

Abstract
Background and Purpose
Adenosine A2A receptor stimulation promotes the synthesis of collagen type I and type III (Col1 and Col3), mediators of fibrosis and scarring. The A2A receptor modulates collagen balance via cAMP/PKA/p38‐MAPK/Akt pathways. Wnt signalling is important in fibrosis and the cAMP and Wnt pathways converge. Because the A2A receptor is Gs‐linked and increases cAMP, we determined whether A2A receptors and Wnt signalling interact.
Experimental Approach
Total β‐catenin, de‐phosphorylated β‐catenin (canonical activation, de‐phospho β‐catenin) and phosphorylated β‐catenin at Ser552 (non‐canonical activation, p‐Ser552 β‐catenin) levels were determined in primary human dermal fibroblasts, cytosol and nucleus, by western blot analysis and fluorescence microscopy, before and after stimulation by A2A receptor‐selective agonist CGS21680, with/without A2A receptor‐selective antagonist (SCH56261) pretreatment. β‐Catenin was knocked down by transfection with scrambled‐siRNA or specific‐siRNA, and Col1 and Col3 levels determined by western blots.
Key Results
CGS21680 stimulation rapidly (15 min) increased cellular β‐catenin levels. Both de‐phospho β‐catenin and p‐Ser552 β‐catenin levels were also increased. CGS21680 stimulated the translocation of total de‐phospho and p‐Ser552 β‐catenin to the nucleus. A2A receptor‐stimulation increased Col1 synthesis similarly in β‐catenin knockeddown and scrambled cells. However, β‐catenin knockdown abolished the increase in Col3 synthesis induced in A2A receptor‐stimulated fibroblasts.
Conclusions and Implications
A2A receptor stimulation promotes Col3 synthesis via the activation of canonical and non‐canonical β‐catenin, consistent with a role for A2A receptors in dermal fibrosis and scarring.
Abbreviations
- Col1
collagen type I
- Col3
collagen type III
- CBP
cofactor CREB‐binding protein
- CREB
cAMP response element binding protein
- CTGF
connective tissue growth factor
- NHDFs
normal human dermal fibroblasts
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| A2A receptor | Akt (PKB) |
| Other protein targets b | ERK1 |
| CBP (CREB binding protein) | ERK2 |
| GSK3β | |
| p38 MAPK | |
| PKA | |
| Smad |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Cutaneous wound healing is initially characterized by inflammation followed by the formation of new vessels and accumulation of matrix elements. Later, re‐epithelialization, scar contraction and remodelling occur (Andrews et al., 2016). However, the wound healing process can be diverted or become overly active leading to excessive matrix production. Dermal fibrosis, which is the pathological remodelling of skin characterized by the excessive accumulation of collagen and other matrix elements, underlies a wide range of clinically important diseases including hypertrophic scarring, keloid and morphea (Andrews et al., 2016; Shaikh and Cronstein, 2016). Scleroderma and other diffuse fibrosing conditions, although uncommon, cause multiple morbidities in affected people, including disfigurement and a diminished range of movements as well as premature mortality as a result of renal and cardiac dysfunctions (Balbir‐Gurman and Braun‐Moscovici, 2012).
Adenosine is a purine nucleoside generated extracellularly from adenine nucleotides, such as ATP and ADP, and is released by cells as a result of direct stimulation, hypoxia, injury or metabolic stress (Eltzschig and Eckle, 2011; Borea et al., 2016). Adenosine levels in the milieu of ischaemic or necrotic tissue can increase several orders of magnitude, from nanomolar to millimolar concentrations (Montesinos et al., 2002). Adenosine activates adenylyl cyclase leading to an increase in intracellular cAMP by stimulation of a family of Gs‐coupled protein receptors; A2A and A2B receptors being the principal ones involved with respect to fibrosis (Hasko et al., 2008; Fredholm et al., 2011). The increase in cAMP, in turn, activates PKA and other downstream targets that influence the expression of genes associated with wound healing and fibrosis (Hasko and Cronstein, 2004; Chan et al., 2006; Mediero et al., 2013; Borea et al., 2016).
The overproduction of A2A receptors, resulting from a deficiency in adenosine deaminase, leads to marked fibrosis (Fernandez et al., 2008). Deletion or blockade of A2A receptors has been shown to completely prevent hepatic and dermal fibrosis in murine models (Perez‐Aso et al., 2012). Furthermore, reducing the capacity to produce extracellular adenosine as a result of CD73 or CD39 deletion also markedly reduces fibrosis in murine models (Fernandez et al., 2013). A2A receptor activation has been shown to stimulate fibroblasts directly and indirectly to produce collagen type I and type III (Col1 and Col3) through different mechanisms (Chan et al., 2013; Perez‐Aso et al., 2013). In addition, different levels of receptor engagement have been shown to promote different types of collagen synthesis (Chan et al., 2013; Perez‐Aso et al., 2013). For example, at low levels of receptor stimulation, Col1 expression is promoted in a pathway involving Akt phosphorylation and activation. However, higher levels of receptor stimulation induce the expression of Col3 via p38 MAPK‐dependent signalling (Perez‐Aso et al., 2013; 2014). The different levels of receptor stimulation required for production of these different collagen subtypes probably reflects, in part, the fact that Col3 expression is negatively regulated by ERK1 and ERK2 and higher levels of receptor activation are required to overcome this inhibition (Perez‐Aso et al., 2014). Thus, A2A receptor‐mediated collagen production contributes to normal wound healing and scar formation by multiple signalling pathways and also participates in the pathogenesis of fibrotic conditions such as scleroderma and cirrhosis (Fernandez et al., 2008; Perez‐Aso et al., 2012).
The role of Wnt/β‐catenin signalling in wound healing and fibrosis has also been extensively investigated (Cheon et al., 2006; Poon et al., 2009; Wei et al., 2011). Canonical Wnts are lipoglycoproteins that are secreted and involved in embryological development, cellular fating and normal homeostasis by activating membrane‐bound frizzled receptors (Wodarz and Nusse, 1998; Reya et al., 2003; van Es et al., 2005; Clevers, 2006). Wnt‐mediated signalling results in the activation of a transcription complex that contains the cytosolic protein β‐catenin and its DNA‐binding partners, lymphocyte enhancer factor (LEF)/T‐cell factor (TCF) (Clevers, 2006). Wnt binding to the frizzled receptor stimulates a rise in cytosolic β‐catenin through inhibition of a multi‐subunit protein complex known as the ‘β‐catenin destruction complex’, which includes the key regulatory enzyme GSK3β. In the absence of Wnt binding to the frizzled receptor, GSK3β constitutively phosphorylates β‐catenin, leading to its ubiquitination and destruction by the proteasome (Clevers, 2006). By inhibiting this phosphorylation, β‐catenin accumulates in the cytoplasm and translocates to the nucleus, where it can complex with the LEF/TCF family of proteins and localize to its gene promoters (Clevers, 2006). Other non‐canonical Wnt signalling pathways have also been described (Komiya and Habas, 2008).
The activation of β‐catenin‐dependent Wnt signalling is one of the initial cellular responses to tissue injury. Wnt/β‐catenin signalling in fibroblast cultures enhances the proliferation, migration and extracellular matrix production of these cells (Wei et al., 2011). Furthermore, cutaneous wound healing studies in mice have demonstrated that β‐catenin signalling is activated after injury, and a mutated form of β‐catenin resistant to ubiquitin‐mediated degradation leads to excessive collagen synthesis and hyperplastic scarring (Cheon et al., 2002). Additionally, β‐catenin induces the contraction of collagen lattices, an important step in the maturation of scars (Poon et al., 2009). Crosstalk between Wnt/β‐catenin and another important mediator of fibrosis, TGF‐β1, has also been demonstrated to occur (Zhou et al., 2012). Wnt/β‐catenin signalling has been shown to up‐regulate the expression of TGF‐β1, and TGF‐β1 promotes β‐catenin signalling (Cheon et al., 2002; Zhou et al., 2012). Mice lacking Smad3, a downstream target of TGF‐β1 signalling, have been demonstrated to have lower levels of β‐catenin activation after injury, and TGF‐β1‐induced proliferation of fibroblasts is reduced in β‐catenin knockedout cells (Cheon et al., 2002). Nevertheless, the relationship between β‐catenin and TGF‐β1 signalling is complex, and there is also evidence of antagonism between the two pathways (Liang et al., 2008).
Recent evidence indicates a convergence of cAMP, the main effector of A2A receptor stimulation, and Wnt/ β‐catenin signalling, which prompted us to analyse the crosstalk between these two signalling pathways (Hino et al., 2005). It has also been shown that direct phosphorylation of β‐catenin by Akt promotes β‐catenin transcriptional activity, representing a possible A2A receptor‐mediated non‐canonical pathway for its activation (Fang et al., 2007). Additionally, although both TGF‐β1 and adenosine promote fibrosis, they follow different signalling pathways wherein A2A receptor stimulation promotes collagen production via cAMP and Akt but independently of Smad2/3 (Perez‐Aso et al., 2014). Thus, Wnt/β‐catenin signalling might represent a final common pathway for these two important mediators of fibrosis, and the results of this study suggest that directly modifying these signalling events might represent a novel approach for diminishing scarring and fibrosis without affecting cutaneous wound healing.
Methods
Stimulation and preparation of cellular extracts and western blots
NHDF cells were cultured in DMEM containing 10% FBS and 1% penicillin/streptomycin (DMEM) and used for experiments from the first to fifth passages. β‐catenin siRNA transfected NHDF cells (siRNA to β‐catenin or scrambled siRNA as control) and NHDF cells were stimulated with the A2A receptor‐selective agonist CGS21680 at increasing doses ranging from 0.1 to 10 μM for the indicated time period (dose–response) or at an indicated dose for different time periods (time–course). When used, the A2A receptor‐selective antagonist SCH58261 (1 μM) was introduced 30 min before CGS21680. For all the experiments with antagonists, a dose–response curve from 0.1 to 10 μM of CGS21680 was run at the same time. After being stimulated, cells were washed with cold PBS and lysed in RIPA buffer containing protease inhibitor cocktail and phosphatase inhibitor cocktail to isolate total cell protein content. The extraction of cytoplasmic and nuclear fraction proteins was carried out using NE‐PER nuclear and cytoplasmic extraction reagent kits. Protein concentration was quantified by BCA protein assay (Mediero et al., 2014). Next, 4 μg of protein extract for β‐catenin, de‐phospho‐β‐catenin (Ser33/37/Thr41) and phospho‐β‐catenin (Ser552), 3 μg for Col1 and 10 μg for Col3 were separated by use of SDS‐PAGE and transferred to nitrocellulose membranes for immunoblotting. After being blocked in Tris‐buffered saline with 0.1% Tween 20 (TBST) plus 3% BSA (Sigma‐Aldrich) for 1 h at room temperature, membranes were incubated overnight at 4°C with primary antibodies against Col1 (1:500), Col3 (1:500), β‐catenin (1:1000), de‐phospho‐β‐catenin (Ser33/37/Thr41) (1:1000), phospho‐β‐catenin (Ser552) (1:1000), p84 (1:1000) and actin (1:1000). The membranes were then washed with TBST and incubated with goat anti‐rabbit IRDye 800CW (1:5000) and goat anti‐mouse IRDye 680RD (1:5000), or donkey anti‐goat IRDye 800CW (1:5000) and donkey anti‐mouse IRDye 680RD (1:5000) for 1 h at room temperature in the dark, and images were visualized with Li‐Cor Odyssey equipment (Li‐Cor Biosciences) where near‐infrared fluorescent signals were detected, because each secondary antibody emits a signal at a different frequency. A specific nuclear signal was determined using mouse monoclonal anti‐nuclear matrix protein p84. The intensities of the respective bands were quantified by densitometric analysis using the Image Studio 2.0.38 software (Li‐Cor Biosciences) (Mediero et al., 2014).
Band quantification was first normalized to actin or p84, and then, the percentage was calculated against the non‐stimulated control blotted on the same membrane. Data obtained from densitometric analyses were normalized to account for unequal loading of samples across the lanes on a gel and for differences in transfer efficiency across a blot.
RNA interference (siRNA)
Double‐stranded siRNAs against β‐catenin (4390824 (s436)) and scrambled siRNAs (4390844) as a negative control were purchased from Ambion (Life Technologies, Grand Island, NY, USA). NHDF cells were transfected with siRNA (50 pmol for β‐catenin knockdown with the negative control at the respective concentrations), using Lipofectamine RNAiMAX (Invitrogen, Life Technologies). After 24 h, CGS21680 (1 μM) was added for 24 h, and cellular extracts were prepared as described above.
Immunocytochemistry
To identify the activation and nuclear translocation of β‐catenin, NHDF cells were plated on glass coverslips (Lab‐Tek II chamber slide, 8‐well; Thermo Fisher Scientific) in DMEM. When they reached 75% confluence, cells were fixed with cold 4% paraformaldehyde (Hatfield, PA, USA) in PBS for 15 min at room temperature. After being washed with PBS for 10 min twice and PBS containing BSA 2% for 10 min once, the cells were blocked with PBS BSA 2% Triton X‐100 0.5% FBS 5% for 1 h at room temperature. Next, cells were incubated overnight at 4°C with primary antibodies against β‐catenin (1:400), de‐phospho‐β‐catenin (Ser33/37/Thr41) (1:400) and phospho‐β‐catenin (Ser552) (1:400). Then, cells were washed with PBS containing BSA 2% and incubated with goat anti‐rabbit IgG (whole molecule)‐FITC for 1 h at room temperature in the dark. After being washed with PBS containing BSA 2% for 10 min twice and PBS for 10 min once, cells were counterstained with DAPI and each sample was examined using a fluorescence microscope (Nikon Eclipse NI‐U fluorescence uplight microscope, Tokyo, Japan).
Statistical analysis
Results are presented as mean ± SEM. Data were analysed by one or two‐way ANOVA followed by Bonferroni post hoc test if F achieved P < 0.05, and there was no significant variance in‐homogeneity or by Student's t‐test. All statistical analyses were performed with graphpad prism software v. 6.0 (Graphpad Software, La Jolla, CA, USA). The α nominal level was set at 0.05 in all cases. A P value of <0.05 was the considered significant. Randomization was not performed, as this experiment did not involve animals or human subjects. Additionally, explicit blinding was not performed; however, squares of identical areas were used to measure pixel density to eliminate bias across experimental results. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Antibodies, reagents and cell line
Normal human dermal fibroblasts (NHDFs) were purchased from Lonza (Walkersville, MD, USA). DMEM (high glucose), FBS, penicillin/streptomycin, NE‐PER nuclear and cytoplasmic extraction reagents kit, and bicinchoninic acid (BCA) Protein Assay Reagent were purchased from Thermo Fisher Scientific (Grand Island, NY, USA). CGS21680 and SCH58261 were purchased from Tocris Bioscience (Ellisville, MO, USA). Rabbit monoclonal antibodies to β‐catenin and de‐phospho‐β‐catenin (Ser33/37/Thr41) and phospho‐β‐catenin (Ser552) were purchased from Cell Signalling Technology (Danvers, MA, USA). Goat antibodies to Col1 and Col3 were purchased from SouthernBiotech (Birmingham, AL, USA). Mouse monoclonal anti‐nuclear matrix protein p84 was purchased from Abcam (Cambridge, MA, USA). Mouse monoclonal antibody to actin (C‐2) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat anti‐rabbit IRDye 800CW, goat anti‐mouse IRDye 680 RD, donkey anti‐goat IRDye 800CW and donkey anti‐mouse IRDye 680 RD were purchased from Li‐Cor Biosciences (Lincoln, NE, USA). DAPI mounting medium, goat anti‐rabbit IgG (whole molecule)‐FITC, the RIPA buffer, protease inhibitor cocktail and phosphatase inhibitor cocktail were purchased from Sigma‐Aldrich (St. Louis, MO, USA).
Results
A2A receptor activation rapidly increases cellular β‐catenin levels
To explore the effects of A2A receptor stimulation on β‐catenin levels in dermal fibroblasts, we first determined the time–course of the effects of stimulation with the A2A receptor‐selective agonist, CGS21680, on β‐catenin levels in NHDFs. We found that after stimulation with CGS21680 (1 μM), cellular β‐catenin levels rapidly increased before achieving a peak at 15 min (174 ± 23% of non‐stimulated control, P < 0.05, n = 6) and 30 min (176 ± 16% of non‐stimulated control, P < 0.05, n = 6) (Figure 1A). The A2A receptor‐mediated increase in β‐catenin levels in NHDFs was dose‐dependent (Figure 1B). We observed that CGS21680 stimulation at concentrations ranging from nanomolar to micromolar increased cellular β‐catenin levels, and pretreatment of NHDFs with the highly selective A2A receptor antagonist SCH58261 (1 μM) completely reversed the effect of CGS21680 on β‐catenin levels (Figure 1B). These results are consistent with the hypothesis that A2A receptor activation prevents β‐catenin degradation, thereby rapidly increasing total cellular β‐catenin levels.
Figure 1.
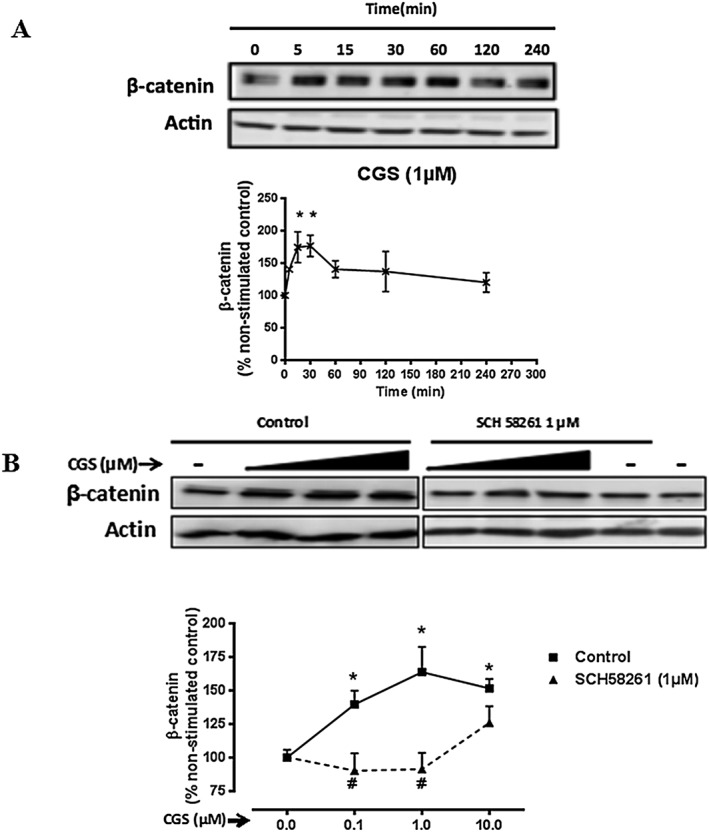
Activation of A2A receptors rapidly increases cellular β‐catenin levels. (A) NHDF cells were incubated with CGS21680 (1 μM) for periods of up to 4 h and collected at the indicated time‐points. (B) NHDF cells were incubated with increasing concentrations of CGS21680 (0.1–10 μM) for 15 min, with or without SCH58261 (1 μM) pre‐incubation. Cellular β‐catenin levels were measured. Representative images of western blots for β‐catenin and actin are shown. Data represent mean ± SEM of six independent experiments as determined by densitometry relative to actin. *P < 0.05 versus non‐stimulated control; #P < 0.05, SCH58261 + CGS26180 versus CGS26180. CGS indicates CGS26180.
Activation of A2A receptors increases cellular β‐catenin levels by both canonical and Akt‐mediated activation
To better define how A2A receptor activation increases β‐catenin expression in dermal fibroblasts, we investigated the levels of β‐catenin after CGS21680 stimulation in NHDFs. Because previous studies have demonstrated that both canonical and non‐canonical activation of β‐catenin leads to an up‐regulation of the expression of its gene, we examined the expression of both de‐phosphorylated β‐catenin (canonical β‐catenin activation, de‐phospho β‐catenin) and phosphorylated β‐catenin at Ser552 (non‐canonical β‐catenin activation, site of β‐catenin activation by AKT, p‐Ser552 β‐catenin) after CGS21680 stimulation for time periods up to 4 h (Figure 2A).
Figure 2.
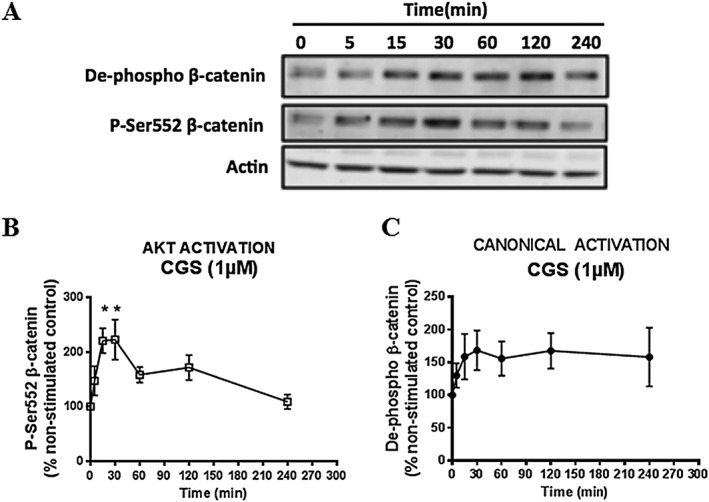
Activation of A2A receptors increases cellular β‐catenin levels via canonical activation and Akt activation. NHDF cells were incubated with CGS21680 (1 μM) for periods of up to 4 h and collected at the indicated time‐points. (A) Representative images of western blots for de‐phospho‐β‐catenin, p‐Ser552‐β‐catenin and actin are shown. Effects of CGS21680 stimulation on (B) p‐Ser552‐β‐catenin and (C) de‐phospho‐β‐catenin were analysed. Data represent mean ± SEM of six independent experiments as determined by densitometry relative to actin. *P < 0.05 versus non‐stimulated control. CGS indicates CGS26180.
After A2A receptor stimulation by CGS21680 (1 μM), cellular p‐Ser552 β‐catenin levels rapidly increased more than twofold at 15 min (220 ± 22% of non‐stimulated control, P < 0.05, n = 6) and at 30 min (222 ± 36% of non‐stimulated control, P < 0.05, n = 6) before gradually decreasing (Figure 2B). The point of maximum increase was consistent with that of total cellular β‐catenin levels after CGS21680 stimulation (Figure 1A). A2A receptor stimulation by CGS21680 (1 μM) also rapidly enhanced cellular de‐phospho β‐catenin levels, although this increase did not achieve statistical significance (Figure 2C). Our data indicate that activation of A2A receptors increases cellular β‐catenin levels via both canonical activation and non‐canonical activation.
Activation of A2A receptors induces nuclear translocation of β‐catenin
Because activation of the Wnt/β‐catenin signalling pathway ultimately results in the nuclear translocation of stabilized β‐catenin, we next examined the β‐catenin levels in the nuclear and cytoplasmic fraction of NHDFs after CGS21680 stimulation. Nuclear translocation of β‐catenin was observed by fluorescence microscopy after CGS21680 (1 μM) (Figure 3A) stimulation and the nuclear translocation of both p‐Ser552 β‐catenin and de‐phospho β‐catenin was blocked by pretreatment with the A2A receptor‐selective antagonist SCH58261 (1 μM) (Figure 3B and C). We observed that, after CGS21680 (1 μM) stimulation, β‐catenin levels in the cytosol fraction increased and peaked after 1 h (158 ± 36% of non‐stimulated control, P < 0.05, n = 5). Thereafter, β‐catenin levels in the cytosolic fraction dramatically decreased and this was accompanied by a concomitant increase in β‐catenin levels in the nuclear fraction with a maximum at 2 h (151 ± 13% of non‐stimulated control, P < 0.05, n = 5) (Figure 4A). Next, we examined the levels of active β‐catenin in the nuclear fraction after 15 min of CGS21680 (1 μM) stimulation. There was a statistically significant increase in the levels of both de‐phospho β‐catenin (132 ± 10% of non‐stimulated control, P < 0.05, n = 5) and p‐Ser552 β‐catenin (165 ± 8% of non‐stimulated control, P < 0.05, n = 5) (Figure 4B). The increase in β‐catenin levels in the nuclear fraction is consistent with the translocation of β‐catenin from the cytosol to the nucleus after A2A receptor activation. Taken together, these results demonstrate that activation of the A2A receptor induces the nuclear translocation of β‐catenin.
Figure 3.
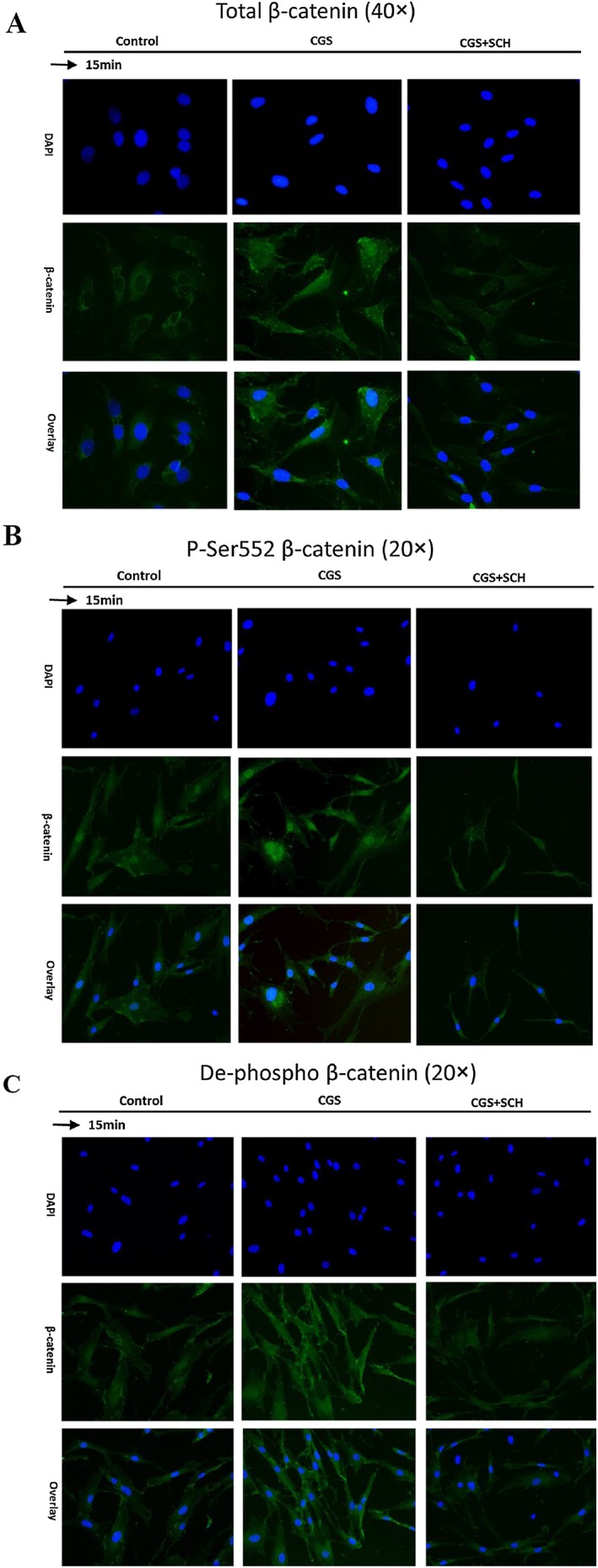
Nuclear translocation of β‐catenin in NHDFs after CGS21680 (1 μM) stimulation for 15 min. NHDFs were processed and immunohistological staining was carried out. Nuclei are shown in blue (DAPI) and β‐catenin in green. CGS21680 (1 μM) stimulation induced nuclear translocation of (A) total β‐catenin, (B) p‐Ser552 β‐catenin and (C) de‐phospho β‐catenin. SCH58261 (1 μM) blocked the nuclear translocation of β‐catenin induced by CGS21680 stimulation. CGS indicates CGS26180 and SCH indicates SCH58261.
Figure 4.
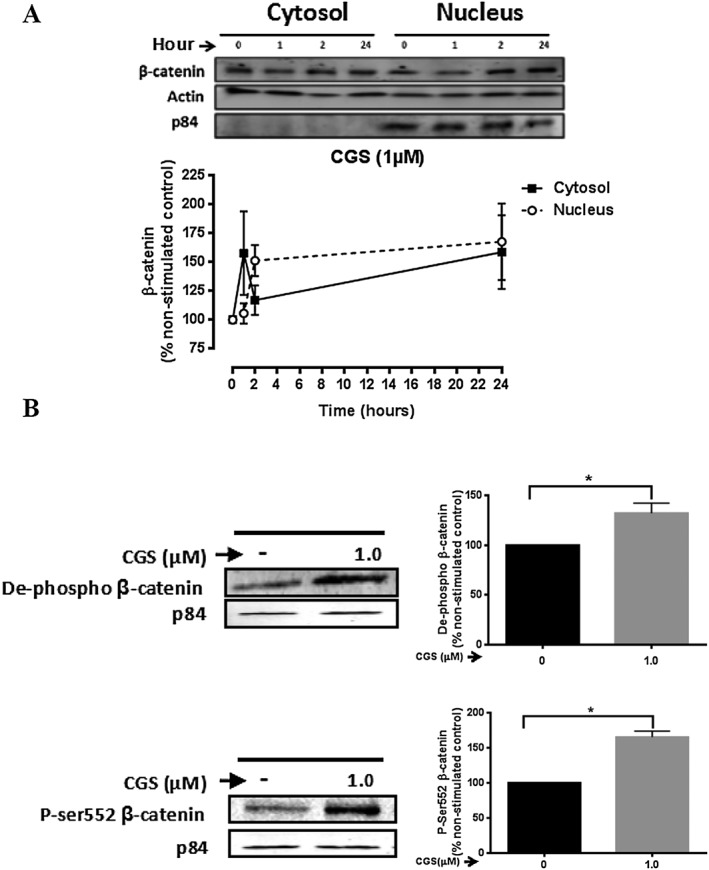
Stimulation of A2A receptors leads to nuclear translocation of β‐catenin. (A) NHDF cells were incubated with CGS21680 (1 μM) for periods up to 24 h and collected at the indicated time‐points. Cytosolic and nuclear fractionation was performed as described in the Methods section. Representative images of western blots for β‐catenin, actin and the nuclear marker p84 are shown. Effects of CGS21680 stimulation on β‐catenin levels in the cytosol and nucleus were analysed. Data represent mean ± SEM of five independent experiments as determined by densitometry relative to actin or the nuclear marker p84. (B) NHDF cells were incubated with CGS21680 (1 μM) for 15 min. Representative images of western blots for de‐phospho and p‐Ser552 β‐catenin in the nuclear fraction are shown. Effects of CGS21680 stimulation on de‐phospho and p‐Ser552 β‐catenin levels in the nucleus were analysed. Data represent mean ± SEM of five independent experiments as determined by densitometry relative to the nuclear marker p84. *P < 0.05 versus non‐stimulated control. CGS indicates CGS26180.
β‐catenin increases Col3, but not Col1 synthesis after A2A receptor activation
The results of previous studies have indicated that A2A receptor activation increases the production of collagen in dermal fibroblasts (Chan et al., 2013; Perez‐Aso et al., 2013). Because A2A receptor activation increases cellular β‐catenin levels and promotes its nuclear translocation for transcriptional activation, we further determined whether β‐catenin signalling is involved in collagen production after A2A receptor activation. After β‐catenin had been reduced by use of siRNA (46 ± 5% decrease in β‐catenin vs. scrambled siRNA transfection, P < 0.05, n = 10) (Figure 5A), we observed that the increase in Col1 synthesis induced by A2A receptor stimulation by CGS21680 (1 μM) was unaffected by β‐catenin knockdown (scrambled siRNA 63 ± 22% increase of Col1 vs. β‐catenin‐siRNA 53 ± 17% increase of Col1, P > 0.05, n = 7). In contrast, β‐catenin knockdown inhibited A2A receptor‐mediated Col3 synthesis by 73% (scrambled siRNA 66 ± 14% increase of Col3 vs. β‐catenin siRNA 18 ± 16% increase of Col3, P < 0.05, n = 8) (Figure 5B). These results indicate that β‐catenin signalling is involved in the synthesis of Col3 but not Col1 evoked by activation of A2A receptors.
Figure 5.
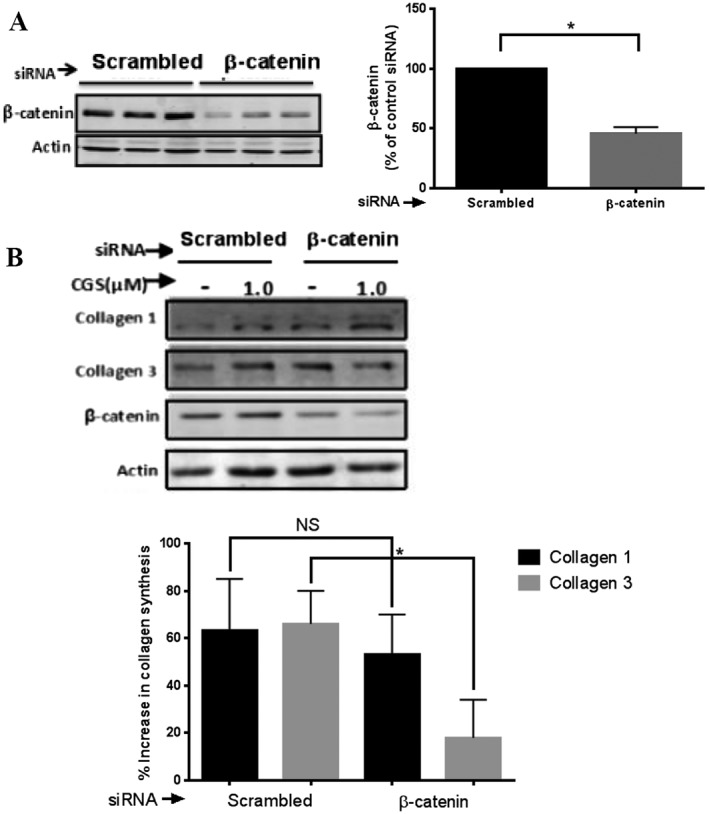
β‐Catenin knockdown prevents A2A receptor‐mediated increase in Col3 but not Col1. NHDF cells were stably transfected with scrambled siRNA or β‐catenin siRNA. (A) Cellular β‐catenin levels were measured. Representative western blots of β‐catenin and actin are shown. Data represent mean ± SEM of 10 independent experiments as determined by densitometry relative to actin. (B) CGS21680 (1 μM) was added for 24 h, after β‐catenin knockdown. A2A receptor stimulation increased Col1 synthesis and was unaffected by β‐catenin knockdown. In contrast, A2A receptor‐mediated increase in Col3 synthesis was reduced with β‐catenin knockdown. Representative western blots of Col1, Col3, β‐catenin and actin are shown. Data represent mean ± SEM of seven (Col1) and eight (Col3) independent experiments as determined by densitometry relative to actin. NS, P > 0.05; *P < 0.05, β‐catenin siRNA versus scrambled siRNA. CGS indicates CGS26180.
Discussion and conclusions
In the present study we showed that the stimulation of A2A receptors rapidly increases total cellular β‐catenin levels via both canonical and Akt‐mediated activation. Moreover, consistent with its effects on cellular β‐catenin levels, activation of the A2A receptor leads to the nuclear translocation of both phosphorylated serine‐552 and de‐phosphorylated β‐catenin at 15 min. Subsequently, there is a further increase in this nuclear translocation up to a maximum of 24 h consistent with the previously reported biphasic nuclear shuttling of β‐catenin following Wnt signalling (Xie et al., 2008; Zhang et al., 2011; Jang et al., 2014; Tapia‐Rojas et al., 2015). The finding that silencing the expression of β‐catenin prevents the synthesis of Col3 mediated by A2A receptors, but not that of Col1, sheds light on the signalling downstream from the A2A receptor involved in stimulating fibrosis and scarring.
Adenosine, by acting on A2A and A2B receptors, increases both Col1 and Col3 synthesis in vitro and in animal models (Perez‐Aso et al., 2013, 2014). Adenosine receptors are differentially expressed in different tissues and signal via distinct pathways downstream from the secondary messenger cAMP (Shaikh and Cronstein, 2016). For example, the A2B receptor mediates renal fibrosis in the setting of hypoxia by inducing the expression of endothelin‐1 (Sorokin and Kohan, 2003; Kong et al., 2006). This pathway is also important in promoting pulmonary interstitial disease (Karmouty‐Quintana et al., 2012). The A2A receptor is the principal adenosine receptor involved in dermal fibrosis (Chan et al., 2006; Perez‐Aso et al., 2013). In normal skin, the ratio of Col1 to Col3 is approximately 4:1; however, in granulation tissue and immature scars, where local adenosine concentrations are elevated, the increased synthesis of Col3 leads to a reduction in the Col1 to Col3 ratio to 2:1 (Perez‐Aso et al., 2013). High levels of A2A receptor stimulation, with correspondingly higher cAMP concentrations, lead to the induction of Epac2 and promote Col3 synthesis (Perez‐Aso et al., 2013). In addition, A2A receptor ligation stimulates an increase in the secretion of connective tissue growth factor (CTGF), which promotes collagen secretion by diminishing the expression of the transcriptional regulator Fli1, a constitutive repressor of CTGF expression (Chan et al., 2013).
The results presented here demonstrate crosstalk between A2A receptors and Wnt/β‐catenin signalling, which probably occurs at several levels (Figure 6). For example, Fli1 inhibits β‐catenin/TCF‐mediated transcription, and CTGF leads to the accumulation and nuclear translocation of β‐catenin as well as increased TCF/LEF transcriptional activity (Navarro et al., 2010; Rooney et al., 2011). PKA can also directly stabilize β‐catenin by inhibiting its ubiquitination (Hino et al., 2005). Downstream, PKA activates cAMP response element‐binding protein (CREB) by phosphorylation, which can modulate gene expression directly through interaction with gene promoters or indirectly by competing with NFκB and other transcription factors for the cofactor CREB‐binding protein (CBP) (Hasko et al., 2008). CBP is a transcriptional co‐activator of β‐catenin, and CBP/β‐catenin‐mediated transcription is critical for cellular proliferation (Hasko et al., 2008; Henderson et al., 2010). Furthermore, selective inhibition of the CBP/β‐catenin interaction attenuates bleomycin‐induced lung fibrosis and reverses established fibrosis (Takemaru and Moon, 2000; Henderson et al., 2010).
Figure 6.
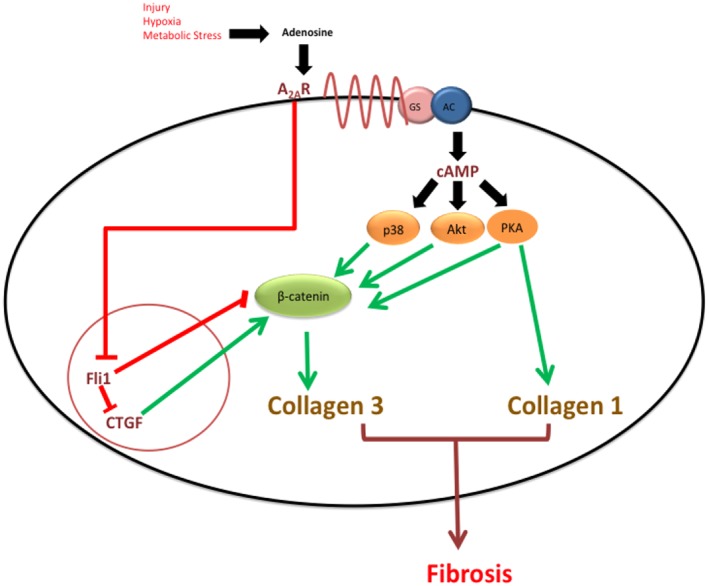
Crosstalk between A2A receptors and Wnt/β‐catenin signalling pathways in human dermal fibroblasts. Adenosine levels increase by several orders of magnitude during cellular injury, hypoxia or metabolic stress. Stimulation of A2A receptors activates a Gs protein leading to a rise in cAMP via adenylyl cyclase and activation of downstream targets. MAPK‐p38, PKA and CTGF have all been shown to directly promote canonical β‐catenin signalling, and Fli1 represses canonical β‐catenin signalling. Akt, which activates β‐catenin non‐canonically at Ser552, also promotes its transcriptional activity (see text). Together, these pathways promote normal wound healing but also pathological fibrosis via Col3 synthesis.
At low levels of A2A receptor activation, PKA represses Col3 synthesis. However, interestingly, at higher levels of receptor engagement, with a correspondingly higher level of cAMP, Col3 levels are increased. This is probably due to the induction of Epac2 and the positive cooperativity of cAMP binding to PKA (Perez‐Aso et al., 2013). We hypothesize that if the A2A receptor/PKA/β‐catenin pathway is important for Col3 synthesis this reflects PKA's unique role as a molecular switch that can integrate multiple upstream signals and can activate different downstream pathways depending on cell‐specific conditions.
It has also been shown that the effects of A2A receptor stimulation on Col3 synthesis are, in part, mediated by p38‐MAPK (Perez‐Aso et al., 2013). Activation of p38‐MAPK also interacts with canonical Wnt/β‐catenin signalling (Bikkavilli et al., 2008). However, although inhibition of p38‐MAPK interrupts Wnt/β‐catenin signalling, its knockdown does not completely abolish the signalling. Rather, p38‐MAPK activation operates as a parallel pathway feeding into the Wnt/β‐catenin pathway by inhibiting GSK3β, a key regulatory enzyme in canonical signalling (Bikkavilli et al., 2008). Finally, direct phosphorylation of β‐catenin at the serine‐552 residue by Akt promotes its transcriptional activity without altering its stability and phosphorylation level by GSK3β. Thus, A2A receptor ligation represents another parallel, non‐canonical pathway for activation of β‐catenin (Fang et al., 2007).
Our results show that β‐catenin activation and translocation are necessary for A2A receptor‐mediated Col3 synthesis but not Col1 synthesis. Interestingly, β‐catenin signalling is required for Smad‐dependent TGF‐β1‐induced Col1 but not Col3 synthesis (Baarsma et al., 2011). Although both TGF‐β1 and adenosine receptors can induce Col1 and Col3 synthesis, we hypothesize that A2A receptor‐dependent β‐catenin signalling is preferentially activated in early wound healing and granulation tissue formation, whereas TGF‐β1‐mediated β‐catenin signalling may be involved in scar maturation.
The A2A receptor is also likely to interact with Wnt/β‐catenin signalling in other tissues. For example, in bone, A2A receptor activation inhibits the differentiation and function of osteoclasts (Mediero et al., 2013). As a consequence, A2A receptor‐deficient mice have markedly increased numbers of osteoclasts and increased bone resorption with diminished bone density (Mediero et al., 2013). Similarly, in mature osteoblasts and osteocytes, β‐catenin plays a role in suppressing osteoclast differentiation (Chen and Long, 2013). In humans, loss‐of‐function mutations of β‐catenin lead to early onset osteoporosis (Chen and Long, 2013). One pathway by which A2A receptor stimulation regulates bone turnover is via PKA‐dependent inhibition of the nuclear translocation of NFκB (Mediero et al., 2013). Activation of PKA directly stabilizes β‐catenin and has been shown to inhibit NFκB activation and translocation to the nucleus through a physical interaction (Takahashi et al., 2002). Similarly, the PI3‐kinase‐Akt signalling pathway, previously shown to activate β‐catenin in a non‐canonical fashion, facilitates osteoblast differentiation, bone growth and mineralization (Saidak et al., 2015).
In conclusion, we have shown that A2A receptor activation leads to activation of both canonical and non‐canonical Wnt/β‐catenin signalling, which is required for Col3 but not Col1 synthesis in primary human dermal fibroblasts. The molecular crosstalk between these two signalling pathways probably occurs at multiple levels in different tissues. Wnt/β‐catenin signalling also represents a final common pathway for both TGF‐β1 and A2A receptor signalling pathways, both of which are essential in wound healing and fibrosis. Thus, selectively modifying this pathway represents an attractive therapeutic target in fibrotic diseases.
Author contributions
G.S., J.Z., M.P‐A., A.M. and B.C. contributed to the conception and design of the experiments. G.S., J.Z., A.M. and M.P‐A. conducted the experiments, collected and assembled the data, and performed data analysis and interpretation. G.S., J.Z. and B.C. contributed to manuscript preparation.
Conflict of interest
A.M. and B.C. have filed a patent on use of adenosine A2AR agonists to prevent prosthesis loosening (pending). A.M. and B.C. have filled a patent on the use of Antibodies against Netrin‐1 for the treatment of bone diseases. G.S., J.Z. M.P‐A. do not have any disclosures. B.C. holds patents numbers 5,932,558; 6,020,321; 6,555,545; 7,795,427; adenosine A1R and A2BR antagonists to treat fatty liver (pending); adenosine A2AR agonists to prevent prosthesis loosening (pending). B.C. is a consultant for Bristol‐Myers Squibb, AstraZeneca, Novartis, CanFite Biopharmaceuticals, Cypress Laboratories, Regeneron (Westat, DSMB), Endocyte, Protalex, Allos, Inc., Savient, Gismo Therapeutics, Antares Pharmaceutical, Medivector, King Pharmaceutical, Celizome, Tap Pharmaceuticals, Prometheus Laboratories, Sepracor, Amgen, Combinatorx, Kyowa Hakka, Hoffman‐LaRoche and Avidimer Therapeutics. BNC has stock in CanFite Biopharmaceuticals.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgments
This work was funded by a National Institutes of Health Grant (Award #2R56AR056672‐06).
Shaikh, G. , Zhang, J. , Perez‐Aso, M. , Mediero, A. , and Cronstein, B. (2016) Adenosine A2A receptor promotes collagen type III synthesis via β‐catenin activation in human dermal fibroblasts. British Journal of Pharmacology, 173: 3279–3291. doi: 10.1111/bph.13615.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J (2016). Keloids: the paradigm of skin fibrosis ‐ pathomechanisms and treatment. Matrix Biol 51: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baarsma HA, Menzen MH, Halayko AJ, Meurs H, Kerstjens HA, Gosens R (2011). beta‐Catenin signaling is required for TGF‐beta1‐induced extracellular matrix production by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 301: L956–L965. [DOI] [PubMed] [Google Scholar]
- Balbir‐Gurman A, Braun‐Moscovici Y (2012). Scleroderma ‐ new aspects in pathogenesis and treatment. Best Pract Res Clin Rheumatol 26: 13–24. [DOI] [PubMed] [Google Scholar]
- Bikkavilli RK, Feigin ME, Malbon CC (2008). p38 Mitogen‐activated protein kinase regulates canonical Wnt‐beta‐catenin signaling by inactivation of GSK3beta. J Cell Sci 121: 3598–3607. [DOI] [PubMed] [Google Scholar]
- Borea PA, Gessi S, Merighi S, Varani K (2016). Adenosine as a multi‐signalling guardian angel in human diseases: when, where and how does it exert its protective effects? Trends Pharmacol Sci 37: 419–434. [DOI] [PubMed] [Google Scholar]
- Chan ES, Fernandez P, Merchant AA, Montesinos MC, Trzaska S, Desai A et al. (2006). Adenosine A2A receptors in diffuse dermal fibrosis: pathogenic role in human dermal fibroblasts and in a murine model of scleroderma. Arthritis Rheum 54: 2632–2642. [DOI] [PubMed] [Google Scholar]
- Chan ES, Liu H, Fernandez P, Luna A, Perez‐Aso M, Bujor AM et al. (2013). Adenosine A(2A) receptors promote collagen production by a Fli1‐ and CTGF‐mediated mechanism. Arthritis Res Ther 15: R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Long F (2013). beta‐Catenin promotes bone formation and suppresses bone resorption in postnatal growing mice. J Bone Miner Res 28: 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon SS, Cheah AY, Turley S, Nadesan P, Poon R, Clevers H et al. (2002). beta‐Catenin stabilization dysregulates mesenchymal cell proliferation, motility, and invasiveness and causes aggressive fibromatosis and hyperplastic cutaneous wounds. Proc Natl Acad Sci U S A 99: 6973–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheon SS, Wei Q, Gurung A, Youn A, Bright T, Poon R et al. (2006). Beta‐catenin regulates wound size and mediates the effect of TGF‐beta in cutaneous healing. FASEB J 20: 692–701. [DOI] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta‐catenin signaling in development and disease. Cell 127: 469–480. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T (2011). Ischemia and reperfusion‐‐from mechanism to translation. Nat Med 17: 1391–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H et al. (2007). Phosphorylation of beta‐catenin by AKT promotes beta‐catenin transcriptional activity. J Biol Chem 282: 11221–11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P, Trzaska S, Wilder T, Chiriboga L, Blackburn MR, Cronstein BN et al. (2008). Pharmacological blockade of A2A receptors prevents dermal fibrosis in a model of elevated tissue adenosine. Am J Pathol 172: 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P, Perez‐Aso M, Smith G, Wilder T, Trzaska S, Chiriboga L et al. (2013). Extracellular generation of adenosine by the ectonucleotidases CD39 and CD73 promotes dermal fibrosis. Am J Pathol 183: 1740–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE (2011). International union of basic and clinical pharmacology. LXXXI nomenclature and classification of adenosine receptors‐‐an update. Pharmacol Rev 63: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasko G, Cronstein BN (2004). Adenosine: an endogenous regulator of innate immunity. Trends Immunol 25: 33–39. [DOI] [PubMed] [Google Scholar]
- Hasko G, Linden J, Cronstein B, Pacher P (2008). Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7: 759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson WR Jr, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B et al. (2010). Inhibition of Wnt/beta‐catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A 107: 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hino S, Tanji C, Nakayama KI, Kikuchi A (2005). Phosphorylation of beta‐catenin by cyclic AMP‐dependent protein kinase stabilizes beta‐catenin through inhibition of its ubiquitination. Mol Cell Biol 25: 9063–9072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J, Ha JH, Chung SI, Yoon Y (2014). Beta‐catenin regulates NF‐kappaB activity and inflammatory cytokine expression in bronchial epithelial cells treated with lipopolysaccharide. Int J Mol Med 34: 632–638. [DOI] [PubMed] [Google Scholar]
- Karmouty‐Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD et al. (2012). The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J 26: 2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya Y, Habas R (2008). Wnt signal transduction pathways. Organogenesis 4: 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP (2006). HIF‐dependent induction of adenosine A2B receptor in hypoxia. FASEB J 20: 2242–2250. [DOI] [PubMed] [Google Scholar]
- Liang MH, Wendland JR, Chuang DM (2008). Lithium inhibits Smad3/4 transactivation via increased CREB activity induced by enhanced PKA and AKT signaling. Mol Cell Neurosci 37: 440–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mediero A, Perez‐Aso M, Cronstein BN (2013). Activation of adenosine A(2A) receptor reduces osteoclast formation via PKA‐ and ERK1/2‐mediated suppression of NFkappaB nuclear translocation. Br J Pharmacol 169: 1372–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mediero A, Perez‐Aso M, Cronstein BN (2014). Activation of EPAC1/2 is essential for osteoclast formation by modulating NFkappaB nuclear translocation and actin cytoskeleton rearrangements. FASEB J 28: 4901–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos MC, Desai A, Chen JF, Yee H, Schwarzschild MA, Fink JS et al. (2002). Adenosine promotes wound healing and mediates angiogenesis in response to tissue injury via occupancy of A(2A) receptors. Am J Pathol 160: 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro D, Agra N, Pestana A, Alonso J, Gonzalez‐Sancho JM (2010). The EWS/FLI1 oncogenic protein inhibits expression of the Wnt inhibitor DICKKOPF‐1 gene and antagonizes beta‐catenin/TCF‐mediated transcription. Carcinogenesis 31: 394–401. [DOI] [PubMed] [Google Scholar]
- Perez‐Aso M, Chiriboga L, Cronstein BN (2012). Pharmacological blockade of adenosine A2A receptors diminishes scarring. FASEB J 26: 4254–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Aso M, Mediero A, Cronstein BN (2013). Adenosine A2A receptor (A2AR) is a fine‐tune regulator of the collagen1:collagen3 balance. Purinergic Signal 9: 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Aso M, Fernandez P, Mediero A, Chan ES, Cronstein BN (2014). Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3. FASEB J 28: 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon R, Nik SA, Ahn J, Slade L, Alman BA (2009). Beta‐catenin and transforming growth factor beta have distinct roles regulating fibroblast cell motility and the induction of collagen lattice contraction. BMC Cell Biol 10: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K et al. (2003). A role for Wnt signalling in self‐renewal of haematopoietic stem cells. Nature 423: 409–414. [DOI] [PubMed] [Google Scholar]
- Rooney B, O'Donovan H, Gaffney A, Browne M, Faherty N, Curran SP et al. (2011). CTGF/CCN2 activates canonical Wnt signalling in mesangial cells through LRP6: implications for the pathogenesis of diabetic nephropathy. FEBS Lett 585: 531–538. [DOI] [PubMed] [Google Scholar]
- Saidak Z, Le Henaff C, Azzi S, Marty C, Da Nascimento S, Sonnet P et al. (2015). Wnt/beta‐catenin signaling mediates osteoblast differentiation triggered by peptide‐induced alpha5beta1 integrin priming in mesenchymal skeletal cells. J Biol Chem 290: 6903–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh G, Cronstein B (2016). Signaling pathways involving adenosine A and A receptors in wound healing and fibrosis. Purinergic Signal 12: 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin A, Kohan DE (2003). Physiology and pathology of endothelin‐1 in renal mesangium. Am J Physiol Renal Physiol 285: F579–F589. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Tetsuka T, Uranishi H, Okamoto T (2002). Inhibition of the NF‐kappaB transcriptional activity by protein kinase A. Eur J Biochem 269: 4559–4565. [DOI] [PubMed] [Google Scholar]
- Takemaru KI, Moon RT (2000). The transcriptional coactivator CBP interacts with beta‐catenin to activate gene expression. J Cell Biol 149: 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia‐Rojas C, Schuller A, Lindsay CB, Ureta RC, Mejias‐Reyes C, Hancke J et al. (2015). Andrographolide activates the canonical Wnt signalling pathway by a mechanism that implicates the non‐ATP competitive inhibition of GSK‐3beta: autoregulation of GSK‐3beta in vivo. Biochem J 466: 415–430. [DOI] [PubMed] [Google Scholar]
- van Es JH, Jay P, Gregorieff A, van Gijn ME, Jonkheer S, Hatzis P et al. (2005). Wnt signalling induces maturation of paneth cells in intestinal crypts. Nat Cell Biol 7: 381–386. [DOI] [PubMed] [Google Scholar]
- Wei J, Melichian D, Komura K, Hinchcliff M, Lam AP, Lafyatis R et al. (2011). Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum 63: 1707–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Nusse R (1998). Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol 14: 59–88. [DOI] [PubMed] [Google Scholar]
- Xie H, Tranguch S, Jia X, Zhang H, Das SK, Dey SK et al. (2008). Inactivation of nuclear Wnt‐beta‐catenin signaling limits blastocyst competency for implantation. Development 135: 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H et al. (2011). FoxM1 promotes beta‐catenin nuclear localization and controls Wnt target‐gene expression and glioma tumorigenesis. Cancer Cell 20: 427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang H et al. (2012). Interactions between beta‐catenin and transforming growth factor‐beta signaling pathways mediate epithelial‐mesenchymal transition and are dependent on the transcriptional co‐activator cAMP‐response element‐binding protein (CREB)‐binding protein (CBP). J Biol Chem 287: 7026–7038. [DOI] [PMC free article] [PubMed] [Google Scholar]