journal of Leukocyte Biology. 2015;97:627–634. doi: 10.1189/jlb.1HI0714-351R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, Anandasabapathy N, Schlesinger S, Caskey M, Liu K, et al. Circulating precursors of human CD1c+ and CD141+ dendritic cells. Journal of Experimental Medicine. 2015;212:401–413. doi: 10.1084/jem.20141441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong C, Idoyaga J, Do Y, Pack M, Park SH, Lee H, Kang Y-S, Choi J-H, Kim JY, Bonito A, et al. Production of monoclonal antibodies that recognize the extracellular domain of mouse langerin/CD207. J Immunol Methods. 2007;324:48–62. doi: 10.1016/j.jim.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, Hollander den NS, Kant SG, et al. Transcription Factor E2-2 Is an Essential and Specific Regulator of Plasmacytoid Dendritic Cell Development. Cell. 2008;135:37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros J, Cagnard N, Woollard K, Patey N, Zhang S-Y, Senechal B, Puel A, Biswas SK, Moshous D, Picard C, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–386. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, Chuang E, Sanborn RE, Lutzky J, Powderly J, et al. Induction of Antigen-Specific Immunity with a Vaccine Targeting NY-ESO-1 to the Dendritic Cell Receptor DEC-205. Science Translational Medicine. 2014;6:232ra51. doi: 10.1126/scitranslmed.3008068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossum E, Grødeland G, Terhorst D, Tveita AA, Vikse E, Mjaaland S, Henri S, Malissen B, Bogen B. Vaccine molecules targeting Xcr1 on cross-presenting DCs induce protective CD8+ T-cell responses against influenza virus. Eur J Immunol. 2015;45:624–635. doi: 10.1002/eji.201445080. [DOI] [PubMed] [Google Scholar]
- Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B. Continuous Expression of the Transcription Factor E2-2 Maintains the Cell Fate of Mature Plasmacytoid Dendritic Cells. Immunity. 2010;33:905–916. doi: 10.1016/j.immuni.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, Miron M, Kumar BV, Griesemer A, Ho S-H, et al. Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity. 2017;46:504–515. doi: 10.1016/j.immuni.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, Gilliet M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. Journal of Experimental Medicine. 2010;207:2921–2930. doi: 10.1084/jem.20101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. 1997;185:1101–1111. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Dutertre C-A, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity. 2016;45:669–684. doi: 10.1016/j.immuni.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanč P, Schulz O, Fischbach H, Martin SR, Kjær S, Reis e Sousa C. A pH- and ionic strength-dependent conformational change in the neck region regulates DNGR-1 function in dendritic cells. Embo J. 2016;35:2484–2497. doi: 10.15252/embj.201694695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang X-N, Malinarich F, Malleret B, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. doi: 10.1016/j.immuni.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidkamp GF, Sander J, Lehmann CHK, Heger L, Eissing N, Baranska A, Lühr JJ, Hoffmann A, Reimer KC, Lux A, et al. Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Sci Immunol. 2016;1:eaai7677. doi: 10.1126/sciimmunol.aai7677. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huysamen C, Willment JA, Dennehy KM, Brown GD. CLEC9A Is a Novel Activation C-type Lectin-like Receptor Expressed on BDCA3+ Dendritic Cells and a Subset of Monocytes. J Biol Chem. 2008;283:16693–16701. doi: 10.1074/jbc.M709923200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, Mucida D, Merad M, Steinman RM. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest. 2013;123:844–854. doi: 10.1172/JCI65260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idoyaga J, Lubkin A, Fiorese C, Lahoud MH, Caminschi I, Huang Y, Rodriguez A, Clausen BE, Park CG, Trumpfheller C, et al. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc Natl Acad Sci USA. 2011;108:2384–2389. doi: 10.1073/pnas.1019547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, Briere F, Chaussabel D, Zurawski G, Palucka AK, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29:497–510. doi: 10.1016/j.immuni.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin Y, Mortha A, Rahman A, Merad M. Regulation of macrophage development and function in peripheral tissues. Nature Reviews Immunology. 2015;15:731–744. doi: 10.1038/nri3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luber CA, Cox J, Lauterbach H, Ben Fancke Selbach M, Tschopp J, Akira S, Wiegand M, Hochrein H, Keeffe MO, et al. Quantitative Proteomics Reveals Subset-Specific Viral Recognition in Dendritic Cells. Immunity. 2010;32:279–289. doi: 10.1016/j.immuni.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Matsui T, Connolly JE, Michnevitz M, Chaussabel D, Yu CI, Glaser C, Tindle S, Pypaert M, Freitas H, Piqueras B, et al. CD2 Distinguishes Two Subsets of Human Plasmacytoid Dendritic Cells with Distinct Phenotype and Functions. J Immunol. 2009;182:6815–6823. doi: 10.4049/jimmunol.0802008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, Patel R, Gautier EL, Hugues S, Longhi MP, et al. The Heterogeneity of Ly6C(hi) Monocytes Controls Their Differentiation into iNOS(+) Macrophages or Monocyte-Derived Dendritic Cells. Immunity. 2016;45:1205–1218. doi: 10.1016/j.immuni.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. Journal of Experimental Medicine. 2012;209:1153–1165. doi: 10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag D, Proietto AI, Loudovaris T, Mannering SI, Vremec D, Shortman K, Wu L, Harrison LC. Human dendritic cell subsets from spleen and blood are similar in phenotype and function but modified by donor health status. The Journal of Immunology. 2011;186:6207–6217. doi: 10.4049/jimmunol.1002632. [DOI] [PubMed] [Google Scholar]
- Morse MA, Chapman R, Powderly J, Blackwell K, Keler T, Green J, Riggs R, He L-Z, Ramakrishna V, Vitale L, et al. Phase I study utilizing a novel antigen-presenting cell-targeted vaccine with Toll-like receptor stimulation to induce immunity to self-antigens in cancer patients. Clin. Cancer Res. 2011;17:4844–4853. doi: 10.1158/1078-0432.CCR-11-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka K, Banchereau J. Dendritic-Cell-Based Therapeutic Cancer Vaccines. Immunity. 2013;39:38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011;29:163–183. doi: 10.1146/annurev-immunol-031210-101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds G, Haniffa M. Human and Mouse Mononuclear Phagocyte Networks: A Tale of Two Species? Front. Immunol. 2015;6:330. doi: 10.3389/fimmu.2015.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samusik N, Good Z, Spitzer MH, Davis KL, Nolan GP. Automated mapping of phenotype space with single-cell data. Nat Meth. 2016;13:493–496. doi: 10.1038/nmeth.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho D, Mourão-Sá D, Joffre OP, Schulz O, Rogers NC, Pennington DJ, Carlyle JR, Reis e Sousa C. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J Clin Invest. 2008;118:2098–2110. doi: 10.1172/JCI34584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, KC W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. Journal of Experimental Medicine. 2012;209:1135–1152. doi: 10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AWS, See P, Shin A, Wasan PS, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraml BU, van Blijswijk J, Zelenay S, Whitney PG, Filby A, Acton SE, Rogers NC, Moncaut N, Carvajal JJ, Sousa CRE. Genetic Tracing via DNGR-1 Expression History Defines Dendritic Cells as a Hematopoietic Lineage. Cell. 2013;154:843–858. doi: 10.1016/j.cell.2013.07.014. [DOI] [PubMed] [Google Scholar]
- See P, Dutertre C-A, Chen J, Günther P, McGovern N, Irac SE, Gunawan M, Beyer M, Händler K, Duan K, et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science. 2017;356:eaag3009. doi: 10.1126/science.aag3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. Journal of Experimental Medicine. 2012;209:653–660. doi: 10.1084/jem.20111457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal K, Dhodapkar KM, Dhodapkar MV. Targeting human dendritic cells in situ to improve vaccines. Immunology Letters. 2014;162:59–67. doi: 10.1016/j.imlet.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo S-L, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang C-L, et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity. 2017;46:148–161. doi: 10.1016/j.immuni.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpfheller C, Longhi MP, Caskey M, Idoyaga J, Bozzacco L, Keler T, Schlesinger SJ, Steinman RM. Dendritic cell-targeted protein vaccines: a novel approach to induce T cell immunity. J Intern Med. 2011;271:183–192. doi: 10.1111/j.1365-2796.2011.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullett KM, Leal Rojas IM, Minoda Y, Tan PS, Zhang J-G, Smith C, Khanna R, Shortman K, Caminschi I, Lahoud MH, et al. Targeting CLEC9A delivers antigen to human CD141+ DC for CD4+ and CD8+T cell recognition. JCI Insight. 2016;1:e87102. doi: 10.1172/jci.insight.87102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356:eaah4573. doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Gregorio JD, Iwahori T, Zhang X, Choi O, Tolentino LL, Prestwood T, Carmi Y, Engleman EG. A distinct subset of plasmacytoid dendritic cells induces activation and differentiation of B and T lymphocytes. Proc Natl Acad Sci USA. 2017;114:1988–1993. doi: 10.1073/pnas.1610630114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.