

Abstract
Background and Purpose
In benign prostatic hyperplasia, increased prostate smooth muscle tone and prostate volume may contribute alone or together to urethral obstruction and voiding symptoms. Consequently, it is assumed there is a connection between smooth muscle tone and growth in the prostate, but any molecular basis for this is poorly understood. Here, we examined effects of Src family kinase (SFK) inhibitors on prostate contraction and growth of stromal cells.
Experimental Approach
SFK inhibitors, AZM475271 and PP2, were applied to human prostate tissues to assess effects on smooth muscle contraction, and to cultured stromal (WPMY‐1) and c‐Src‐deficient cells to examine effects on proliferation, actin organization and viability.
Key Results
SFKs were detected by real time PCR, western blot and immunofluorescence in human prostate tissues, some being located to smooth muscle cells. AZM475271 (10 μM) and PP2 (10 μM) inhibited SFK in prostate tissues and WPMY‐1 cells. Both inhibitors reduced α1‐adrenoceptor‐mediated and neurogenic contraction of prostate strips. This may result from cytoskeletal deorganization, which was observed in response to AZM475271 and PP2 in WPMY‐1 cells by staining of actin filaments with phalloidin. This was paralleled by reduced proliferation of wildtype but not of c‐Src‐deficient cells; cytotoxicity was mainly observed at higher concentrations (>50 μM).
Conclusions and Implications
In human prostate, smooth muscle tone and growth are both controlled by an SFK‐dependent process, which may explain their common role in bladder outlet obstruction. Targeting prostate smooth muscle tone and prostate growth simultaneously by a single compound may, in principal, be possible.
Abbreviations
- ATCC
American Type Culture Collection
- AZM475271
N‐(2‐chloro‐5‐methoxyphenyl)‐6‐methoxy‐7‐((1‐methylpiperidin‐4‐yl)methoxy)quinazolin‐4‐amine
- BOO
bladder outlet obstruction
- BPH
benign prostatic hyperplasia
- CCK‐8
cell counting kit 8
- ECL
enhanced chemiluminescence
- EdU
5‐ethynyl‐2′‐deoxyuridine
- EFS
electric field stimulation
- LUTS
lower urinary tract symptoms
- PBS‐T
PBS containing 0.1% Tween 20
- PF573228
3,4‐dihydro‐6‐[[4‐[[[3‐(methylsulfonyl)phenyl]methyl]amino]‐5‐(trifluoromethyl)‐2‐pyrimidinyl]amino]‐2(1H)‐quinolinone
- PP2
3‐(4‐chlorophenyl)1‐(1,1‐dimethylethyl)‐1H‐pyrazolol[3,4d]pyrimidin‐4‐amine
- PSA
prostate‐specific antigen
- RIPA
radioimmunoprecipitation assay
- RT‐PCR
real time PCR
- TURP
transurethral resection of the prostate
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes b |
| α1‐adrenoceptor | FAK family |
| Src family |
| LIGANDS |
|---|
| (‐)‐noradrenaline |
| Phenylephrine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Prostate smooth muscle contraction and prostate growth may cause voiding symptoms in patients with benign prostate hyperplasia, by impairing the bladder outlet due to urethral obstruction (Oelke et al., 2013; Hennenberg et al., 2014). This condition has been termed bladder outlet obstruction (BOO) and it is estimated that male lower urinary tract symptoms (LUTS) affected around 1.3 billion people worldwide in 2013, in many cases due to BOO (Irwin et al., 2011; Gratzke et al., 2015a). Complying with their critical role in the aetiology of LUTS, contraction and growth in the prostate are important targets for medical therapy (Oelke et al., 2013). However, despite their common role in BOO, contraction and growth of the prostate are still regarded separately during treatment of voiding symptoms and in basic research. In fact, combination therapies are usually required to target prostate smooth muscle tone and prostate size simultaneously in patients (Fullhase et al., 2013; Oelke et al., 2013).
α1‐Adrenoceptor antagonists are routinely applied to improve symptoms by inducing prostate smooth muscle relaxation, while prostate size can be reduced by treatment with 5α‐reductase inhibitors; this reduces the occurrence of acute urinary retention and the need for surgery (Oelke et al., 2013; Hennenberg et al., 2014). Notwithstanding the outstanding popularity of both medications, it becomes increasingly clear that their efficacy is limited (Hennenberg et al., 2014). Maximum improvements in the objective micturition parameters and of patients' perceived symptom scores are below 50%, while up to 30% may be observed with placebos (Oelke et al., 2013; Hennenberg et al., 2014). In fact, the symptoms of 36–45% of patients are not satisfactorily alleviated by the combined treatment with α1‐blockers and 5α‐reductase inhibitors, and these patients still experience considerable impairments in their quality of life, despite the cost of more than 4.2 billion USD for α1‐blockers and 5α‐reductase inhibitors worldwide in 2009 (Ventura et al., 2011; Fullhase et al., 2013). Within the first 12 months of therapy with α1‐blockers, up to 65% of patients discontinue their treatment (Cindolo et al., 2015). Given the high prevalence of male LUTS, together with the age‐dependent incidence, the expected demographic transition in industrial nations and limits of present pharmacotherapy, new options with higher efficacy would be much appreciated in the view of patients and physicians (Gratzke et al., 2015b). This can only be achieved by acquiring a detailed understanding of the molecular aetiology of LUTS, and by the identification of new targets (Soler et al., 2013).
Based on their shared contributions to the obstructive symptoms of BOO, it is reasonable to assume there is a connection between the control of smooth muscle contraction and growth in the prostate. However, the putative mechanisms connecting both components with each other, and any molecular links involved in this connection remain to be understood. Src family kinases (SFKs) are non‐receptor tyrosine kinases involved in the regulation of central cellular functions in different cell types and organs (Amata et al., 2014). SFKs can promote proliferation and cell cycle in various cell types, including non‐prostatic smooth muscle cells (Krymskaya et al., 2005; Yang et al., 2005; Hsieh et al., 2009; Li et al., 2010; Ishigaki et al., 2011). In parallel, smooth muscle contraction in the cardiovascular system, airways, gastrointestinal tract and uterus have been found to be affected by SFK inhibitors (Che and Carmines, 2005; Weigand et al., 2006; Oguma et al., 2007; Ross et al., 2007; Lu et al., 2008; Phillippe et al., 2009; El‐Daly et al., 2014). Taken together, these findings have prompted us to investigate a putative role for SFKs in the hyperplastic, human prostate.
Methods
Human prostate tissues
Human prostate tissues were obtained from patients undergoing radical prostatectomy for prostate cancer by open surgery. Prostates from patients receiving transurethral resection of the prostate (TURP) prior to radical prostatectomy were not included to this study, as TURP may affect availability and quality of the periurethral tissue. The research was carried out in accordance with the Declaration of Helsinki of the World Medical Association, and was approved by the ethics committee of the Ludwig‐Maximilians University, Munich, Germany. Patients provided informed consent for use of their tissue. Tissues were taken from the periurethral zone, while most prostate tumours are located in the peripheral zone (Pradidarcheep et al., 2011; Shaikhibrahim et al., 2012). Tissue samples did not exhibit histological signs of neoplasia, cancer or inflammation. Benign prostatic hyperplasia (BPH) is present in ~80% of patients with prostate cancer (Alcaraz et al., 2009; Orsted and Bojesen, 2013). Samples were taken immediately after prostatectomy, followed by macroscopical examination by a pathologist. Organ bath studies were performed immediately after sampling, while samples for molecular analyses were shock frozen in liquid nitrogen and stored at −80°C.
Real time PCR
RNA from frozen prostate tissues was isolated using the RNeasy Mini kit (Qiagen, Hilden, Germany). For isolation, 30 mg of tissue was homogenized using the FastPrep®‐24 system with matrix A (MP Biomedicals, Illkirch, France). RNA concentrations were measured spectrophotometrically. Reverse transcription to cDNA was performed with 1 μg of isolated RNA using the Reverse Transcription System (Promega, Madison, WI, USA). Real time PCR (RT‐PCR) for SFKs and GAPDH was performed with a Roche Light Cycler (Roche, Basel, Switzerland) using primers provided by Qiagen (Hilden, Germany) as ready‐to‐use mixes (Table 1). PCR reactions were performed in a volume of 25 μL containing 5 μL LightCycler® FastStart DNA MasterPlus SYBR Green I (Roche, Basel, Switzerland), 1 μL template, 1 μL primer and 18 μL water. Denaturation was performed for 10 min at 95°C, and amplification with 45 cycles of 15 s at 95°C followed by 60 s at 60°C. The specificity of primers and amplification was demonstrated by subsequent analysis of melting points, which revealed single peaks for each target. Results are expressed based on the number of cycles (Ct), at which the fluorescence signal exceeded a defined threshold.
Table 1.
Primers used for RT‐PCR
| Gene | RefSeq number | Reference position | Catalogue number |
|---|---|---|---|
| SRC | NM_005417 | 1366 | PPH00103C |
| NM_198291 | 1291 | ||
| LCK | NM_001042771 | 924 | PPH00185E |
| FYN | NM_002037 | 628 | PPH00147A |
| LYN | NM_001111097 | 1575 | PPH01635A |
| FGR | NM_001042729 | 1076 | PPH00093F |
| NM_001042747 | 1135 | ||
| NM_005248 | 1092 | ||
| HCK | NM_001172129 | 1758 | PPH00160A |
| FRK (syn. Rak) | NM_002031 | 1871 | PPH05726A |
| YES | NM_005433 | 1842 | PPH00255F |
| BLK | NM_001715 | 1691 | PPH00085A |
| GAPDH | NM_002046 | n. a. | QT00079247 |
RefSeq numbers refer to transcripts with reliable sequence, which are contained in the ‘National Center for Biotechnology Informations’ (NCBI) Reference Sequence database, and correspond to the indicated gene. The reference position is a position contained within the sequence of the amplicon relative to the start of the relevant RefSeq sequence. Catalogue number indicates the manufacturer's order number.
Western blot analysis
Frozen prostate tissues were homogenized in a buffer containing 25 mM Tris/HCl, 10 μM PMSF, 1 mM benzamidine and 10 μg·mL−1 leupeptine hemisulfate, using the FastPrep®‐24 system with matrix A (MP Biomedicals, Illkirch, France). After centrifugation (20 000 g, 4 min), supernatants were assayed for protein concentration using the Dc‐Assay kit (Biorad, Munich, Germany) and boiled for 10 min with SDS sample buffer (Roth, Karlsruhe, Germany). Samples of WPMY‐1, SYF‐2459, or SYF‐2498 cells were prepared as described below. Samples of prostate homogenates (20 μg per lane) or WPMY‐1 cells (40 μg per lane) were subjected to SDS‐PAGE, and proteins were blotted on Protran® nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). Membranes were blocked with PBS containing 5% milk powder (Roth, Karlsruhe, Germany) overnight, and incubated with rabbit anti phospho‐Src family (Tyr416) (#6943) (reacts with different SFK members, including c‐Src, Lyn, Fyn, Lck, Yes, Hck, when phosphorylated at equivalent sites) (New England Biolabs, Ipswich, MA, USA), rabbit anti c‐Src (#2110) (New England Biolabs, Ipswich, MA, USA), rabbit anti Lck (sc‐13), rabbit anti Fyn (sc‐16), rabbit anti Lyn (sc‐15), rabbit anti Fgr (sc‐17), rabbit anti Hck (sc‐72), goat anti Rak (sc‐6377), rabbit anti Yes (sc‐14), rabbit anti Blk (sc‐938), mouse anti pan‐cytokeratin (sc‐8018), mouse anti calponin 1/2/3 (sc‐136987), mouse anti prostate‐specific antigen (PSA) (sc‐7316) or mouse anti β‐actin antibody (sc‐47778) (if not stated otherwise, all compounds were from Santa Cruz Biotechnology, Santa Cruz, CA, USA). Subsequently, membranes were washed with PBS containing 0.1% Tween 20 (PBS‐T), incubated with secondary biotinylated goat anti rabbit, horse anti mouse or horse anti goat IgG (BA‐1000, BA‐2000, BA‐9500) (Vector Laboratories, Burlingame, CA, USA), washed again with PBS‐T, incubated with avidin and biotinylated horseradish peroxidase from the ‘Vectastain ABC kit’ (Vector Laboratories, Burlingame, CA, USA) both diluted 1:200 in PBS, and washed again with PBS‐T. Finally, blots were developed with enhanced chemiluminescence (ECL) using ECL Hyperfilm (GE Healthcare, Freiburg, Germany).
Immunofluorescence
Human prostate specimens, embedded in optimal cutting temperature (OCT) compound, were snap‐frozen in liquid nitrogen and kept at −80°C. Sections (8 μm) were cut in a cryostat and collected on Superfrost® microscope slides. Sections were post‐fixed in methanol at −20°C and blocked in 1% BSA before incubation with primary antibody overnight at room temperature. For double labelling, the following primary antibodies were used: rabbit anti c‐Src (#2110) (New England Biolabs, Ipswich, MA, USA), rabbit anti Lck (sc‐13), rabbit anti Fyn (sc‐16), rabbit anti Lyn (sc‐15), mouse anti tyrosine hydroxylase (sc‐374 048), mouse anti pan‐cytokeratin (sc‐8018) and mouse anti calponin 1/2/3 (sc‐136 987). Binding sites were visualized using Cy3‐ and Cy5‐conjugated secondary antibodies (goat anti mouse, AP124C, Millipore, Billerica, MA, USA; and goat anti rabbit, ab6564, Abcam, Cambridge, UK). Nuclei were counterstained with DAPI (Invitrogen, Camarillo, CA, USA). Immunolabelled sections were analysed using a laser scanning microscope (Leica SP2, Wetzlar, Germany). Fluorescence was recorded with separate detectors. Control stainings without primary antibodies did not yield any signals.
Tension measurements
Prostate strips (6 × 3 × 3 mm) were mounted in 10 mL aerated (95% O2 and 5% CO2) tissue baths (Föhr Medical Instruments, Seeheim, Germany), containing Krebs–Henseleit solution (37°C, pH 7.4). Preparations were stretched to 4.9 mN and left to equilibrate for 45 min. In the initial phase of the equilibration period, spontaneous decreases in tone were usually observed. Therefore, tension was adjusted three times during the equilibration period, until a stable resting tone (4.9 mN) was attained. After the equilibration period, maximum contraction induced by 80 mM KCl was assessed. Subsequently, chambers were washed three times with Krebs–Henseleit solution for a total of 30 min. Cumulative concentration response curves for noradrenaline or phenylephrine, or frequency‐ response curves induced by electric field stimulation (EFS) were created after addition of SFK inhibitors, or DMSO for controls. Application of EFS simulates action potentials, resulting in the release of endogenous neurotransmitters, including noradrenaline. For calculation of agonist‐ or EFS‐induced contractions, tensions were expressed as % of KCl‐induced contractions, as this may correct for different stromal/epithelial ratios, different smooth muscle content, varying degree of BPH or any other heterogeneity between prostate samples and patients.
Cell culture
WPMY‐1 cells are an immortalized cell line obtained from human prostate stroma without prostate cancer. Cells were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA), and kept in RPMI 1640 (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin at 37°C with 5% CO2. Before addition of AZM475271 (10 μM) or PP2 (10 μM), the medium was changed to a FCS‐free medium. For western blot analysis, cells were lyzed using radioimmunoprecipitation assay (RIPA) buffer (Sigma‐Aldrich, St. Louis, MO, USA), and removed from flasks after 15 min of incubation on ice. Cell debris was removed by centrifugation (10 000 g, 10 min, 4°C), and different aliquots of supernatants were either subjected to protein determination or boiled with SDS sample buffer. C‐Src‐knockout (c‐Src−) cells (SYF‐2459) and corresponding wildtype cells (c‐Src+ and SYF‐2498) are immortalized fibroblast cell lines obtained from mouse embryos. Cells were purchased from ATCC, and kept in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% FCS and 1% penicillin/streptomycin at 37°C with 5% CO2. Before addition of AZM475271 (10 μM) or PP2 (10 μM), the medium was changed to a FCS‐free medium. For western blot analysis, cells were lyzed as described for WPMY‐1 cells.
Phosphorylation assessments
For analysis of SFK phosphorylation, WPMY‐1 cells were grown in T75 flasks. After 48 h, AZM475271 (final concentration 10 μM), PP2 (10 μM) or solvent (DMSO) was added. After 2 h, cells were lyzed with RIPA buffer, and removed from flasks after a 15 min incubation on ice. Cell debris was removed by centrifugation (10 000 g, 10 min, 4°C), and supernatants were subjected to western blot analysis for phospho‐Src, total Src and β‐actin. For experiments with tissues, samples were prepared as small strips (2–3 mm × 1 mm) and allocated to dishes of a six‐well plate containing Custodiol® solution (Köhler, Bensheim, Germany). Custodiol is an organ‐protective solution, intended for use during organ transplantation. During experiments, plates were kept at 37°C under continuous shaking. AZM475271 (final concentration 10 μM), PP2 (10 μM) or solvent (DMSO) were added simultaneouly to separate wells, and samples were shock frozen after 60 min. Samples were stored at −80°C until western blot analysis was performed. On each western blot, samples from the same patient or the same experiment were compared (inhibitors vs. controls). Intensities of resulting bands were quantified densitometrically using ‘Image J’ (NIH, Bethesda, Maryland, USA). For semiquantitative calculation, samples without inhibitors (controls) were set to 100%, and samples with inhibitors were expressed as % of controls. This normalization is inevitable due to the large variations of the control values obtained from densitometric quantification (providing ‘arbitrary units’). These variations result not only mostly from variations of SFK levels between prostates from different patients (compare Figure 1B) but also from detection (slight differences in incubation periods, exposure times), and from different adjustments during scanning and digitization of blots (required due to varying background or due to different SFK content). These variations of the control values cannot practically be avoided; therefore, normalization of controls was required, although this excludes any comparisons by statistical tests.
Figure 1.
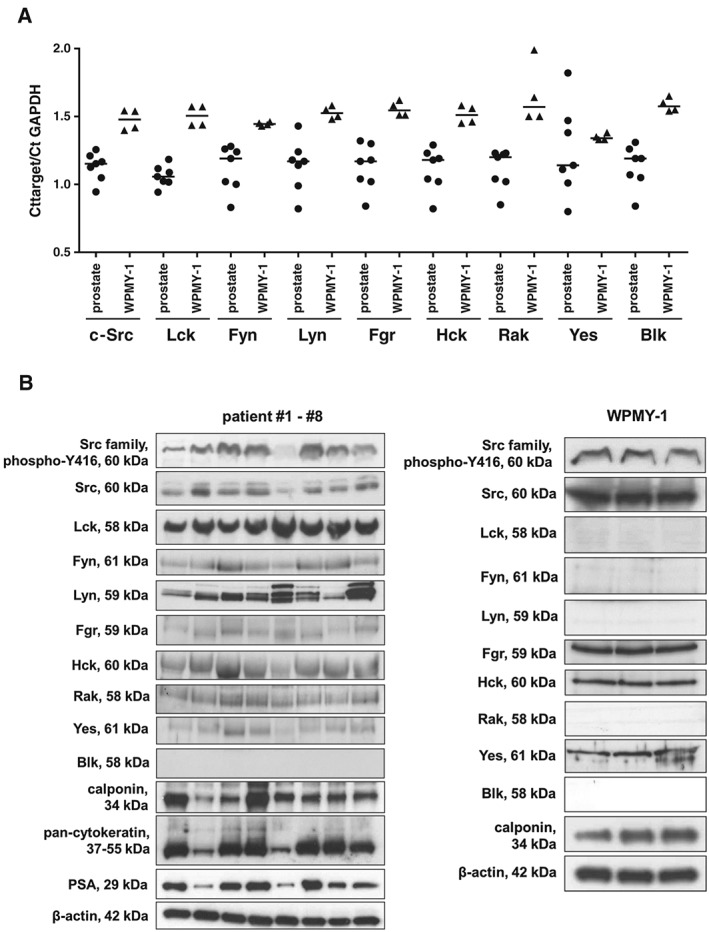
Detection of SFKs, Y416‐phosphorylated SFKs, markers (calponin for smooth muscle, pan‐cytokeratin for glandular epithelial cells and PSA for degree of hyperplasia), α1A‐adrenoceptors, and controls (β‐actin and GAPDH) at mRNA and protein level by RT‐PCR (A) and western blot (B). Phosphorylation at Y416 (c‐Src) or equivalent sites was assessed using a site‐ and phospho‐specific antibody reacting with different SFK members, including c‐Src and Lck. Varying content of PSA in tissues reflects different degree of BPH in patients undergoing radical prostatectomy (B). Expression of calponin and α1A‐adrenoceptors (WPMY‐1 cells) are important features of prostate smooth muscle cells (B). Indicated molecular weights reflect the expected sizes of proteins. Blots without bands cover regions from 50–70 kDa. Shown are all data from tissues from n = 7 (RT‐PCR) or n = 8 (western blot) patients, and from samples of WPMY‐1 cells from n = 4 (RT‐PCR) or n = 3 (western blot) independent experiments with WPMY‐1 cells (A, B).
Phalloidin staining
For fluorescence staining with phalloidin, cells were grown on Lab‐Tek Chamber slides (Thermo Fisher, Waltham, MA, USA) with inhibitors, or solvent. Staining was performed using 100 μM FITC‐labelled phalloidin (Sigma‐Aldrich, Munich, Germany), according to the manufacturer's instructions. Labelled cells were analysed using a laser scanning microscope (Leica SP2, Wetzlar, Germany).
Cytotoxicity assay
Cytotoxicity of Src inhibitors was assessed using the cell counting kit 8 (CCK‐8) (Sigma‐Aldrich, St. Louis, MO, USA). Cells were grown in 96‐well plates (20 000 cells per well) for 24 h, before AZM475271, PP2 or solvent were added in indicated concentrations (10, 50 and 100 μM). Subsequently, cells were grown for different periods (24, 48 and 72 h). Separate controls were performed for each period. At the end of this period, 10 μL of [2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H–tetrazolium monosodium salt (WST‐8) from CCK‐8 was added, and absorbance in each well was measured at 450 nm after incubation for 2 h at 37°C.
Cell proliferation assay
WPMY‐1, SYF‐2459 and SYF‐2498 cells were plated with cell line‐specific densities (50 000 cells per well for WPMY‐1; 30 000 cells per well for SYF‐2459 and SYF‐2498) on 16‐well chambered coverslips (Thermo Scientific, Waltham, MA, USA). After 24 h, cells were treated with AZM475271 (10 μM), PP2 (10 μM) or solvent. After a further 24, 48 or 72 h, the medium was changed to a 10 mM 5‐ethynyl‐2′‐deoxyuridine (EdU) solution in FCS‐free medium containing Src inhibitors or solvent. Twenty hours later, cells were fixed with 3.7% formaldehyde. EdU incorporation was determined using the ‘EdU‐Click 555’ cell proliferation assay (Baseclick, Tutzing, Germany) according to the manufacturer's instructions. In this assay, incorporation of EdU into DNA is assessed by detection with fluorescing 5‐carboxytetramethylrhodamine. Counterstaining of all nuclei was performed with DAPI. Cells were analysed by fluorescence microscopy (excitation: 546 nm; emission: 479 nm).
Drugs and nomenclature
AZM475271 and PP2 are structurally unrelated inhibitors of SFKs. 3,4‐Dihydro‐6‐[[4‐[[[3‐(methylsulfonyl)phenyl]methyl]amino]‐5‐(trifluoromethyl)‐2‐pyrimidinyl]amino]‐2(1H)‐quinolinone (PF573228) is an inhibitor of FAK. Stock solutions (10 mM) were prepared in DMSO, and kept at −20°C until use. Phenylephrine ((R)‐3‐[−1‐hydroxy‐2‐(methylamino)ethyl]phenol) is a selective agonist of α1‐adrenoceptors. Aqueous stock solutions of phenylephrine and noradrenaline (10 mM) were freshly prepared before each experiment. AZM475271, PP2 and PF573228 were obtained from Tocris (Bristol, UK); phenylephrine and noradrenaline were obtained from Sigma (Munich, Germany).
Data and analysis
Data are presented as means ± SEM with the indicated number (n) of experiments. Emax and pEC50 values were calculated by curve fitting of single experiments using GraphPad Prism 6 (Statcon, Witzenhausen, Germany. Student's two‐tailed t‐test, or one‐way anova and multivariate anova were used for paired or unpaired observations. Two‐way anova was used for comparison of EFS‐induced contractions. P values <0.05 were considered statistically significant. Data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
c‐Src and Lck in prostate tissues and WPMY‐1 cells
Using RT‐PCR and western blot analysis, we detected the expression of most SFK members in prostate tissues, taken from the periurethral zone from patients undergoing radical prostatectomy (Figure 1A,B). Almost all SFKs, including c‐Src, Lck, Fyn, Lyn, Fgr, Hck, Rak and Yes, were detectable at mRNA or protein level in prostate homogenates (Figure 1A,B). Only Blk was detectable by RT‐PCR, but not in Western blot analysis (Figure 1A,B). Detection of PSA confirmed the presence of hyperplasia in these samples (Figure 1B).
By co‐labelling of prostate sections with the smooth muscle marker calponin and the epithelial marker pan‐cytokeratin in fluorescence stainings, we observed c‐Src immunoreactivity in stromal smooth muscle cells and in glandular epithelial cells (Figure 2). Immunoreactivity for Lck was limited to non‐smooth muscle cells in sections with prostate architecture composed of glands and stroma, while it appeared in smooth muscle cells of areas showing extended stromal content (Figure 2). Immunoreactivity for Fyn was strong in smooth muscle cells, but weaker in glandular epithelial cells (Figure 3). Immunoreactivity for Lyn was moderate in smooth muscle cells, but lacking in glandular epithelial cells (Figure 3). Immunoreactivity for Fyn and Lyn, but not Lck, colocalized with the marker for catecholaminerigc nerves, TH.
Figure 2.
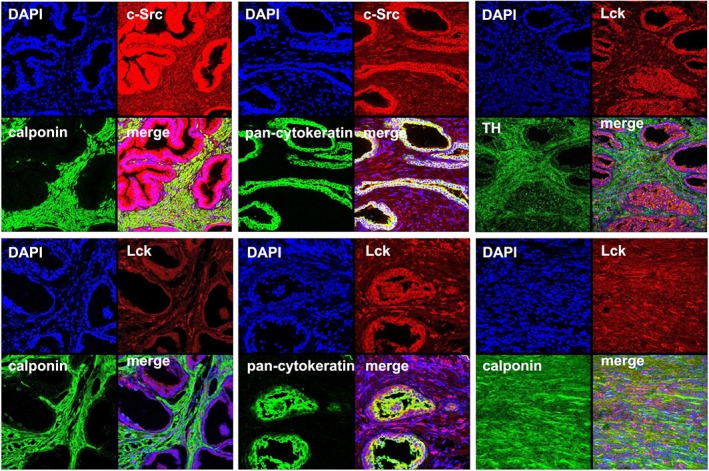
Fluorescence stainings of human prostate tissues for c‐Src, Lck and markers. Sections of human prostate tissues were double labelled with antibodies for c‐Src or Lck, together with antibodies for calponin, TH or pan‐cytokeratin. Besides typical prostate architecture composed of stroma and glands, tissues from the periurethral zone may show extended stromal areas without glands, but showing immunoreactivity for the smooth muscle marker calponin. Shown are representative stainings from a series with tissues from n = 5 patients for each combination.
Figure 3.
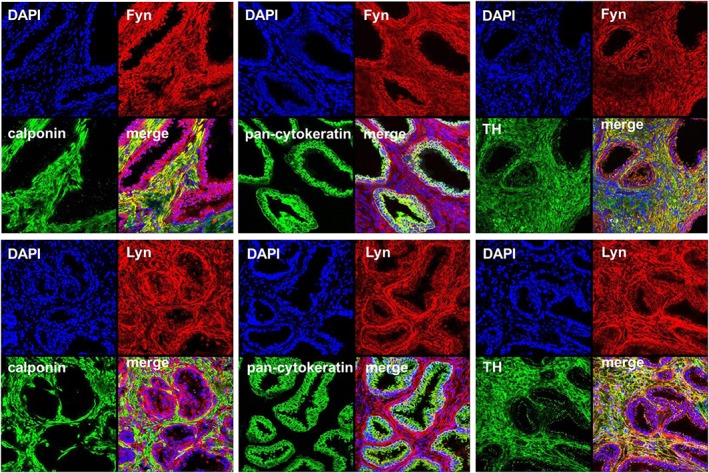
Fluorescence stainings of human prostate tissues for Fyn, Lyn and markers. Sections of human prostate tissues were double‐labelled with antibodies for Fyn or Lyn, together with antibodies for calponin, TH or pan‐cytokeratin. Shown are representative stainings from a series with tissues from n = 5 patients for each combination.
In WPMY‐1 cells, an immortalized cell line from the stroma of human prostate without prostate cancer, we detected SFKs and calponin by western blot analysis (Figure 1B). C‐Src, Fgr, Hck and Yes were detectable by RT‐PCR and western blots, whereas remaining SFKs were only detectable at mRNA level (Figure 1A,B).
SFK activation requires autophosphorylation, including tyrosine 416 (Y416) of c‐Src, or equivalent sites of different SFK members (Roskoski, 2005). Using a phospho‐ and site‐specific antibody reacting with different SFK members, SFKs phosphorylated at Y416 or corresponding positions was detectable in prostate tissue from patients, and in WPMY‐1 cells (Figure 4). In separate experiments, we applied AZM475271 and PP2, which are small molecule inhibitors with high SFK specificity, to prostate tissues and WPMY‐1 cells. Both inhibitors (10 μM each) reduced Y416 phosphorylation by about 50%, reflecting SFK inhibition by AZM475271 and PP2 (Figure 4). Because c‐Src may work in concert with focal adhesion kinase (FAK), which is also activated by phosphorylation (Kunit et al., 2014), we examined FAK phosphorylation in prostate tissues and WPMY‐1 cells incubated with or without SFK inhibitors. Neither AZM475271, nor PP2 affected FAK phosphorylation in tissues or WPMY‐1 cells (Figure 4).
Figure 4.
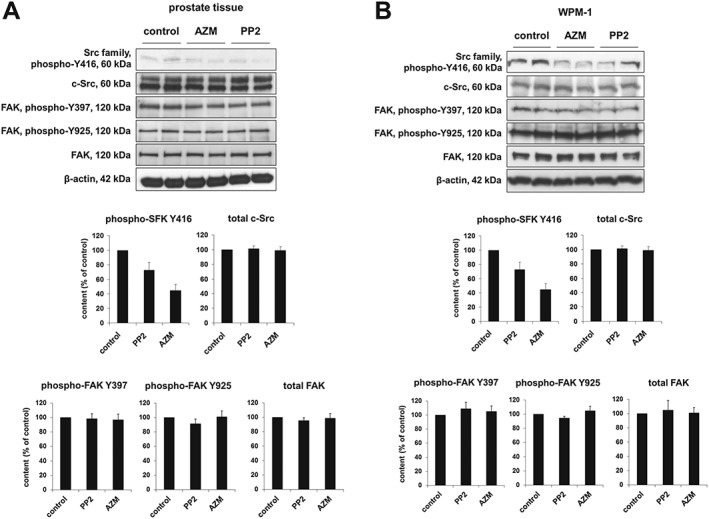
Effects of AZM475271 and PP2 on SFK and FAK phosphorylation. Detection of Y416‐phosphorylated SFKs, total c‐Src, Y397‐ and Y925‐phosphorylated FAK, total FAK and β‐actin was performed by western blot using site‐ and phospho‐specific antibodies. Content of phosphorylated and total proteins was semiquantitatively compared between tissues or cells incubated with DMSO (control) or inhibitors. Application of AZM475271 or PP2 (10 μM each) reduced SFK phosphorylation at tyrosine 416 in prostate tissues and WPMY‐1 cells (A). In contrast, AZM475271 or PP2 (10 μM each) were without effect on FAK phosphorylation at both positions (Y397 and Y925) in prostate tissues (B). Shown are representative western blots and quantification of all experiments (series with n = 6 independent experiments and different prostates, respectively, or with n = 6 independent experiments with WPMY‐1 cells) (means ± SEM).
Inhibition of smooth muscle contraction by SFK inhibitors
In organ bath studies, we assessed the effects of AZM475271 (10 μM) and PP2 (10 μM) on contraction of human prostate tissues (Figure 5). Smooth muscle contractions of prostate preparations were induced concentration‐ or frequency‐dependently by noradrenaline (0.1–100 μM), by the α1‐adrenoceptor agonist phenylephrine (0.1–100 μM), or by EFS (2–32 hz). EFS causes neuronal action potentials, leading to contraction by release of endogenous neurotransmitters. Contractions induced by noradrenaline, phenylephrine and EFS were significantly inhibited by each of both inhibitors at different concentrations of agonists and different frequencies during EFS (Figure 5). This inhibition was confirmed after calculation of Emax values by curve fitting, which was performed for agonist‐induced contractions (Table 2). Both inhibitors significantly reduced Emax values for noradrenaline and phenylephrine (Table 2). In contrast, neither AZM475271 nor PP2 changed pEC50 values for noradrenaline‐ or phenylephrine‐induced contractions of prostate strips (Table 2). A two‐way anova was conducted to compare effects of inhibitors with those of DMSO on contractions, indicating significant inhibition of noradrenaline‐, phenylephrine, and EFS‐induced contractions by AZM475271 and PP2. Multivariate analysis showed that noradrenaline‐induced contractions were significantly inhibited at 3, 10, 30 and 100 μM by AZM475271, and at 10, 30 and 100 μM by PP2. Similarly, phenylephrine‐induced contractions were significantly inhibited at 10, 30 and 100 μM by AZM475271, and at 10, 30 and 100 μM by PP2. EFS‐induced contractions were significantly inhibited by both inhibitors at 16 and 32 Hz.
Figure 5.
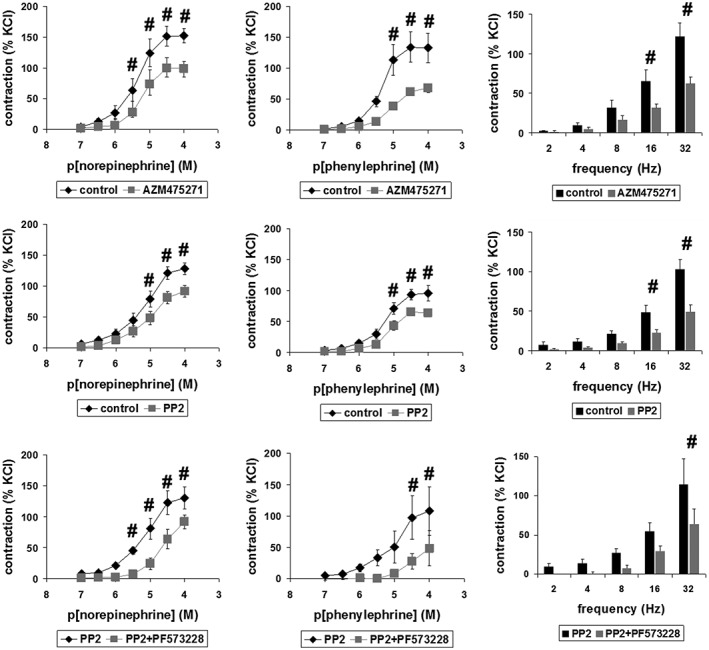
Effects of AZM475271 and PP2 on contraction of human prostate strips. In an organ bath, contraction in response to noradrenaline, phenylephrine or EFS was assessed after pre‐incubation with AZM475271 (10 μM), PP2 (10 μM) or solvent (DMSO) (upper and middle panels). In separate sets of experiments, the combination of PP2 with the FAK inhibitor PF573228 (100 μM) was compared with effects of PP2 alone (lower panels). Tensions after application of contractile stimuli have been expressed as % of contraction by high molar KCl, which was assessed before application of inhibitors or solvents. This reference allows comparisons despite different conditions of tissues or patients, for example due to varying degrees of BPH, different content of smooth muscle or any other heterogeneity. Shown are means ± SEM from experiments with tissues from n = 7 (AZM475271/noradrenaline), n = 10 (AZM475271/phenylephrine), n = 9 (AZM475271/EFS), n = 8 (PP2/noradrenaline), n = 6 (PP2/phenylephrine), n = 7 (PP2/EFS), n = 6 (PP2 + PF573228/noradrenaline), n = 6 (PP2 + PF573228/phenylephrine) or n = 5 (PP2 + PF573228/EFS) patients (# P < 0.05 for DMSO vs. inhibitor in multivariate analysis), with tissues from each patient being allocated to both groups in each diagram.
Table 2.
pEC50 and Emax values for noradrenaline and phenylephrine in the presence of AZM475271 (10 μM), PP2 (10 μM), PF573228 (100 μM) or solvent
| pEC50 (M) | Emax (% KCl) | |||
|---|---|---|---|---|
| noradrenaline | phenylephrine | noradrenaline | phenylephrine | |
| DMSO | 5.285 ± 0.170 | 5.291 ± 0.109 | 174 ± 9.9 | 155 ± 28.4 |
| AZM475271 | 5.014 ± 0.213 | 4.974 ± 0.114 | 129 ± 11.1* | 83 ± 8.9* |
| DMSO | 5.16 ± 0.185 | 5.201 ± 0.085 | 149 ± 10.5 | 108 ± 11.6 |
| PP2 | 5.028 ± 0.161 | 5.073 ± 0.122 | 108 ± 12.2* | 77 ± 7.3* |
| PP2 | 5.048 ± 0.120 | 4.746 ± 0.111 | 147 ± 18.6 | 127 ± 40.7 |
| PP2 + PF573228 | 4.252 ± 0.196** | 4.469 ± 0.199 | 163 ± 13.2 | 99 ± 70.8 |
Values were obtained by curve fitting of each single experiment. Values in the table represent means ± SEM from all experiments (* P < 0.05 vs. corresponding DMSO control, ** P < 0.05 vs. corresponding PP2 series). Number of experiments for each series is identical to those in Figure 5.
Because c‐Src may act cooperatively with focal adhesion kinase (FAK) (Kunit et al., 2014), we examined effects of combinations containing SFK and FAK inhibitors. Agonist‐ and EFS‐induced tensions of prostate strips were significantly lower after combined application of SFK and the FAK inhibitor PF573228 than tensions observed after SFK inhibitors alone (Figure 5). Two‐way anova indicated that addition of PF573228 significantly enhanced PP2‐induced inhibition of contractions evoked by noradrenaline, phenylephrine or EFS. Multivariate analysis showed that noradrenaline‐induced tensions were significantly lower after addition of the combination of PP2 with PF573228 compared with PP2 alone after 3, 10, 30 and 100 μM. Similarly, phenylephrine‐induced tensions were significantly lower after addition of the combination of PP2 with PF573228 compared with PP2 alone after 30 and 100 μM. EFS‐induced contractions were significantly lower after addition of the combination of PP2 with PF573228 compared with PP2 alone at 32 Hz. We concluded that SFKs act (at least partially) independently from FAK to promote prostate smooth muscle tone. This may be supported by Emax values, which was lower for phenylephrine after addition of the combination, or by pEC50 values, being lower for both agonists after addition of the combination (Table 2).
Disorganization of actin by SFK inhibitors in WPMY‐1 cells
By phalloidin staining, we visualized effects of AZM475271 (10 μM) and PP2 (10 μM) on actin filaments in WPMY‐1 cells (Figure 6). In solvent‐treated control cells, actin filaments were arranged to bundles, forming long and thin protrusions, with elongations from adjacent cells overlapping each other (Figure 6). AZM475271 or PP2 (24 h) caused complete breakdown of this filament organization, including shortening of filaments and regressing organization to bundles, resulting in a rounded cell shape without any protrusions (Figure 6).
Figure 6.
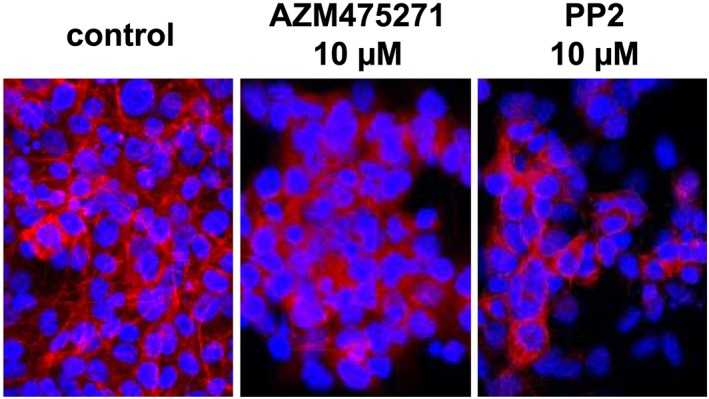
Effects of AZM475271 and PP2 on actin organization in WPMY‐1 cells. Actin filaments were visualized by staining with FITC‐coupled phalloidin, after incubation of WPMY‐1 cells with solvent, AZM475271 (10 μM) or PP2 (10 μM) for 24 h. Shown are representative images from a series with five independent experiments, with similar results.
Cytotoxicity of SFK inhibitors in WMPY‐1 cells
Survival of WPMY‐1 cells was assessed by CCK‐8 assay, and turned out to be concentration‐ and time‐dependent after application of AZM475271 or PP2 (Figure 7). Twenty‐four hours after incubation with 10 μM of AZM475271 or PP2, survival was still 88 ± 4.5% or 82 ± 3.3% respectively (P < 0.05 for control vs. PP2) (Figure 7). The same concentration reduced survival to 34 ± 3.2% or 28 ± 2.3% after 72 h of incubation (P < 0.05 for control vs. AZM475271 or PP2), while 100 μM of AZM475271 or PP2 reduced survival to 17 ± 1.2% or 49 ± 3.6% already after 24 h (P < 0.05 for control vs. AZM475271 or PP2) (Figure 7).
Figure 7.
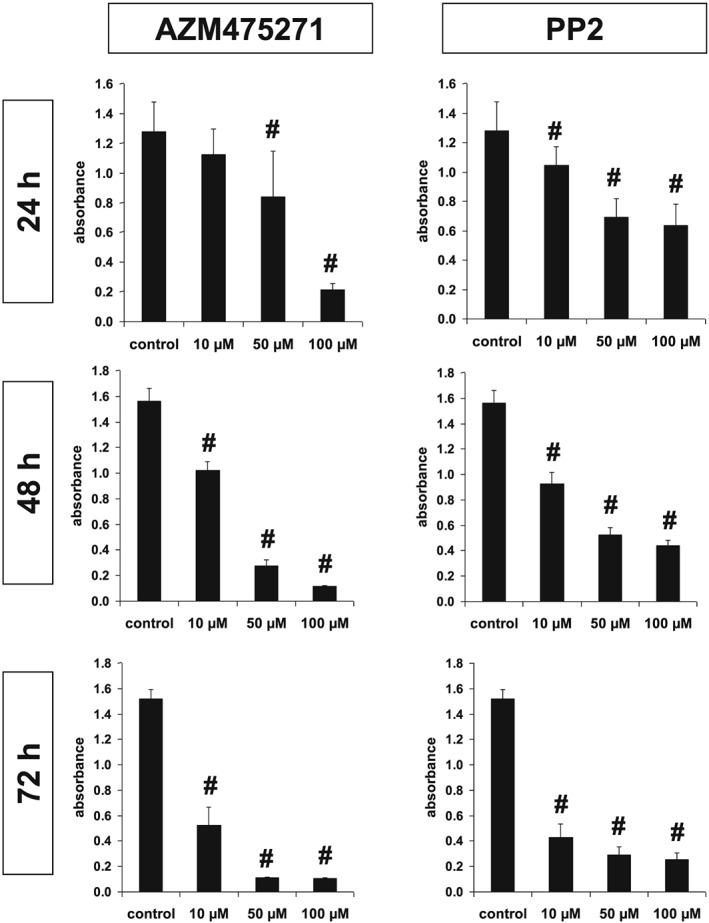
Cytotoxicity of AZM475271 and PP2 in WPMY‐1 cells. Survival of WPMY‐1 cells was assessed using the CCK‐8 assay, after incubation with solvent, AZM475271 (10–100 μM) or PP2 (10–100 μM) for 24–72 h. Shown are means ± SEM from series with nine independent experiments for each setting (# P < 0.05 vs. control).
Inhibition of proliferation by SFK inhibitors in WMPY‐1 cells
Parallel to reduced survival, AZM475271 (10 μM) and PP2 (10 μM) reduced the relative proliferation rate in the remaining populations of WPMY‐1 cells. The number of proliferating cells was assessed by an EdU assay, and referred to the total number of cells being visualized with DAPI (Figure 8). Compared with solvent‐treated cells, the relative number of proliferating cells was significantly lower after treatment with 10 μM of AZM475271 or PP2 (Figure 8). This decline of proliferation rate in response to AZM475271 or PP2 was progressive over time (24–72 h; P < 0.05 for 24 vs. 48 h, and 48 vs. 72 h), while it stayed constant in solvent‐treated controls (Figure 8).
Figure 8.
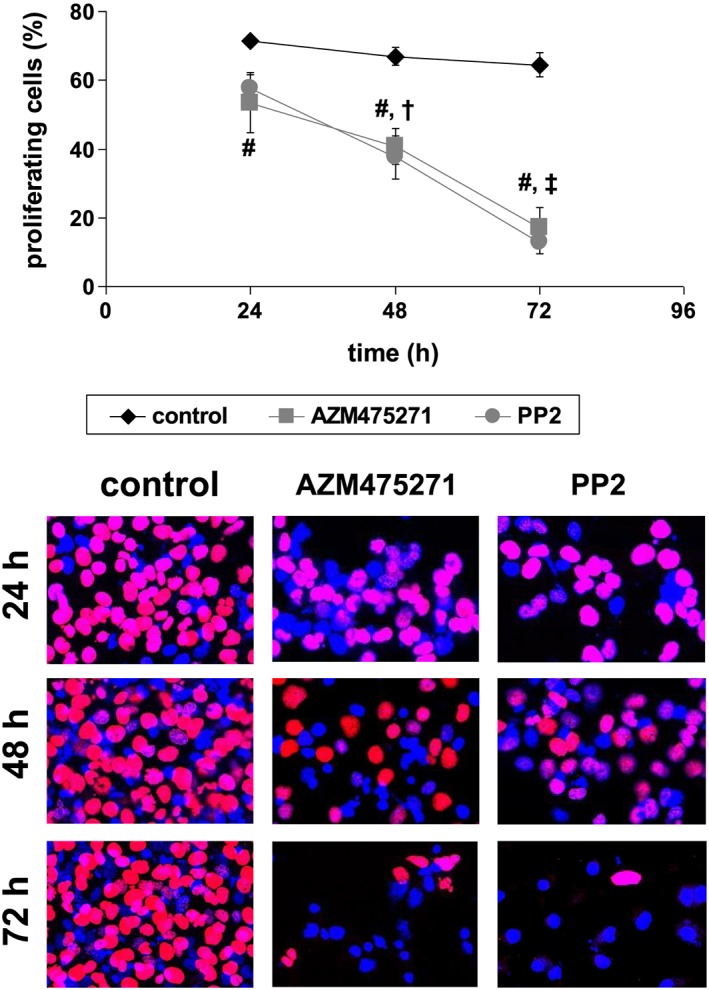
Effects of AZM475271 and PP2 on proliferation of WPMY‐1 cells. Proliferation rate was assessed by EdU assay after incubation with solvent, AZM475271 (10 μM) or PP2 (10 μM) for 24–72 h. The number of cells showing proliferation (red nuclei) was referred to the total number of cells (red + blue nuclei), to correct for reduced number of cells after longer incubation periods (compare with Figure 5). Shown are representative images and means ± SEM, from series with nine independent experiments for each setting (# P < 0.05 for AZM475271 and PP2 vs. corresponding control, † P < 0.05 vs. 24 h for AZM475271 and PP2, ‡ P < 0.05 vs. 48 h for AZM475271 and PP2).
Effects of SFK inhibitors in SYF‐2459 and SYF‐2498 cells
Western blot analysis confirmed the knockout of c‐Src in SYF‐2459 cells: Bands with the expected size for c‐Src were detectable by western blot analysis in c‐Src+ SYF‐2498 cells, while no bands were detectable in c‐Src− SYF‐2459 cells (Figure 9A). Hck, Lyn and Fgr were detectable in both cell lines as well, with levels of Hck being notably higher in c‐Src− SYF‐2459 cells compared with SYF‐2498 cells (Figure 9A). Remaining SFKs were undetectable in each of both cell lines (Figure 9A).
Figure 9.
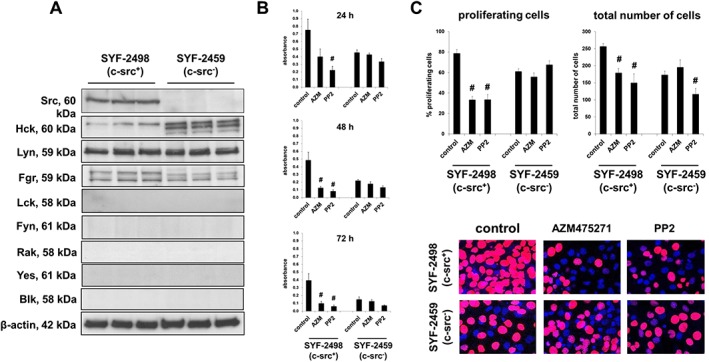
Effects of AZM475271 and PP2 on SYF‐2498 and SYF‐2459 cells. Knockout of c‐Src in SYF‐2459 cells was confirmed by comparison of c‐Src content in wildtype cells (SYF‐2498) by western blot analyses (A). Survival was assessed using the CCK‐8 assay, after incubation of SYF‐2498 and SYF‐2459 cells with solvent (control), AZM475271 (10 μM) or PP2 (10 μM) for 24–72 h (means ±SEM from series with five independent experiments for each setting; # P < 0.05 vs. control) (B). Proliferation rate was assessed by EdU assay after incubation of SYF‐2498 and SYF‐2459 cells with solvent, AZM475271 (10 μM) or PP2 (10 μM) for 24 h. The number of cells showing proliferation (red nuclei) was referred to the total number of cells (red + blue nuclei), to correct for reduced number of cells after longer incubation periods, and for reduced number of SYF‐2459 cells. Shown are representative images and means ± SEM, from a series with five to seven independent experiments for each setting (# P < 0.05 for AZM475271 and PP2 vs. corresponding control) (C).
Effects of AZM475271 and PP2 (10 μM) on survival of SYF‐2459 and SYF‐2498 cells was assessed by CCK‐8 assay (Figure 9B). Results were clearly divergent in both cell lines (Figure 9B). In the wildtype cell line, SYF‐2498, both inhibitors induced marked decreases of survival: survival was reduced to 28 ± 3.8% after 48 h or to 25 ± 4.6% after 72 h by 10 μM AZM475271 (P < 0.05 for control vs. AZM475271), and to 16 ± 2% after 48 h or to 15 ± 3.1% after 72 h by 10 μM PP2 (P < 0.05 for control vs. PP2) (Figure 9B). In contrast, decreases in survival were lacking or less pronounced in c‐Src− SYF‐2459 cells: 48 h or 72 h after AZM475271, survival was still 84 ± 16.1% or 96 ± 17.2% of controls, and 59 ± 8.6% or 55 ± 10.4% of controls 48 h or 72 h after addition of PP2 (Figure 9B).
Effects of AZM475271 and PP2 (10 μM, 24 h) on proliferation of SYF‐2459 and SYF‐2498 cells was assessed by EdU assay (Figure 9C). Results were clearly divergent in both cell lines (Figure 9C): while the wildtype cell line (SYF‐2498) reacted to AZM475271 and PP2 with significant decreases in the proliferation rate, no response in proliferation was noted in c‐Src− SYF‐2459 cells (Figure 9C). Moreover, SYF‐2459 cells showed impaired growth, being reflected by a significantly reduced total number of cells despite in the control group (Figure 9C).
Discussion
In patients with BPH, smooth muscle contraction and prostate enlargement due to stromal growth may both contribute to urethral obstruction, impaired bladder outlet and consequent voiding symptoms (Oelke et al., 2013; Hennenberg et al., 2014). Despite this shared role for aetiology of BOO and voiding symptoms in countless patients, smooth muscle contraction and stromal growth in the prostate are still regarded separately in clinical management of LUTS. This is contrasted by efficacy of available options for medical treatment of voiding symptoms, which is still limited (Oelke et al., 2013; Hennenberg et al., 2014). Moreover, combination therapies are required to target smooth muscle contraction and growth in the prostate at once (Oelke et al., 2013; Hennenberg et al., 2014). Recent progress from basic research provided rising evidence for possible relationships between the control of smooth muscle tone and regulation of stromal growth in the human prostate (Wang et al., 2015). On the basis of our present findings, a connection between contraction and growth in the prostate may be assumed, being accomplished by SFKs. In fact, this study suggests a dual role of SFKs in smooth muscle of the hyperplastic human prostate. First, SFKs appear to regulate prostate smooth muscle tone by promoting contraction. Secondly, SFKs may be critical for prostate growth, by triggering proliferation in the stroma. We observed that SFK inhibitors were capable to inhibit contraction of human prostate tissues and growth of stromal cells.
Our findings were obtained using human prostate tissues, and cultured WPMY‐1 cells, which are established models for studying prostate contraction and stromal growth. Prostate tissues were taken from the periurethral zone from patients undergoing radical prostatectomy for prostate cancer. As prostate tumours are mostly localized to the peripheral zone, periurethral samples are widely tumour‐free (Pradidarcheep et al., 2011; Shaikhibrahim et al., 2012). Hyperplasia to different degree may be indicated by varying content of PSA, and is found at least in 80% of patients with prostate tumour (Levitt and Slawin, 2007; Alcaraz et al., 2009; Orsted and Bojesen, 2013). Consequently, samples in our study may represent hyperplastic tissues without tumours. WPMY‐1 cells are an immortalized cell line from human prostatic stroma, which were initially characterized as myofibroblasts (Webber et al., 1999). Myofibroblasts may resemble smooth muscle cells, as they show the ability to contract, and share common markers with smooth muscle cells (Webber et al., 1999). In fact, smooth muscle cells can undergo phenotype transition to myofibroblasts, for example during cell culture (Webber et al., 1999). Our findings confirm that WPMY‐1 cells may closely resemble prostatic smooth muscle cells, as we observed the expression of the smooth muscle marker calponin. Nevertheless, it needs to be stressed that important features of native cells may get lost during culture and that such culture‐related alterations in phenotype may be regarded as possible limitations.
Numerous previous studies addressing SFKs in the prostate focussed on oncologic context (Cai et al., 2011; Montero et al., 2011; Varkaris et al., 2014; Vlaeminck‐Guillem et al., 2014). In prostate cancer cells, SFKs may promote migration, which depends on actin dynamics and cytoskelet organization (Slack et al., 2001; Sabbota et al., 2010). Only few studies considered SFKs in non‐oncologic prostate cells, including reports about c‐Src in normal prostate epithelial cells, or the role of Lyn for prostate development (Allard et al., 1997; Zhou et al., 2003; Pavone et al., 2011). Recently, we observed expression of c‐Src in hyperplastic prostate tissue (Kunit et al., 2014). This finding, together with reports describing a role of SFKs in prostate tumour cells, cell cycle, and for smooth muscle contraction outside the lower urinary tract prompted us to examine effects of SFK inhibitors on contraction of prostate smooth muscle and proliferation of stromal cells.
Findings were confirmed using two structurally unrelated small molecule inhibitors with presumably high SFK specificity. PP2 may be specific for some SFK members and the related c‐Src kinase up to the lower micromolar range, with highest specificity for c‐Src, Lck and Fyn, and has been commonly used for investigation of SFK functions (Hanke et al., 1996; Bain et al., 2003; Ischenko et al., 2007). AZM475271 has been developed because oral bioavailability and potency of previous SFK inhibitors were limited (Yezhelyev et al., 2004). It has been available for several years and inhibits c‐Src, Lck and c‐Yes, but has been used in less than a handful of published studies and was obviously never applied to study smooth muscle contraction before (Yezhelyev et al., 2004; Ischenko et al., 2007; Ischenko et al., 2010). Because c‐Src and Lck represent the intersection of the kinase spectrum being targeted by AZM475271 and PP2, we assume that they account for control of smooth muscle tone and cell growth observed in prostate tissue or WPMY‐1 cells. However, immunoreactivity for Lck was lacking in smooth muscle cells of prostate tissues with normal architecture, and Lck in WPMY‐1 cells was undetectable by Western blot analysis, so that we speculate that effects of AZM475271 and PP2 resulted from inhibition of c‐Src. Finally, a considerable degree of specificity of both inhibitors for c‐Src may be suggested by our findings from c‐Src knockout cells (SYF‐2459), which were compared with wildtype cells (SYF‐2498). While wildetype cells reacted with marked decreases in proliferation rate and survival to AZM475271 and PP2, c‐Src‐deficient cells did not react any more to any of both inhibitors, although they may contain other SFKs than c‐Src. This may provide further clues that unspecific inhibition from AZM475271 or PP2 may be limited. Notably, inhibitor concentrations in these experiments were the same as those in the organ bath (10 μM). Finally, the lacking effects of AZM475271 and PP2 in c‐Src knockout cells supports the idea that c‐Src is critical for proliferation and cell cycle.
We observed that both inhibitors, AZM475271 and PP2, inhibited the contraction of prostate strips. To the best of our knowledge, this study is the first showing that SFK inhibitors effectively reduce contraction in any smooth muscle preparations of human origin, or in the prostate of any species. Previous studies addressed effects on smooth muscle contraction in the cardiovascular system, airways, gastrointestinal tract or uterus of rodents and pigs (Che and Carmines, 2005; Weigand et al., 2006; Oguma et al., 2007; Ross et al., 2007; Lu et al., 2008; Phillippe et al., 2009; El‐Daly et al., 2014). In prostate tissues and in WMPY‐1 cells, AZM475271 and PP2 reduced SFK phosphorylation by about 50%, what may reflect incomplete inhibition of c‐Src activity. Limited degree of inhibition and lacking knowledge on how much activity is inhibited may be regarded as a possible limitation, but might explain why we observed inhibition of contractions by around 50%. On the other hand, reduction of phosphorylation may appear incomplete, if residual phosphorylation is left despite full kinase inhibition; kinase inhibition may reduce the phosphorylation rate, but not the dephosphorylation rate. Src may cooperate with FAK in some instances (Kunit et al., 2014). Our results from combined application of AZM475271 or PP2 together with an FAK inhibitor indicate that SFK functions in prostate smooth muscle cells occur at least partially independently of FAK. FAK inhibitors alone were described to reduce prostate contraction (Kunit et al., 2014). If FAK and SFKs cooperated to promote contraction, no additive effects FAK and SFK inhibitors (as it may be suggested by our findings) should be observed. The conclusion that both kinases do not cooperate during prostate smooth muscle contraction is additionally supported by our observation that neither AZM475271 nor PP2 affected phosphorylation of FAK at two different positions. Definitely, our data do not prove a cooperation of SFKs with FAK in the human prostate. However, this conclusion may be limited to such SFKs, which are sensitive to AZM475271 and PP2. Other SFKs, being less or not sensitive to these inhibitors may indeed interact with FAK in prostate. On the other hand, it may not be excluded, that a lacking effect of AZM475271 and PP2 on FAK phosphorylation was attributed to the degree of SFK inhibition, which was incomplete and ranged around 50%. Finally, relationships between SFKs and FAK may differ between different organs, as our observations in the prostate may diverge from reports using vascular smooth muscle (Min et al., 2012).
Because cytotoxicity of 10 μM AZM475271 or PP2 in WPMY‐1 cells was minor within the first 24 h, we exclude that inhibition of contraction of prostate strips in the organ bath was due to immediate cell death. Rather, reduced contractility may be explained by cytoskeletal deorganization in response to AZM475271 and PP2, which was observed by phalloidin staining of actin filaments in WPMY‐1 cells. It appears likely that similar disorganization of actin filaments occurred in prostate tissues when AZM475271 or PP2 were applied in the organ bath. In fact, polymerization of actin, and correct assembly of actin filaments are mandatory for smooth muscle contraction (Hennenberg et al., 2014).
Contraction and proliferation have in common that they both depend on correct organization of the cytoskeleton. Segregation of chromosomes during mitosis requires assembly and correct placement of the spindle apparatus, which is mainly composed of cytoskeletal components (Wittmann et al., 2001). Development of cleavage furrows and division during cytokinesis are tightly regulated by actin filaments (Glotzer, 2005). Thus, it seems comprehensible that disorganization of actin filaments by SFK inhibitors in WPMY‐1 cells was paralleled by reduced proliferation. A similar role of SFKs for proliferation was previously suggested for non‐prostatic smooth muscle cells, including airway and vascular smooth muscle cells (Krymskaya et al., 2005; Yang et al., 2005; Hsieh et al., 2009; Li et al., 2010; Ishigaki et al., 2011).
Apart from any translational value, our findings strongly suggest that control of smooth muscle contractility and stromal growth in the human prostate may be tightly connected with each other. This link may include SFKs, which may affect contractility and growth concurrently by regulation of actin dynamics. Notably, other compounds may affect contraction and growth at once in the prostate as well, including inhibitors for Rho kinase, Rac GTPases or p21‐activated kinases, what may additionally point to this link (Rees et al., 2003; Wang et al., 2015; Wang et al., 2016). Together, the idea of such a connection may appear attractive, as both processes, that is contraction and growth, are important targets for medical therapy of BOO (Oelke et al., 2013). At present, separate medications are required to hit smooth muscle tone and enlargement of the prostate for treatment of voiding symptoms, or to reduce occurrence of acute urinary retention and need for surgery in millions of patients with BPH (Oelke et al., 2013). Our present study demonstrates that it is principally possible to address contraction and growth in the hyperplastic prostate at once using single compounds. Whether or not SFK inhibitors may be effective in reducing LUTS may only be found out in vivo. However, it should be kept in mind, that prostate tone in vivo depends not only on adrenergic contraction but also on non‐adrenergic mediators (e.g. thromboxane A2 and endothelins), which were not regarded in the present study (Hennenberg et al., 2014). Finally, the concept that SFKs synchronize and promote critical functions (such as growth and contraction) in BPH may be reinforced by findings in other organs, where SFKs are important players in cellular hypertrophy or hyperplastic processes (Walcher et al., 2006; Li et al., 2007; Liu et al., 2011; Cordero et al., 2014; Ho et al., 2015).
Conclusions
Our findings suggest that the control of smooth muscle contraction and growth are linked with each other in the human prostate. Such a connection may be provided by SFKs. SFK inhibitors were found to inhibit prostate smooth muscle contraction and the growth of prostate cells. Hence, targeting contraction and growth in the prostate simultaneously by a single compound is, in principal, possible.
Author contributions
M.H. created the research design. Y.W., C.G., A.T., B.R., A.C., F.S., A.H., S.J., R.W., C.L., C.G.S. and MH were involved in acquisition of data. Y.W., A.T., B.R., A.C., A.H., S.J. and M.H. analysed data. Y.W., C.G., C.L., C.G.S. and M.H. performed interpretation of data. Y.W. and M.H. drafted the paper. A.C., C.G., A.T., B.R., A.C., F.S., R.W., C.L. and C.G.S. critically revised the manuscript. All authors approved the submitted and final version. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Prof. Dr. E. Noessner and her coworkers for support with immunofluorescence microscopy. We thank Prof. Dr. T. Kirchner (Institute of Pathology, Ludwig‐Maximilians University, Munich) and his coworkers Dr. V. Mai and Dr. C. Faber for asservation of tissue samples from prostates. This study was supported by grants from the ‘Deutsche Forschungsgemeinschaft’ (grants HE 5825/2‐1 and GR 3333/2‐1). Dr. Y. Wang received a scholarship from the ‘Chinese Scholarship Council’ (CSC) and the ‘Deutscher Akademischer Austauschdienst’ (DAAD) (ST‐34, no. 91601899).
Wang, Y. , Gratzke, C. , Tamalunas, A. , Rutz, B. , Ciotkowska, A. , Strittmatter, F. , Herlemann, A. , Janich, S. , Waidelich, R. , Liu, C. , Stief, C. G. , and Hennenberg, M. (2016) Smooth muscle contraction and growth of stromal cells in the human prostate are both inhibited by the Src family kinase inhibitors, AZM475271 and PP2. British Journal of Pharmacology, 173: 3342–3358. doi: 10.1111/bph.13623.
References
- Alcaraz A, Hammerer P, Tubaro A, Schroder FH, Castro R (2009). Is there evidence of a relationship between benign prostatic hyperplasia and prostate cancer? Findings of a literature review. Eur Urol 55: 864–873. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard P, Atfi A, Landry F, Chapdelaine A, Chevalier S (1997). Estradiol activates p60src, p53/56lyn and renatured p50/55 protein tyrosine kinases in the dog prostate. Mol Cell Endocrinol 126: 25–34. [DOI] [PubMed] [Google Scholar]
- Amata I, Maffei M, Pons M (2014). Phosphorylation of unique domains of Src family kinases. Front Genet 5: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003). The specificities of protein kinase inhibitors: an update. Biochem J 371: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Smith DA, Memarzadeh S, Lowell CA, Cooper JA, Witte ON (2011). Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci USA 108: 6579–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che Q, Carmines PK (2005). Src family kinase involvement in rat preglomerular microvascular contractile and [Ca2+]i responses to ANG II. Am J Physiol Renal Physiol 288: F658–F664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cindolo L, Pirozzi L, Fanizza C, Romero M, Tubaro A, Autorino R et al. (2015). Drug adherence and clinical outcomes for patients under pharmacological therapy for lower urinary tract symptoms related to benign prostatic hyperplasia: population‐based cohort Study. Eur Urol 68: 418–425. [DOI] [PubMed] [Google Scholar]
- Cordero JB, Ridgway RA, Valeri N, Nixon C, Frame MC, Muller WJ et al. (2014). c‐Src drives intestinal regeneration and transformation. EMBO J 33: 1474–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Daly M, Saifeddine M, Mihara K, Ramachandran R, Triggle CR, Hollenberg MD (2014). Proteinase‐activated receptors 1 and 2 and the regulation of porcine coronary artery contractility: a role for distinct tyrosine kinase pathways. Br J Pharmacol 171: 2413–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullhase C, Chapple C, Cornu JN, De Nunzio C, Gratzke C, Kaplan SA et al. (2013). Systematic review of combination drug therapy for non‐neurogenic male lower urinary tract symptoms. Eur Urol 64: 228–243. [DOI] [PubMed] [Google Scholar]
- Glotzer M (2005). The molecular requirements for cytokinesis. Science 307: 1735–1739. [DOI] [PubMed] [Google Scholar]
- Gratzke C, Bachmann A, Descazeaud A, Drake MJ, Madersbacher S, Mamoulakis C et al. (2015a). EAU guidelines on the assessment of non‐neurogenic male lower urinary tract symptoms including benign prostatic obstruction. Eur Urol 67: 1099–1109. [DOI] [PubMed] [Google Scholar]
- Gratzke C, Bachmann A, Descazeaud A, Drake MJ, Madersbacher S, Mamoulakis C et al. (2015b). EAU guidelines on the assessment of non‐neurogenic male lower urinary tract symptoms including benign prostatic obstruction. Eur Urol 67: 1099–1109. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ et al. (1996). Discovery of a novel, potent, and Src family‐selective tyrosine kinase inhibitor. Study of Lck‐ and FynT‐dependent T cell activation. J Biol Chem 271: 695–701. [DOI] [PubMed] [Google Scholar]
- Hennenberg M, Stief CG, Gratzke C (2014). Prostatic alpha1‐adrenoceptors: new concepts of function, regulation, and intracellular signaling. NeurourolUrodyn 33: 1074–1085. [DOI] [PubMed] [Google Scholar]
- Ho DM, Pallavi SK, Artavanis‐Tsakonas S (2015). The notch‐mediated hyperplasia circuitry in drosophila reveals a Src‐JNK signaling axis. Elife 4: e05996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh HL, Tung WH, Wu CY, Wang HH, Lin CC, Wang TS et al. (2009). Thrombin induces EGF receptor expression and cell proliferation via a PKC(delta)/c‐Src‐dependent pathway in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 29: 1594–1601. [DOI] [PubMed] [Google Scholar]
- Irwin DE, Kopp ZS, Agatep B, Milsom I, Abrams P (2011). Worldwide prevalence estimates of lower urinary tract symptoms, overactive bladder, urinary incontinence and bladder outlet obstruction. BJU Int 108: 1132–1138. [DOI] [PubMed] [Google Scholar]
- Ischenko I, Guba M, Yezhelyev M, Papyan A, Schmid G, Green T et al. (2007). Effect of Src kinase inhibition on metastasis and tumor angiogenesis in human pancreatic cancer. Angiogenesis 10: 167–182. [DOI] [PubMed] [Google Scholar]
- Ischenko I, Seeliger H, Camaj P, Kleespies A, Guba M, Eichhorn ME et al. (2010). Src tyrosine kinase inhibition suppresses lymphangiogenesis in vitro and in vivo. Curr Cancer Drug Targets 10: 546–553. [DOI] [PubMed] [Google Scholar]
- Ishigaki T, Imanaka‐Yoshida K, Shimojo N, Matsushima S, Taki W, Yoshida T (2011). Tenascin‐C enhances crosstalk signaling of integrin alphavbeta3/PDGFR‐beta complex by SRC recruitment promoting PDGF‐induced proliferation and migration in smooth muscle cells. J Cell Physiol 226: 2617–2624. [DOI] [PubMed] [Google Scholar]
- Krymskaya VP, Goncharova EA, Ammit AJ, Lim PN, Goncharov DA, Eszterhas A et al. (2005). Src is necessary and sufficient for human airway smooth muscle cell proliferation and migration. FASEB J 19: 428–430. [DOI] [PubMed] [Google Scholar]
- Kunit T, Gratzke C, Schreiber A, Strittmatter F, Waidelich R, Rutz B et al. (2014). Inhibition of smooth muscle force generation by focal adhesion kinase inhibitors in the hyperplastic human prostate. Am J Physiol Renal Physiol 307: F823–F832. [DOI] [PubMed] [Google Scholar]
- Levitt JM, Slawin KM (2007). Prostate‐specific antigen and prostate‐specific antigen derivatives as predictors of benign prostatic hyperplasia progression. Curr Urol Rep 8: 269–274. [DOI] [PubMed] [Google Scholar]
- Li W, Marshall C, Mei L, Gelfand J, Seykora JT (2007). Srcasm corrects Fyn‐induced epidermal hyperplasia by kinase down‐regulation. J Biol Chem 282: 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Levesque LO, Anand‐Srivastava MB (2010). Epidermal growth factor receptor transactivation by endogenous vasoactive peptides contributes to hyperproliferation of vascular smooth muscle cells of SHR. Am J Physiol Heart Circ Physiol 299: H1959–H1967. [DOI] [PubMed] [Google Scholar]
- Liu G, Hitomi H, Hosomi N, Shibayama Y, Nakano D, Kiyomoto H et al. (2011). Prorenin induces vascular smooth muscle cell proliferation and hypertrophy via epidermal growth factor receptor‐mediated extracellular signal‐regulated kinase and Akt activation pathway. J Hypertens 29: 696–705. [DOI] [PubMed] [Google Scholar]
- Lu R, Alioua A, Kumar Y, Kundu P, Eghbali M, Weisstaub NV et al. (2008). c‐Src tyrosine kinase, a critical component for 5‐HT2A receptor‐mediated contraction in rat aorta. J Physiol 586: 3855–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Reznichenko M, Poythress RH, Gallant CM, Vetterkind S, Li Y et al. (2012). Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol 227: 3585–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero JC, Seoane S, Ocana A, Pandiella A (2011). Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res 17: 5546–5552. [DOI] [PubMed] [Google Scholar]
- Oelke M, Bachmann A, Descazeaud A, Emberton M, Gravas S, Michel MC et al. (2013). EAU guidelines on the treatment and follow‐up of non‐neurogenic male lower urinary tract symptoms including benign prostatic obstruction. Eur Urol 64: 118–140. [DOI] [PubMed] [Google Scholar]
- Oguma T, Ito S, Kondo M, Makino Y, Shimokata K, Honjo H et al. (2007). Roles of P2X receptors and Ca2+ sensitization in extracellular adenosine triphosphate‐induced hyperresponsiveness in airway smooth muscle. Clin Exp Allergy 37: 893–900. [DOI] [PubMed] [Google Scholar]
- Orsted DD, Bojesen SE (2013). The link between benign prostatic hyperplasia and prostate cancer. Nat Rev Urol 10: 49–54. [DOI] [PubMed] [Google Scholar]
- Pavone LM, Cattaneo F, Rea S, De Pasquale V, Spina A, Sauchelli E et al. (2011). Intracellular signaling cascades triggered by the NK1 fragment of hepatocyte growth factor in human prostate epithelial cell line PNT1A. Cell Signal 23: 1961–1971. [DOI] [PubMed] [Google Scholar]
- Phillippe M, Sweet LM, Bradley DF, Engle D (2009). Role of nonreceptor protein tyrosine kinases during phospholipase C‐gamma 1‐related uterine contractions in the rat. Reprod Sci 16: 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradidarcheep W, Wallner C, Dabhoiwala NF, Lamers WH (2011). Anatomy and histology of the lower urinary tract. Handb Exp Pharmacol : 117–148. [DOI] [PubMed] [Google Scholar]
- Rees RW, Foxwell NA, Ralph DJ, Kell PD, Moncada S, Cellek S (2003). Y‐27632, a Rho‐kinase inhibitor, inhibits proliferation and adrenergic contraction of prostatic smooth muscle cells. J Urol 170: 2517–2522. [DOI] [PubMed] [Google Scholar]
- Roskoski R Jr (2005). Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun 331: 1–14. [DOI] [PubMed] [Google Scholar]
- Ross GR, Kang M, Shirwany N, Malykhina AP, Drozd M, Akbarali HI (2007). Nitrotyrosylation of Ca2+ channels prevents c‐Src kinase regulation of colonic smooth muscle contractility in experimental colitis. J Pharmacol Exp Ther 322: 948–956. [DOI] [PubMed] [Google Scholar]
- Sabbota AL, Kim HR, Zhe X, Fridman R, Bonfil RD, Cher ML (2010). Shedding of RANKL by tumor‐associated MT1‐MMP activates Src‐dependent prostate cancer cell migration. Cancer Res 70: 5558–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikhibrahim Z, Lindstrot A, Ellinger J, Rogenhofer S, Buettner R, Perner S et al. (2012). The peripheral zone of the prostate is more prone to tumor development than the transitional zone: is the ETS family the key? Mol Med Rep 5: 313–316. [DOI] [PubMed] [Google Scholar]
- Slack JK, Adams RB, Rovin JD, Bissonette EA, Stoker CE, Parsons JT (2001). Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene 20: 1152–1163. [DOI] [PubMed] [Google Scholar]
- Soler R, Andersson KE, Chancellor MB, Chapple CR, de Groat WC, Drake MJ et al. (2013). Future direction in pharmacotherapy for non‐neurogenic male lower urinary tract symptoms. Eur Urol 64: 610–621. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG (2014). Src signaling pathways in prostate cancer. Cancer Metastasis Rev 33: 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura S, Oliver V, White CW, Xie JH, Haynes JM, Exintaris B (2011). Novel drug targets for the pharmacotherapy of benign prostatic hyperplasia (BPH. Br J Pharmacol 163: 891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaeminck‐Guillem V, Gillet G, Rimokh R (2014). SRC: marker or actor in prostate cancer aggressiveness. Front Oncol 4: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcher D, Babiak C, Poletek P, Rosenkranz S, Bach H, Betz S et al. (2006). C‐Peptide induces vascular smooth muscle cell proliferation: involvement of SRC‐kinase, phosphatidylinositol 3‐kinase, and extracellular signal‐regulated kinase 1/2. Circ Res 99: 1181–1187. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gratzke C, Tamalunas A, Wiemer N, Ciotkowska A, Rutz B et al. (2016). P21‐activated kinase inhibitors FRAX486 and IPA3: inhibition of prostate stromal cell growth and effects on smooth muscle contraction in the human prostate. PLoS One 11: e0153312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kunit T, Ciotkowska A, Rutz B, Schreiber A, Strittmatter F et al. (2015). Inhibition of prostate smooth muscle contraction and prostate stromal cell growth by the inhibitors of Rac, NSC23766 and EHT1864. Br J Pharmacol 172: 2905–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber MM, Trakul N, Thraves PS, Bello‐DeOcampo D, Chu WW, Storto PD et al. (1999). A human prostatic stromal myofibroblast cell line WPMY‐1: a model for stromal‐epithelial interactions in prostatic neoplasia. Carcinogenesis 20: 1185–1192. [DOI] [PubMed] [Google Scholar]
- Weigand L, Sylvester JT, Shimoda LA (2006). Mechanisms of endothelin‐1‐induced contraction in pulmonary arteries from chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 290: L284–L290. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Hyman A, Desai A (2001). The spindle: a dynamic assembly of microtubules and motors. Nat Cell Biol 3: E28–E34. [DOI] [PubMed] [Google Scholar]
- Yang CM, Lin MI, Hsieh HL, Sun CC, Ma YH, Hsiao LD (2005). Bradykinin‐induced p42/p44 MAPK phosphorylation and cell proliferation via Src, EGF receptors, and PI3‐K/Akt in vascular smooth muscle cells. J Cell Physiol 203: 538–546. [DOI] [PubMed] [Google Scholar]
- Yezhelyev MV, Koehl G, Guba M, Brabletz T, Jauch KW, Ryan A et al. (2004). Inhibition of SRC tyrosine kinase as treatment for human pancreatic cancer growing orthotopically in nude mice. Clin Cancer Res 10: 8028–8036. [DOI] [PubMed] [Google Scholar]
- Zhou J, Scholes J, Hsieh JT (2003). Characterization of a novel negative regulator (DOC‐2/DAB2) of c‐Src in normal prostatic epithelium and cancer. J Biol Chem 278: 6936–6941. [DOI] [PubMed] [Google Scholar]