

Abstract
Background and Purpose
Everolimus is an allosteric inhibitor of the mechanistic target of rapamycin complex 1 (mTORC1) widely known for its potent autophagy stimulating properties. Because everolimus shows poor solubility and stability in aqueous solutions, long‐term in vivo administration in preclinical models is challenging. The aim of the present study was to evaluate the effects of short‐term and long‐term everolimus administration on mTORC1 inhibition and autophagy induction in mice.
Experimental Approach
We developed a vehicle in which everolimus was solubilized and stable at 37°C for at least 1 month. Using osmotic minipumps, GFP microtubule‐associated protein light chain 3 transgenic mice were treated continuously either with vehicle or everolimus (1.5 mg·kg−1 per day) for 3 or 28 days. Alternatively, a regimen consisting of intermittent everolimus administration (every other day) for 56 days by oral gavage was used. Autophagy markers and mTORC1 activation status were investigated in the liver.
Key Results
As expected, everolimus inhibited mTORC1 and stimulated autophagy in the liver after 3 days of treatment. However, continuous administration for 28 days resulted in hyperactivation of the Akt1‐mTORC1 pathway accompanied by a remarkable decrease in autophagy markers. Everolimus given intermittently for 56 days partially rescued mTORC1 sensitivity to the drug but without inducing autophagy. The failure to induce autophagy following long‐term everolimus administration was due to uncoupling of the mTORC1 substrate unc‐51 like autophagy activating kinase 1.
Conclusions and Implications
Our data encourage the use of intermittent everolimus regimens to prevent tolerance and to extend its activity.
Abbreviations
- ACTB
β‐actin
- LC3
microtubule‐associated protein light chain 3
- mTORC1/2
mechanistic target of rapamycin, complex 1/2
- RCC
renal cell carcinoma
- p70S6K
ribosomal protein S6 kinase B1
- S6rp
S6 ribosomal protein
- SQSTM1
sequestosome 1
- ULK1
unc‐51 like autophagy activating kinase
Tables of Links
| LIGANDS |
|---|
| Chloroquine |
| Everolimus |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Everolimus (RAD001) is one of several semisynthetic derivatives of rapamycin, collectively known as rapalogs, with superior pharmacokinetics and pharmacodynamics as compared with the parent compound (Maurizio Salvadori, 2011; Lamming et al., 2013). The drug is widely used in immunosuppressive and cancer therapy as well as in drug‐eluting stents (Wander et al., 2011; De Meyer et al., 2015). Like all rapalogs, everolimus binds to its intracellular target FK‐506 binding protein 1A to form a complex that inhibits the mechanistic target of rapamycin (mTOR) in an allosteric manner (Hay and Sonenberg, 2004; Dowling et al., 2010). mTOR is a large (~270 kDa) serine/threonine kinase highly conserved in all eukaryotes that can be found in two distinct complexes: mTOR complex 1 (mTORC1) and mTORC2. Rapalogs are mainly mTORC1 inhibitors (Martinet et al., 2014a; Kim and Guan, 2015; Kurdi et al., 2016). Inhibition of mTORC1 leads to several downstream events including the inhibition of cellular proliferation, cap‐dependent translation and stimulation of autophagy, a catabolic mechanism involving self‐digestion of organelles and long‐lived proteins (Sehgal, 2003; Dowling et al., 2010). Because autophagy is considered a cell survival mechanism (Ohsumi, 2014; Martinet et al., 2014a), a growing body of evidence links impaired autophagy to many pathophysiological processes, most of which are chronic and dependent on long‐term treatment (Jiang and Mizushima, 2014). Therefore, major efforts are being made to stimulate autophagy in several disease models (Rubinsztein et al., 2007; Liao et al., 2012; Wirawan et al., 2012; Nixon, 2013). Nutrient deprivation is a strong stimulus of the autophagic process, but because this approach is unsuitable for chronic treatment in vivo, pharmacological interventions remain the most feasible choice. Rapalogs are therefore frequently used for their ability to promote autophagy (Verheye et al., 2007; Crazzolara et al., 2009; Zhai et al., 2014; Kim and Guan, 2015). Remarkably, even though several methods have been applied to administer everolimus chronically in mice, little or no information is available regarding autophagy induction after long‐term mTORC1 inhibition in vivo. Osmotic minipumps represent an elegant method to administer the drug continuously in preclinical in vivo studies. The devices are easy to use, provide constant drug plasma levels for various periods depending on the chosen model and induce only minimal stress in the animals (Cameron et al., 2014). Unlike other widely implemented techniques such as intraperitoneal injections or oral gavage, osmotic minipumps, once implanted, require little attention from the investigator. However, sometimes the manufacturer's recommendations on the compatibility of some solvents with the osmotic minipump are neglected and organic solvents are used in far higher concentrations than approved. This incorrect use may account for unreliable pumping rates and imprecise data interpretation. Consequently, the first objective of the present study was to develop a vehicle that ensures maximal everolimus stability at body temperature over long periods while not compromising the physical integrity of the osmotic minipump. The second aim was to investigate the effect of everolimus treatment following short‐term (3 days) and continuous chronic administration (28 days) on mTORC1 signalling and autophagy in mice. To this end, we have chosen to analyse liver tissue of treated animals because this organ is widely known to react in a quick and robust manner to induction of autophagy (Mizushima et al., 2004; Martinet et al., 2013).
Methods
Everolimus solution
Everolimus (Novartis Institutes for Biomedical Research) was dissolved in a vehicle composed of 50% (v/v) DMSO (Acros Organics, 167850010), 40% (v/v) propylene glycol (Fagron, 610948) and 10% (v/v) absolute ethanol (Fisher Chemicals, E/0650DF/17). The mixture was supplemented with 0.4 μL·mL−1 Tween 20 (Sigma‐Aldrich, 1379) to increase the solubility of everolimus and was stored light‐protected at −20°C as a 10 or 20 mM stock solution for up to 1 month. For stability tests, the everolimus stock was incubated at 37°C for different time intervals, then diluted in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum to treat murine J774A.1 macrophages. Four hours after treatment, the phosphorylation status of ribosomal protein S6 kinase B1 (p70S6K) was examined by Western blotting..
Mice
All animal care and experimental procedures were approved by the ethics committee of the University of Antwerp (no. 2012‐54). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). We used female GFP‐microtubule‐associated protein light chain 3 (LC3) transgenic mice (RIKEN BioResource Center, strain GFP‐LC3 #53), containing a GFP fused to rat LC3 under control of the chicken β actin promoter. The mice were housed in a temperature‐controlled environment (six animals per cage) with 12 h light/dark cycles and received food and water ad libitum. These mice are widely used in autophagy research (Mizushima et al., 2004). The mice were selected randomly from the pool of all cages and divided into groups. All analyses were carried out without prior knowledge of the treatments. Mice received a numerical code throughout the whole experiment.
In vivo administration of everolimus by osmotic minipumps
Female GFP‐LC3 mice (13–15 weeks old) were randomly divided into groups (n = 6 per group) and anaesthetised using sevoflurane. Hair was shaved off in the neck area, and a small incision was made between the ears. Using a haemostat, a subcutaneous pocket wide enough for an osmotic minipump was created. The pocket was flushed with saline and an osmotic minipump (Alzet) was inserted. After closing the incision with sterile sutures, a single dose of 7.5 mg·kg−1 ketorolac tromethamine (Ketalar(R), Pfizer, USA) was subcutaneously administered to reduce any postoperative pain. Two types of osmotic minipumps were used to deliver either vehicle or everolimus (Novartis, Switzerland) at 1.5 mg·kg−1 per day. Model 1003D was used to treat mice for 3 days, while model 1004 offered a long‐term delivery of 28 days. Calculations of the required everolimus concentrations were made per individual mouse based on its weight and using the online tool provided by Alzet. At the end of the experiment, mice were injected intraperitoneally with 100 mg·kg−1 chloroquine (Sigma‐Aldrich, 50–63‐5) dissolved in saline. Three hours later, blood was taken via the retro‐orbital plexus of anaesthetised mice (ketamine (Rompun(R), Beyer, Germany) 100 mg·kg−1 and xylazine 10 mg·kg−1, i.p.). The animals were subsequently killed using an overdose of sodium pentobarbital (250 mg·kg−1, I.P.). Samples of the liver were collected for Western blotting, immunohistochemical analysis, fluorescence microscopy and transmission electron microscopy (TEM). Administration of the vehicle alone had no effect on mTORC1 signalling or autophagy in vivo (not shown).
In vivo administration of everolimus by oral gavage
Female GFP‐LC3 transgenic mice (RIKEN BioResource Center, strain GFP‐LC3 #53) were divided into two groups (n = 6 per group, mice 13–15 weeks old) receiving either everolimus or vehicle [25 μL, diluted with 175 μL of Dulbecco's phosphate buffered saline (Gibco, 14 190 094)]. Oral gavage was performed using a 20G flexible catheter after removing the needle (Terumo; outer diameter 1.10 mm, inner diameter 0.80 mm) every other day for a period of 56 days. Following this period, mice were injected with 100 mg·kg−1 chloroquine I.P. and killed 3 h later.
Western blotting
Cells were lysed in an appropriate volume of Laemmli sample buffer (Bio‐Rad, 1610737). Tissue samples were first homogenized in RIPA buffer (Sigma‐Aldrich, R0278) containing protease and phosphatase inhibitors (Roche Diagnostics, 04693159001; 04906845001) using a Precellys 24 homogenizer (Bertin technologies). Samples were then heat‐denatured for 5 min in boiling water and loaded on pre‐casted Novex Bolt 4–12% Bis‐Tris gels (Invitrogen, BG‐04120). After electrophoresis, proteins were transferred to an Immobilon‐FL PVDF membrane (Millipore, IPFL00010) according to standard procedures. Membranes were blocked for 1 h in Odyssey blocking buffer (LI‐COR Biosciences, 972‐40100) diluted 1:5 with PBS. After blocking, membranes were probed overnight at 4°C with primary antibodies diluted in Odyssey blocking buffer, followed by 1 h incubation with IRDye‐labelled secondary antibodies at room temperature. Antibody detection was achieved using an Odyssey SA infrared imaging system (LI‐COR Biosciences). The intensity of the protein bands was quantified using Image Studio software.
The following mouse antibodies were used: anti‐actin, β (ACTB) (clone AC‐15, Sigma‐Aldrich, A5441), anti‐Cdk2 (BD Transduction Laboratories, C18520) and anti‐LC3 (clone 5F10, Nanotools, 0231–100/LC3‐5F10). Rabbit antibodies included anti‐GFP (ab6556) from Abcam and anti‐sequestosome 1 (SQSTM1) (#5114), anti‐phospho‐p70S6K(Thr389) (#9205), anti‐p70S6K (#9202), anti‐phospho‐Akt(Ser473) (#9271), anti‐Akt (#9272), anti‐phospho‐S6 ribosomal protein (S6rp)(Ser235/236) (#2211), anti‐S6rp (#2217), anti‐phospho‐unc‐51 like autophagy activating kinase 1 (ULK1)(Ser757) (#6888), anti‐ULK1 (#8054), anti‐phospho‐mTOR(Ser2448) (#2971) and anti‐mTOR (#2972) from Cell Signalling. IRDye‐labelled secondary antibodies (goat anti‐mouse IgG, 926‐68070 and goat anti‐rabbit IgG, 926‐32211) were purchased from LI‐COR Biosciences.
Immunohistochemistry
Tissue samples were fixed in 4% neutral buffered formalin for 24 h prior to paraffin embedding. Thereafter, 5‐μm‐thick sections were prepared and immunostained for LC3 using mouse anti‐LC3 (clone 5F10, Nanotools, 0231‐100/LC3‐5F10) followed by anti‐mouse Envision+ (Dako, K4001) as previously described (Martinet et al., 2013; Martinet et al., 2014b). Quantification was done by taking five images per section at random, and ImageJ software was used for analysis of the LC3‐positive area.
Fluorescence microscopy
Formalin‐fixed, paraffin‐embedded tissue sections (5 μm thick) were deparaffinized with toluene followed by washing in isopropanol and water. Samples were finished with Vectashield mounting medium containing DAPI (Vector laboratories, H‐1200). Images were taken using an EVOS FL Auto Cell Imaging System (ThermoFisher) and quantification of GFP puncta was done with ImageJ software.
Transmission electron microscopy
Tissues were prepared for TEM analysis as previously described (Martinet et al., 2014a). A FEI Tecnai microscope was used to examine ultrathin sections at 80–120 kV. Quantification of autophagic vacuoles was achieved by manually counting vacuoles in five images per section taken at random.
Everolimus plasma concentration
A sample of 100 μL plasma was mixed with an equal volume of TitriPUR 0.1 M ZnSO4 precipitation reagent (Merck Millipore, 1.08879.1000) and 250 μL [13C2D4]everolimus internal standard solution (5 μg·L−1, Analytical Services International) dissolved in 100% methanol (LCMS reagent, J.T. Baker, 9830–02). Samples were vortexed for 1 min, kept 5 min in an ultrasonic bath and centrifuged for 10 min at 18 400× g. Twenty microlitre of the clear supernatant was injected in an online SPE trapping column (Luna 3 μm C18 10 × 4.6 mm, Phenomenex, 00D‐4251‐E0). By using a 60/40 methanol/water mixture, the analyte was eluted onto an analytical column (Zorbax Eclipse XBD‐C18 1.8 μm 4.6 × 50 mm, Agilent, 925975‐902) in a high‐speed online HPLC tandem MS system (Agilent HPLC 1260 connected to a 6490 Triple Quadrupole Mass Spectrometer). The mobile phase used for chromatography consisted of an isocratic mixture of water/methanol (5/95) in which 2 mM ammonium acetate and 0.5% formic acid were dissolved. The analytical column temperature was kept at 60°C.
For detection, an Agilent 6490 quadrupole mass spectrometer was used in positive mode. Nitrogen was employed as nebulzing and collision gas. The jetstream settings were as follows: drying gas temperature 200°C, drying gasflow 14 L·min−1, sheath gas temperature 250°C with a flow of 11 L·min−1, capillary voltage 4500 V.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as box‐plots (showing medians and interquartile ranges with whiskers representing ranges) and were analysed with the Mann–Whitney U‐test using GraphPad Prism software (version 6.0). A probability value <0.05 was considered significant.
Results
Everolimus stability
Different solvent mixtures were prepared to investigate the stability of everolimus in a vehicle compatible with an osmotic minipump for long periods. As described previously (Simamora et al., 2001), adding a benzoate buffer would increase solubility of everolimus. However, this acidic environment resulted in diminished activity of the drug after only 3 days of incubation at 37°C (data not shown). Addition of water to the mixture equally resulted in accelerated degradation of the drug. Based on the physico‐chemical properties of everolimus, a mixture of 50% DMSO, 40% propylene glycol and 10% ethanol supplemented with 0.4 μL·mL−1 Tween 20 was eventually used to prepare a 10 mM everolimus stock solution. The stock was incubated light‐protected at 37°C, serially diluted in RPMI medium and used to treat J774A.1 murine cells at different time points (0, 10 and 28 days). The stability of everolimus was assessed by its ability to inhibit the phosphorylation of p70S6K at Thr389, a highly sensitive target of mTORC1. From day 0 until day 10, the phosphorylation of p70S6K was fully suppressed by subnanomolar concentrations of everolimus (Figure 1), whereas after 28 days, only concentrations above 1 nM led to complete dephosphorylation of p70S6K. Everolimus did not influence the expression of total p70S6K.
Figure 1.
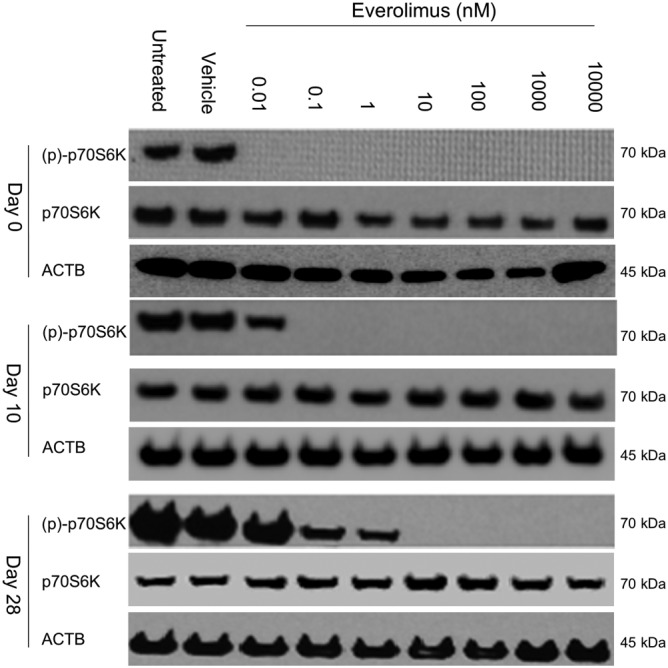
Everolimus is stable at 37°C for at least 28 days in vehicle solution. A 10 mM everolimus stock was made in the proposed vehicle (see text for details) and stored light‐protected at 37°C. At different time points (0, 10 or 28 days), J774A.1 murine macrophage cells were treated with a serial dilution containing 0.01 nM to 1 μM everolimus (three replicates per treatment). After 4 h of treatment, cells were lysed and subjected to Western blotting (detection of p70S6K phosphorylation at Thr389).
Short‐term (3 days) everolimus treatment
An osmotic minipump filled with everolimus solution was inserted subcutaneously in GFP‐LC3 mice.. Mice were intraperitoneally injected with 100 mg·kg−1 chloroquine 3 h prior to kill to block autophagic flux. The minipump delivered the drug for 3 days at a constant rate of 1.5 mg·kg−1 per day, yielding a plasma concentration of 630 ± 34 nM at the time of killing. Western blot analysis of liver tissue from everolimus‐treated GFP‐LC3 mice revealed no effect of short‐term everolimus administration on Akt1 phosphorylation upstream of mTOR while the phosphorylation of both mTOR and its downstream target S6rp was significantly suppressed compared to the vehicle‐treated control animals (Figure 2). This was accompanied with a decrease of the mTORC1‐controlled cell cycle progression regulator cyclin‐dependent kinase 2 (Cdk2) expression, a crucial protein involved in cell proliferation (Woods, 2010), when normalized with the ACTB loading control.
Figure 2.
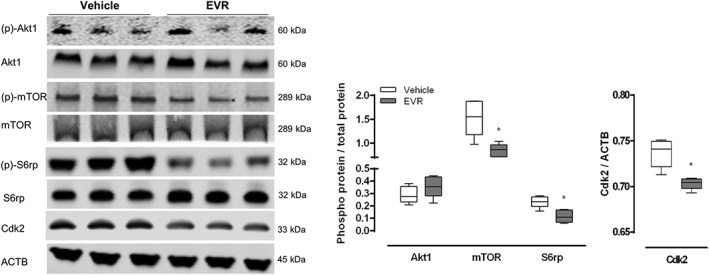
Short‐term (3 days) everolimus treatment results in mTOR inhibition in vivo. Osmotic minipumps releasing either everolimus (EVR, 1.5 mg·kg−1 per day) or vehicle were implanted in GFP‐LC3 mice. After 3 days, mice were given an I.P. injection of 100 mg·kg−1 chloroquine and were killed. Liver samples were collected and homogenized. Western blotting was performed to measure the phosphorylation status of Akt1 (Ser473), mTOR (Ser2448) and S6rp (Ser235/236) as well as the expression of Cdk2. ACTB was used as a loading control. Representative blots are shown (n = 6 for both groups, *P < 0.05, significantly different from vehicle).
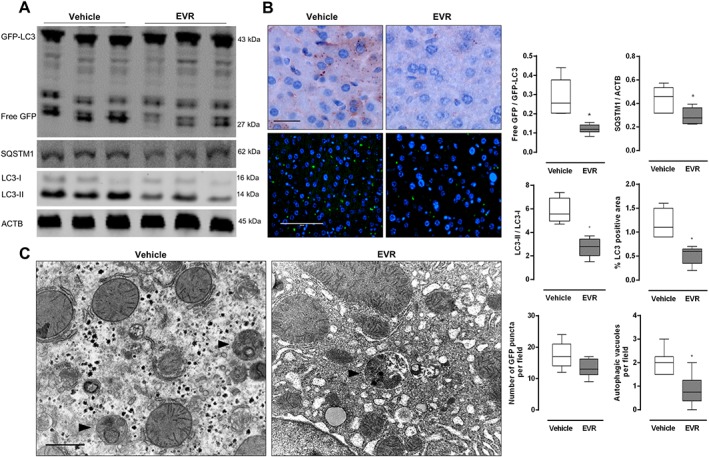
To evaluate the induction of autophagy in vivo, Western blotting was used to assess several autophagy markers such as the accumulation of SQSTM1, the ratio free GFP to GFP‐LC3 and the ratio endogenous LC3‐II to LC3‐I. Ratios of free GFP/GFP‐LC3 and LC3‐II/LC3‐I showed a significant increase following everolimus treatment, indicating effective autophagy induction (Figure 3A). Increased levels of SQSTM1 were also observed. Immunohistochemical analysis of LC3 in formalin‐fixed liver tissue showed a significant increase in LC3 dots in everolimus‐treated animals (Figure 3B, upper panel). Enhanced GFP fluorescence in the everolimus group further indicated autophagy induction (Figure 3B, lower panel). Lastly, TEM analysis confirmed the formation of numerous autophagic vacuoles after administration of everolimus (Figure 3C). It should be noted that the results described above were obtained after injecting mice with chloroquine to prevent protein degradation in autolysosomes and to improve the accumulation of autophagy markers. Results of autophagy induction without chloroquine injection are shown in Figure S1. In the absence of chloroquine, levels of SQSTM1 were decreased in the treated animals while ratios of LC3‐II/LC3‐I and free GFP/GFP‐LC3 were increased implying functional autophagic flux.
Figure 3.

Short‐term (3 days) everolimus treatment results in autophagy stimulation in vivo. Osmotic minipumps releasing either everolimus (EVR, 1.5 mg·kg−1 per day) or vehicle were implanted in GFP‐LC3 mice. After 3 days, mice were given an I.P. injection of 100 mg·kg−1 chloroquine and were killed. Liver samples were collected. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3, LC‐II/LC3‐I and accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analysed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using TEM. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05, significantly different from vehicle).
Long‐term (28 days) continuous everolimus treatment
To administer everolimus chronically, osmotic minipumps were implanted in GFP‐LC3 mice to deliver the drug for 28 days at the same constant rate of 1.5 mg·kg−1 per day. This approach yielded a plasma concentration of 503 ± 58 nM at day 28. This concentration, while lower than the one found after short‐term treatment, most likely due to everolimus degradation, was still far above the 5 nM threshold for in vivo mTORC1 inhibition (Tanaka et al., 2008; Fox et al., 2010; O'Reilly and McSheehy, 2010). Nonetheless, everolimus was unable to dephosphorylate mTOR or S6rp. Akt1 – upstream of mTOR – showed significant hyperphosphorylation while the downstream target Cdk2 was overexpressed in the everolimus‐treated animals (Figure 4). Western blot results also revealed lower expression levels of SQSTM1 accompanied with a decrease in the ratio of both free GFP/GFP‐LC3 and LC3‐II/LC3‐I in the everolimus‐treated group versus controls (Figure 5A), suggesting a decline (rather than an increase) in autophagy. Impaired autophagy was confirmed by immunohistochemistry (lack of LC3 puncta, Figure 5B, upper panel), fluorescence microscopy (decrease in GFP fluorescence, Figure 5B, lower panel) and TEM (decreased number of autophagic vesicles, Figure 5C). Results without chloroquine injection prior to kill also showed a decline in autophagy in everolimus‐treated animals as demonstrated by decreased LC3‐II/LC3‐I and free GFP/GFP‐LC3 ratios as well as a reduction of GFP positive puncta (Figure S2).
Figure 4.
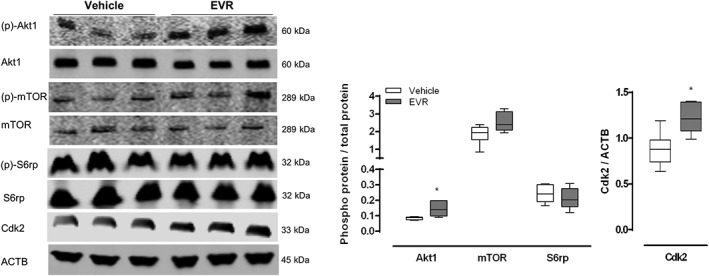
Continuous long‐term (28 days) everolimus treatment induces drug tolerance in vivo. GFP‐LC3 mice received osmotic minipumps releasing either everolimus (EVR, 1.5 mg·kg−1 per day) or vehicle for a period of 28 days. Prior to killing, mice received an i.p.injection of 100 mg·kg−1 chloroquine. Western blotting was performed to measure the phosphorylation status of Akt1 (Ser473), mTOR (Ser2448) and S6rp (Ser) as well as the expression of Cdk2. ACTB was used as a loading control. Representative blots are shown (n = 6 for both groups, *P < 0.05, significantly different from vehicle).
Figure 5.
Continuous long‐term (28 days) everolimus treatment results in a decrease in autophagic markers in vivo. GFP‐LC3 mice received osmotic minipumps releasing either everolimus (EVR, 1.5 mg kg−1 day−1) or vehicle for a period of 28 days. Prior to killing, mice received an i.p. injection of 100 mg kg−1 chloroquine. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3 and LC3‐II/LC3‐I as well as the accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analysed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using TEM. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05, significantly different from vehicle).
Long‐term (56 days) intermittent everolimus treatment
In an attempt to bypass everolimus resistance after continuous everolimus treatment, we decided to perform another set of experiments in which everolimus is administered to mice intermittently. This approach allows a drug‐free period during which liver cells may reactivate mTORC1. The drug, or vehicle, was administered to female GFP‐LC3 mice by oral gavage every other day for a period of 56 days, thereby effectively treating mice for a total of 28 days. Mice were given a dose of 3 mg·kg−1, which resulted in a plasma concentration of 603 ± 112 nM in blood withdrawn 2 h after oral gavage. Treated mice revealed a marked sensitivity to the drug as indicated by the strong dephosphorylation of S6rp. However, Akt1 phosphorylation was slightly increased while mTOR phosphorylation and expression of Cdk2 were unaffected (Figure 6). Autophagy induction was not observed (Figure 7A–C).
Figure 6.
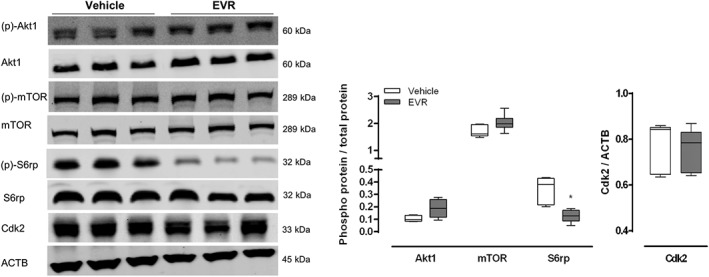
Intermittent long‐term (56 days) everolimus treatment rescues mTORC1 sensitivity to therapy in vivo. GFP‐LC3 mice were administered 3 mg·kg−1 everolimus (EVR) by oral gavage every other day for a period of 56 days. At the end of the experiment, mice were then given an I.P. injection containing 100 mg·kg−1 chloroquine and were killed 3 h later. Western blotting was performed to measure the phosphorylation status of Akt1 (Ser473), mTOR (Ser2448) and S6rp (Ser235/236) as well as the expression of Cdk2. ACTB was used as a loading control. Representative blots are shown (n = 6 for both groups, *P < 0.05, significantly different from vehicle).
Figure 7.

Intermittent long‐term (56 days) everolimus treatment fails to induce autophagy in vivo. GFP‐LC3 mice were administered 3 mg kg−1 everolimus (EVR) by oral gavage every other day for a period of 56 days. At the end of the experiment, mice were then given an I.P. injection containing 100 mg·kg−1 chloroquine and were killed 3 h later. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3 and LC3‐II/LC3‐I as well as the accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analysed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using TEM. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05, significantly different from vehicle).
Because failure to induce autophagy was not due to depletion of autophagy specific proteins such as SQSTM1 or LC3, we investigated the activation status of the critical autophagy initiator ULK1 (Lee and Tournier, 2011; Chan, 2012). Under normal circumstances, mTORC1 inactivates ULK1 by phosphorylating the protein at serine 757, which leads to inhibition of autophagy. Inhibition of mTORC1 dephosphorylates ULK1 and triggers initiation of autophagy (Ganley et al., 2009; Nakagawa et al., 2012). We observed a decrease in ULK1 phosphorylation following short‐term everolimus treatment (Figure 8). However, phosphorylation increased significantly when the drug was given continuously for a long period (Figure 8). Intermittent administration failed to have any effect (Figure 8).
Figure 8.
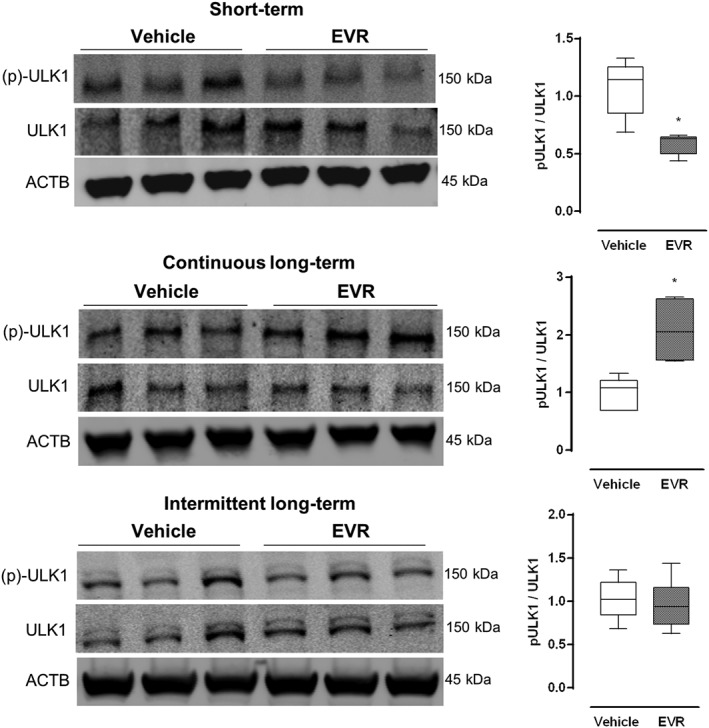
Frequency of everolimus administration affects ULK1 phosphorylation in vivo. Osmotic minipumps releasing either everolimus (1.5 mg·kg−1 per day) or vehicle were implanted in GFP‐LC3 mice for 3 days (short‐term) or 28 days (continuous long‐term). Alternatively, GFP‐LC3 mice were administered 3 mg·kg−1 everolimus by oral gavage every other day for a period of 56 days (intermittent). mTORC1‐specific phosphorylation of ULK1 at Ser757 was measured in the liver by Western blotting. The ratio phosphorylated of protein/total protein was used to assess the degree of phosphorylation. Representative blots are shown (n = 6 for all groups, *P < 0.05, significantly different from vehicle).
Discussion and conclusions
Since being FDA approved, everolimus has been used in immunosuppressive therapy and has recently also been evaluated clinically for the treatment of numerous disorders, in particular cancer (Verheye et al., 2007; Mueller et al., 2008; Wander et al., 2011; Martinet et al., 2014a). One of the crucial in vivo effects of everolimus pertains to its ability to stimulate autophagy. However, despite the chronic nature of most disorders in which the drug has been tested, little is known about its efficacy in inducing autophagy following long‐term treatment. This study was initially designed to compare the in vivo effects of a short‐ and long‐term treatment with everolimus on mTORC1 inhibition and autophagy induction using osmotic minipumps implanted subcutaneously in GFP‐LC3 transgenic mice. Given the chronic nature of the study, we first tested several solvent mixtures to ensure optimal solubility and stability of everolimus at physiological temperatures over long time spans. A vehicle composed of 50% DMSO, 40% propylene glycol and 10% ethanol supplemented with 0.4 μL·mL−1 Tween 20 was found to solubilize everolimus and to keep it stable at 37°C for at least 28 days. Because organic solvents at high concentrations can damage the inner compartments of an osmotic minipump, the concentration of each component in the solvent mixture was carefully chosen to avoid disturbance of the pumping rate and the generation of unreliable data. Even though some activity was lost during the in vitro incubation of everolimus in the above‐mentioned solvent mixture at 37°C, everolimus retained most of its activity, resulting in full mTORC1 inhibition. To our knowledge, no other vehicles have been described in which everolimus remains stable at this temperature for long periods.
Short‐term (3 days) treatment of GFP‐LC3 mice with everolimus led to strong mTORC1 inhibition in the liver as demonstrated by a significant reduction of S6rp and mTOR phosphorylation. Moreover, autophagy was clearly stimulated as shown by Western blotting, immunohistochemistry, fluorescence microscopy and TEM. In contrast, continuous long‐term (28 days) administration of everolimus resulted in diminished autophagy as compared with the non‐treated control group. This finding could be explained by a complete failure of everolimus to inhibit mTORC1 signalling in the liver tissue of these mice, as previously reported following chronic exposure to rapamycin (Drake et al., 2013). Of note, the plasma levels of everolimus following continuous administration were lower than those measured during short‐term treatment (503 ± 58 nM vs. 630 ± 34 nM respectively). This reduction is most likely due to a certain degree of everolimus degradation in the osmotic minipump, or else the result of individual variability in the pharmacokinetics of the drug as shown in humans (Budde et al., 2004). Nonetheless, because everolimus is known to inhibit mTORC1 in vivo at plasma concentrations as low as 5 nM (Tanaka et al., 2008; Fox et al., 2010; O'Reilly and McSheehy, 2010), this reduction could not explain the loss of everolimus efficacy.
A plausible alternative explanation might relate to the resistance to everolimus following long‐term treatment, which is a clinically relevant and well‐described phenomenon in renal cell carcinoma (RCC), prostate cancer and some other tumour types (Tsaur et al., 2011; Hassan et al., 2014; Juengel et al., 2014; Wagle et al., 2014). These tumours, while initially reacting well to everolimus therapy, often progress to a resistant form in the long run. This resistance is only seen following allosteric mTORC1 inhibition leaving tumours still responsive to a novel class of mTOR kinase inhibitors that are currently being tested in clinical trials (Guichard et al., 2015). A number of mechanisms including absence of the mTORC1‐S6rp negative feedback loop, mutations of the allosteric binding site of mTOR or hyperactivation of the Akt1‐mTORC1 pathway have been proposed as possible explanations for the acquired resistance to allosteric mTORC1 inhibitors (Carew et al., 2011; Porta et al., 2014; Saran et al., 2015; Yang et al., 2015). For example, long‐term everolimus treatment of RCC cells in vitro resulted in everolimus resistance, a condition that was characterized by a high phosphorylation status of both Akt1 and S6rp (Juengel et al., 2012; Hassan et al., 2014; Juengel et al., 2014), which are up and downstream of mTORC1 respectively. Moreover, a significant up‐regulation of the cell cycle regulator Cdk2 was observed in RCC cells after prolonged everolimus treatment. Interestingly, we also found these features in the liver samples of GFP‐LC3 mice after continuous, long‐term everolimus treatment. It is therefore tempting to speculate that analogous to a tumour exposed to long‐term mTORC1 inhibition, the high synthesis rate of liver cells (Turley et al., 1981; Shahbazian et al., 1987) drives the initiation of adaptation mechanisms, resulting in hyperactivation of the Akt1‐mTORC1 pathway and drug tolerance. Therefore, we hypothesized that a drug‐free period could relieve pressure on the cells and prevent mTORC1 resistance to everolimus. Indeed, the use of intermittent administration regimens in avoiding resistance against everolimus is supported by a recent case of a patient with malignant insulinomia (Baratelli et al., 2014) and has also been shown to minimize undesirable immunosuppressive effects of everolimus without affecting its activity on tumours (Boulay et al., 2004).
Because peritonitis is a common side effect of intraperitoneal injections with solutions containing DMSO, we delivered everolimus by oral gavage. We decided to use an intermittent regimen delivering everolimus every other day because of the very short half‐life (6 h) in mice (O'Reilly and McSheehy, 2010). The experiment lasted 56 days during which GFP‐LC3 mice effectively received everolimus (or vehicle) for 28 days to allow comparison with the group receiving everolimus continuously. The delivered dose (3 mg·kg−1) yielded plasma concentrations comparable with those obtained when using osmotic minipumps. Strikingly, intermittent treatment resulted in a strong reduction in S6rp phosphorylation in the liver. Compared with continuous administration, a drug‐free period effectively attenuated the development of tolerance and successfully extended the period during which the liver remained (partially) sensitive to treatment with everolimus. These results may add new insights to the field of oncology as efficacy of everolimus in several tumours has been correlated with its ability to inhibit S6rp phosphorylation (O'Reilly and McSheehy, 2010; Li et al., 2014; Masuda et al., 2014). However, there was no sign of autophagy induction in the intermittently treated group despite the apparent mTORC1 sensitivity to everolimus. Because mTORC1 substrates are not equally sensitive to allosteric mTORC1 inhibition (Thoreen and Sabatini, 2009; Thoreen et al., 2009; Benjamin et al., 2011; Yoon and Roux, 2013; Saran et al., 2015), we hypothesized that less sensitive substrates may become uncoupled from this inhibition more rapidly resulting in partial mTORC1 inhibition. For instance, while phosphorylation of the very sensitive substrate S6rp may still be suppressed, the less sensitive ULK1 can explain why autophagy was not induced. ULK1 is the main effector through which both mTORC1 and the AMP‐activated protein kinase are able to initiate autophagy (Kim et al., 2011; Nakagawa et al., 2012; Russell et al., 2013; Saran et al., 2015). Consequently, we investigated the phosphorylation status of ULK1 at the mTORC1 specific Ser757 site. In accordance with its ability to induce autophagy, short‐term administration of everolimus promptly inhibited ULK1 phosphorylation. This effect was lost following long‐term treatment, either continuously or intermittently, resulting in significant up‐regulation of ULK1 phosphorylation and inhibition of autophagy.
Taken together, our data indicate that allosteric short‐term (3 days) mTORC1 inhibition by everolimus in GFP‐LC3 mice triggers dephosphorylation of the mTORC1 target S6rp and induction of autophagy (Figure 9). However, if mice were continuously exposed to everolimus for longer periods (28 days), mTORC1 resistance and autophagy inhibition occurred (Figure 9). Long‐term (56 days) intermittent administration of everolimus led to dephosphorylation of S6rp, but did not rescue the autophagy‐inducing properties of everolimus (Figure 9). These findings are particularly interesting in cancer research since resistance to everolimus is very common in tumours and S6rp dephosphorylation mediated by the drug has been linked to its anti‐tumour activity.
Figure 9.
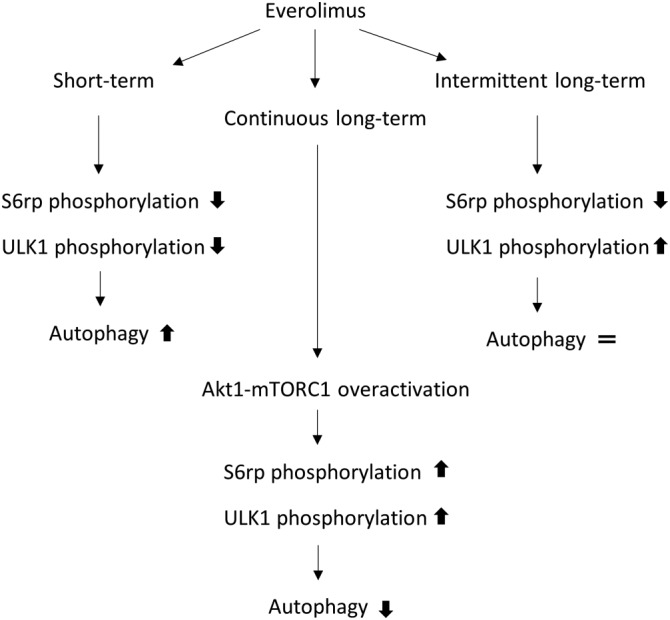
Summary scheme of the main results. Short‐term everolimus administration inhibits phosphorylation of the mTORC1 substrates S6rp and ULK1 resulting in robust induction of autophagy. Continuous long‐term administration of everolimus triggers adaptation mechanisms resulting in hyperphosphorylation of ULK1 and inhibition of autophagy. Long‐term administration of everolimus intermittently rescues responsiveness to the drug only in the most sensitive substrate (S6rp) without any effect on autophagy.
Author contributions
D.M.G.R.Y., M.W., N.H. and K.A. study conception and design; K.A.: All the Western Blotting data, histology data and surgical procedures. D.D.M. and A.S.: All the plasma concentration measurements. L.A. performed the oral gavages. T.J‐P.: All the electron microscopy data, acquisition of data; K.A.: Analysis of the Western Blotting, histology and electron microscopy data. D.D.M. and A.S.: Analysis of the plasma concentration data. M.W. and D.M.G.R.Y.: Interpretation of the data in general. L.K.: Statistical analysis, analysis and interpretation; All authors helped drafting the manuscript. K.A.: Introduction, Methods and Results. L.A., D.D.M., T.J‐P. and A.S.: Results and critical revision. L.K.: Discussion (clinical aspects) and critical revision. N.H.: Discussion/Results (Plasma concentration and clinical implications). M.W. and D.M.G.R.Y.: Discussion and critical revision, drafting of manuscript; All authors approved the final version to be submitted and agreed to be accountable.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Short‐term (3 days) everolimus treatment without chloroquine. Osmotic minipumps releasing either everolimus (EVR, 1.5 mg kg−1 day−1) or vehicle were implanted in GFP‐LC3 mice. Animals were killed after 3 days. Liver samples were collected. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3, LC‐II/LC3‐I and accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analyzed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using transmission electron microscopy. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05 versus vehicle).
Figure S2 Continuous long‐term (28 days) everolimus treatment without chloroquine. GFP‐LC3 mice received osmotic minipumps releasing either everolimus (EVR, 1.5 mg kg−1 day−1) or vehicle for a period of 28 days. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3 and LC3‐II/LC3‐I as well as the accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analyzed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using transmission electron microscopy. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05 versus vehicle).
Supporting info item.
Acknowledgements
This work was supported by the Fund for Scientific Research (FWO)‐Flanders (project G.0160.13 N) and the University of Antwerp (BOF). The FEI Tecnai transmission electron microscope was purchased with support of the Hercules Foundation. The authors thank Dr. Noboru Mizushima (Tokyo Medical and Dental University, Japan) for providing GFP‐LC3 mice and Rita Van Den Bossche, Hermine Fret, Anne‐Elise Van Hoydonck, Lieve Svensson and Francis Terloo for their excellent technical support.
Kurdi, A. , De Doncker, M. , Leloup, A. , Neels, H. , Timmermans, J. ‐P. , Lemmens, K. , Apers, S. , De Meyer, G. R. Y. , and Martinet, W. (2016) Continuous administration of the mTORC1 inhibitor everolimus induces tolerance and decreases autophagy in mice. British Journal of Pharmacology, 173: 3359–3371. doi: 10.1111/bph.13626.
References
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratelli C, Brizzi MP, Tampellini M, Scagliotti GV, Priola A, Terzolo M et al. (2014). Intermittent everolimus administration for malignant insulinoma. Endocrinol Diabetes Metab Case Rep 2014: 140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D, Colombi M, Moroni C, Hall MN (2011). Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov 10: 868–880. [DOI] [PubMed] [Google Scholar]
- Boulay A, Zumstein‐Mecker S, Stephan C, Beuvink I, Zilbermann F, Haller R et al. (2004). Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res 64: 252–261. [DOI] [PubMed] [Google Scholar]
- Budde K, Neumayer HH, Lehne G, Winkler M, Hauser IA, Lison A et al. (2004). Tolerability and steady‐state pharmacokinetics of everolimus in maintenance renal transplant patients. Nephrol Dial Transplant 19: 2606–2614. [DOI] [PubMed] [Google Scholar]
- Cameron AM, Adams DH, Greenwood JE, Anderson PJ, Cowin AJ (2014). A novel murine model of hypertrophic scarring using subcutaneous infusion of bleomycin. Plast Reconstr Surg 133: 69–78. [DOI] [PubMed] [Google Scholar]
- Carew JS, Kelly KR, Nawrocki ST (2011). Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol 6: 17–27. [DOI] [PubMed] [Google Scholar]
- Chan EY (2012). Regulation and function of uncoordinated‐51 like kinase proteins. Antioxid Redox Signal 17: 775–785. [DOI] [PubMed] [Google Scholar]
- Crazzolara R, Bradstock KF, Bendall LJ (2009). RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 5: 727–728. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Meyer GRY, Grootaert MOJ, Michiels CF, Kurdi A, Schrijvers DM, Martinet W (2015). Autophagy in vascular disease. Circ Res 116: 468–479. [DOI] [PubMed] [Google Scholar]
- Dowling RJO, Topisirovic I, Fonseca BD, Sonenberg N (2010). Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta 1804: 433–439. [DOI] [PubMed] [Google Scholar]
- Drake JC, Peelor FF, Biela LM, Watkins MK, Miller RA, Hamilton KL et al. (2013). Assessment of mitochondrial biogenesis and mtorc1 signaling during chronic rapamycin feeding in male and female mice. J Gerontol A Biol Sci Med Sci 68: 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JH, Connor T, Chopra V, Dorsey K, Kama JA, Bleckmann D et al. (2010). The mTOR kinase inhibitor Everolimus decreases S6 kinase phosphorylation but fails to reduce mutant huntingtin levels in brain and is not neuroprotective in the R6/2 mouse model of Huntington's disease. Mol Neurodegener 5: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X (2009). ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284: 12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichard SM, Curwen J, Bihani T, D'Cruz CM, Yates JWT, Grondine M et al. (2015). AZD2014, an inhibitor of mTORC1 and mTORC2, is highly effective in ER+ breast cancer when administered using intermittent or continuous schedules. Mol Cancer Ther 14: 2508–2518. [DOI] [PubMed] [Google Scholar]
- Hassan B, Akcakanat A, Sangai T, Evans KW, Adkins F, Eterovic AK et al. (2014). Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget 5: 8544–8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N (2004). Upstream and downstream of mTOR. Genes Dev 18: 1926–1945. [DOI] [PubMed] [Google Scholar]
- Jiang P, Mizushima N (2014). Autophagy and human diseases. Cell Res 24: 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juengel E, Dauselt A, Makarević J, Wiesner C, Tsaur I, Bartsch G et al. (2012). Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Lett 324: 83–90. [DOI] [PubMed] [Google Scholar]
- Juengel E, Nowaz S, Makarevi J, Natsheh I, Werner I, Nelson K et al. (2014). HDAC‐inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol Cancer 13: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan K‐L (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Guan K‐L (2015). mTOR: a pharmacologic target for autophagy regulation. J Clin Investig 125: 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdi A, De Meyer GRY, Martinet W (2016). Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br J Clin Pharmacol 82: 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Sabatini DM, Baur JA (2013). Rapalogs and mTOR inhibitors as anti‐aging therapeutics. J Clin Investig 123: 980–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E‐J, Tournier C (2011). The requirement of uncoordinated 51‐like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 7: 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kong Y, Si L, Chi Z, Cui C, Sheng X et al. (2014). Phosphorylation of mTOR and S6RP predicts the efficacy of everolimus in patients with metastatic renal cell carcinoma. BMC Cancer 14: 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS et al. (2012). Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15: 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, De Loof H, De Meyer GRY (2014a). mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 233: 601–607. [DOI] [PubMed] [Google Scholar]
- Martinet W, Schrijvers DM, Timmermans JP, Bult H, De Meyer GRY (2013). Immunohistochemical analysis of macroautophagy: recommendations and limitations. Autophagy 9: 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, Timmermans J‐P, De Meyer GRY (2014b). Methods to assess autophagy in situ‐transmission electron microscopy versus immunohistochemistry. Methods Enzymol 543: 89–114. [DOI] [PubMed] [Google Scholar]
- Masuda M, Chen W‐Y, Miyanaga A, Nakamura Y, Kawasaki K, Sakuma T et al. (2014). Alternative mammalian target of rapamycin (mTOR) signal activation in sorafenib‐resistant hepatocellular carcinoma cells revealed by array‐based pathway profiling. Mol Cell Proteomics 13: 1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizio Salvadori EB (2011). Long‐term outcome of everolimus treatment in transplant patients. Transpl Res Risk Manag 3: 77–90. [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J (2008). Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR−/− mice despite severe hypercholesterolemia. Atherosclerosis 198: 39–48. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Nishihara K, Inui KI, Masuda S (2012). Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur J Pharmacol 696: 143–154. [DOI] [PubMed] [Google Scholar]
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997. [DOI] [PubMed] [Google Scholar]
- O'Reilly T, McSheehy PM (2010). Biomarker development for the clinical activity of the mTOR inhibitor everolimus (RAD001): processes, limitations, and further proposals. Transl Oncol 3: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y (2014). Historical landmarks of autophagy research. Cell Res 24: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C, Paglino C, Mosca A (2014). Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol 4: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ (2007). Potential therapeutic applications of autophagy. Nat Rev Drug Discov 6: 304–312. [DOI] [PubMed] [Google Scholar]
- Russell RC, Tian Y, Yuan H, Park HW, Chang Y‐Y, Kim J et al. (2013). ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 15: 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saran U, Foti M, Dufour J‐F (2015). Cellular and molecular effects of the mTOR inhibitor everolimus. Clin Sci (Lond) 129: 895–914. [DOI] [PubMed] [Google Scholar]
- Sehgal SN (2003). Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc 35: S7–S14. [DOI] [PubMed] [Google Scholar]
- Shahbazian FM, Jacobs M, Lajtha A (1987). Rates of protein synthesis in brain and other organs. Int J Dev Neurosci 5: 39–42. [DOI] [PubMed] [Google Scholar]
- Simamora P, Alvarez JM, Yalkowsky SH (2001). Solubilization of rapamycin. Int J Pharm 213: 25–29. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka C, O'Reilly T, Kovarik JM, Shand N, Hazell K, Judson I et al. (2008). Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol 26: 1596–1602. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Sabatini DM (2009). Rapamycin inhibits mTORC1, but not completely. (July): 725–726. [DOI] [PubMed]
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y et al. (2009). An ATP‐competitive mammalian target of rapamycin inhibitor reveals rapamycin‐resistant functions of mTORC1. J Biol Chem 284: 8023–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaur I, Makarević J, Hudak L, Juengel E, Kurosch M, Wiesner C et al. (2011). The cdk1‐cyclin B complex is involved in everolimus triggered resistance in the PC3 prostate cancer cell line. Cancer Lett 313: 84–90. [DOI] [PubMed] [Google Scholar]
- Turley SD, Andersen JM, Dietschy JM (1981). Rates of sterol synthesis and uptake in the major organs of the rat in vivo. J Lipid Res 22: 551–569. [PubMed] [Google Scholar]
- Verheye S, Martinet W, Kockx MM, Knaapen MWM, Salu K, Timmermans J‐P et al. (2007). Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol 49: 706–715. [DOI] [PubMed] [Google Scholar]
- Wagle N, Grabiner BC, Van Allen EM, Amin‐Mansour A, Taylor‐Weiner A, Rosenberg M et al. (2014). Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med 371: 1426–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wander SA, Hennessy BT, Slingerland JM (2011). Next‐generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Investig 121: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P (2012). Autophagy: for better or for worse. Cell Res 22: 43–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods TC (2010). Regulation of cell migration by mTOR is mediated through changes in p27Kip1 phosphorylation. Cell Cycle 9: 2057–2058. [DOI] [PubMed] [Google Scholar]
- Yang G, Murashige DS, Humphrey SJ, James DE (2015). A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep 12: 937–943. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Roux PP (2013). Rapamycin resistance: MTORC1 substrates hold some of the answers. Curr Biol 23: R880–R883. [DOI] [PubMed] [Google Scholar]
- Zhai C, Cheng J, Mujahid H, Wang H, Kong J, Yin Y et al. (2014). Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS One 9: e90563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Short‐term (3 days) everolimus treatment without chloroquine. Osmotic minipumps releasing either everolimus (EVR, 1.5 mg kg−1 day−1) or vehicle were implanted in GFP‐LC3 mice. Animals were killed after 3 days. Liver samples were collected. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3, LC‐II/LC3‐I and accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analyzed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using transmission electron microscopy. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05 versus vehicle).
Figure S2 Continuous long‐term (28 days) everolimus treatment without chloroquine. GFP‐LC3 mice received osmotic minipumps releasing either everolimus (EVR, 1.5 mg kg−1 day−1) or vehicle for a period of 28 days. (A) Western blotting was performed to assess the ratio of free GFP/GFP‐LC3 and LC3‐II/LC3‐I as well as the accumulation of SQSTM1. (B) Liver samples were immunohistochemically stained for LC3 (upper panels) or analyzed with fluorescence microscopy for GFP positive puncta (lower panels). Scale bar = 100 μm. (C) Autophagic vacuoles (arrow heads) were detected in ultrathin sections using transmission electron microscopy. Scale bar = 1 μm (n = 6 for both groups, *P < 0.05 versus vehicle).
Supporting info item.