

Abstract
The present study aimed to identify the level of programmed death-1 (PD-1) expression in infiltrating cluster of differentiation (CD)4+ and CD8+ T cells isolated from lung cancer tissues, and investigated whether the level of PD-1 expression may be directly regulated by lung cancer cells via prostaglandin E2 (PGE2)-associated signaling pathways in patients with lung cancer. A total of 75 patients with lung cancer were enrolled in the present study. The percentage of infiltrating CD4+ and CD8+ T cells was determined by flow cytometry. ELISA was performed to evaluate the concentration of PGE2 in lung cancer tissue homogenate. The correlation between PGE2 and PD-1 expression levels in CD8+ T cells was assessed by Spearman's rank correlation test. The expression levels of PD-1 and PGE2 receptors were determined by reverse transcription-quantitative polymerase chain reaction and western blotting, respectively. The level of PD-1 expression in infiltrating CD8+ T cells was gradually increased as the stage of lung cancer increased. The level of PD-1 expression was also positively associated with the concentration of PGE2 in lung cancer tissues. Furthermore, the level of PD-1 expression was closely associated with the PGE2/EP2 and PGE2/EP4 signaling pathways. The activation of PGE2-associated EP2- and EP4-pathways may positively regulate the level of PD-1 in infiltrating CD8+ T cells, which results in immune tolerance in the lung cancer microenvironment.
Keywords: lung cancer, infiltrating T lymphocytes, programmed cell death-1, prostaglandin E2
Introduction
Lung cancer is the main cause of mortalities, accounting for 18% of all cancer-associated mortalities (1). Lung cancer has the highest incidence and mortality rates amongst all malignancies worldwide (2,3). The pathogenesis of lung cancer remains unclear. However, an inflammatory microenvironment consisting of infiltrating lymphocytes and secretary cytokines is recognized to be a key inducer of tumorigenesis and malignancy (4–6).
As a major cyclooxygenase-2-derived metabolite, prostaglandin E2 (PGE2) is well known as an important inflammatory factor, which is able to induce tumor growth and suppress immune functions by secreting into the tumor microenvironment (7,8). It has been revealed that PGE2 inhibits immune responses by upregulating the level of forkhead transcription factor 3 expression, which is known to promote the development of cluster of differentiation (CD)4+ and CD25+ regulatory T cells (Treg) (9).
Programmed cell death 1 (PD-1) belongs to the CD-28 family and is expressed on T cells, dendritic cells, natural killer cells, macrophages and B-cells. Programmed cell death ligand (PD-L)1 and PD-L2 are two major ligands of PD-1. PD-L1 is reported to be produced by T-cells, B cells and myeloid dendritic cells, and at low levels in the lungs, kidney, liver and heart. The activation of PD-1/PD-L1 signaling in tumors can inhibit T cell function and weaken the immune response, leading to a poor prognosis (10–12). Blocking a combination of PGE2 and PD-1 signaling has been reported to be therapeutic in chronic lymphocytic choriomeningitis virus infection by augmenting the numbers of functional virus-specific cytotoxic T lymphocytes via PGE2 receptors, EP2 and EP4 (9,13). However, whether a direct association exists between PGE2 and PD-1 remains unclear.
In the present study, the level of PD-1 expression in infiltrating CD4+ and CD8+ T cells isolated from lung cancer tissues was analyzed and whether the level of PD-1 expression in T cells may be directly regulated by PGE2 in lung cancer tissue homogenate, which my lead to immune inhibition, was investigated. Clarifying the immune tolerance of infiltrating lymphocytes would be useful for improving the immunotherapy of lung cancer.
Patients and methods
Patients
A total of 75 patients with lung cancer were recruited for the present study from The Third Hospital of Southern Medical University (Guangzhou, China) between August 2014 and October 2015. Patients with lung cancer enrolled in the present study were classified into four groups [stages I (n=20), II (n=25), III (n=17) and IV (n=13)], according to the pathological tumor-node-metastasis stage based upon the 8th edition of the American Joint Committee on Cancer Staging Manual (14). Any participants with systemic disorders or viral infections were excluded from the study.
Written informed consent was obtained from all patients prior to enrollment in the present study, and the experimental protocol was approved by the Ethics Committee of the Third Hospital of Southern Medical University. The demographic and clinical characteristics of patients are presented in Table I.
Table I.
Basic clinical characteristics of the patients in the present study.
| Parameters | Stage I, n=20 | Stage II, n=25 | Stage III, n=17 | Stage IV, n=13 |
|---|---|---|---|---|
| TNM stage | T1aN0M0 (11) | T1bN1M0 (7) | T1N2M0 (6) | T2N2M1a (8) |
| (no. of patients) | T1bN0M0 (9) | T2aN1M0 (10) | T2N2M0 (6) | T3N2M1a (5) |
| T2bN0M0 (8) | T3N1M0 (5) | |||
| Age, yearsa | 56 (42–63) | 58 (46–67) | 57 (44–65) | 56 (45–68) |
| Sex | ||||
| Female/male | 14/6 | 17/8 | 12/5 | 9/4 |
Age is presented as median and range. TNM, tumor-node-metastasis.
Tissues and isolation of lymphocytes
Fresh lung cancer tissue samples were cut from the center of the tumor block (40–50 mg). Following washing in ice-cold PBS, the tumor tissues were cut into 1 mm3 sections. A mechanical trituration method was used to obtain tissue homogenate. Single cells from lung cancer tissue homogenate were prepared by filtrating using a 100-mesh sieve. Subsequently, the cells were suspended in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific Inc., Waltham, MA, USA). Ficoll-Paque Plus (GE Healthcare, Chicago, IL, USA) was then added to the cell suspension to isolate infiltrating lymphocytes in lung cancer tissues using a density-gradient centrifugation method.
Fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (cat. no. 555346; dilution, 1:500), phycoerythrin (PE)-conjugated anti-CD8 (cat. no. 557086; dilution, 1:500) and isotype-matched controls (dilution, 1:500; all from BD Biosciences, San Jose, CA, USA) were used to stain the isolated lymphocytes at 37°C for 30 min. CD4+ T cells and CD8+ T cells were sorted using FACSAria II (BD Biosciences). All data were analyzed using Flow Jo software (version 7.6.2; Tree Star, Inc., Ashland, OR, USA).
Cell culture and ELISA
In order to investigate the association between PGE2 and PD-1, sorted infiltrating CD4+ and CD8+ T cells were cultured at a density of 1×105 cells/well in 24-well plates (Corning Incorporation NY, USA), were pre-coated with CD3/CD28 monoclonal antibodies and were maintained in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 0.1 mg/ml streptomycin. All cell culture media and supplements were purchased from Gibco (Thermo Fisher Scientific, Inc.). EP1 antagonist ONO-8711 (Cayman Chemical Company, Ann Arbor, MI, USA), EP2 antagonist ONO-AE1-259-01, EP3 antagonist ONO-AE5-599 (both from ONO Pharmaceutical Co., Osaka, Japan) and EP4 antagonist GW627368 (MedChem Express Co., Shanghai, China) were used to inhibit the corresponding signaling pathways.
The concentration of PGE2 in the supernatant of lung cancer tissue homogenate was determined by ELISA, according to the manufacturer's instructions (Uscn Life Sciences, Inc., Wuhan, China).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the T cell subsets of patients with lung cancer using TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions. Reverse transcription was performed using the RevertAid First Strand cDNA Synthesis kit (Thermo Fisher Scientific Inc.). The thermocycling conditions were as follows: 37°C for 30 min and 65°C for 10 min.
Gene-specific PCR amplification was performed using Power SYBR Green Master Mix (Thermo Fisher Scientific Inc.). The thermocycling conditions for qPCR were as follows: 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min. The relative level of gene expression was evaluated using the 2−ΔΔCq method (15) following normalization to the level of GAPDH expression. The primers used are presented in Table II.
Table II.
Primer sequences used in quantitative polymerase chain reaction.
| Primer | Primer sequences (5′-3′) |
|---|---|
| PD-1 | |
| Forward | AAGCTTATGTGGGTCCGGC |
| Reverse | GGATCCTCAAAGAGGCC |
| EP1 | |
| Forward | TCGCTTCGGCCTCCACCTTCTTTG |
| Reverse | CGTTGGGCCTCTGGTTGTGCTTAG |
| EP2 | |
| Forward | CCACGATGCTCCTGCTGCTT |
| Reverse | TCCACAAAGGTCAGTCTGTTT |
| EP3 | |
| Forward | CGGGGCTACGGAGGGGATGC |
| Reverse | ATGGCGCTGGCGATGAACAACGAG |
| EP4 | |
| Forward | GGTCATCTTACTCATCGCCACCTCTC |
| Reverse | TCCCACTAACCTCATCCACCAACAG |
| GAPDH | |
| Forward | GGTGGTCTCCTCTGACTTCAACA |
| Reverse | GTGGTCGTTGAGGGCAATG |
PD-1, programmed cell death 1; EP, prostaglandin E receptor.
Western blotting
The cells were lysed in RIPA buffer with protease inhibitors (Roche Diagnostics, Basel, Switzerland) on ice for 30 min and centrifuged for 10 min at 12,000 × g at 4°C. Protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Inc.). Subsequently, 30 µg protein/well were separated on a 10% SDS-PAGE gel and transferred onto polyvinylidene difluoride membranes (GE Healthcare). Membranes were blocked with 5% skimmed dry milk for 30 min at room temperature and were incubated overnight at 4°C with rabbit anti-human PD-1 monoclonal antibody (cat. no. ab214421; dilution, 1:1,000), rabbit anti-human EP-2 polyclonal antibody (cat. no. ab117270; dilution, 1:1,000), rabbit anti-human EP-4 polyclonal antibody (cat. no. ab45295; dilution, 1:1,000) and rabbit anti-human GAPDH monoclonal antibody (cat. no. ab181602; dilution, 1:2,000; all from Abcam, Cambridge, UK). The membranes were subsequently incubated with a goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (cat. no. sc-2004; dilution, 1:1,500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) for 2 h at 37°C. The blots were visualized using the enhanced chemiluminescence detection system (Beyotime Institute of Biotechnology, Haimen, China). The densitometry score was determined using Quantity One software (version 4.6; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
All data were expressed as the mean ± standard deviation. The Kruskal-Wallis and Dunn's multiple comparison tests were used to compare three or more groups of sample data. The comparison between two groups was analyzed by the Mann-Whitney non-parametric test. The correlations between variables were evaluated by the Spearman's rank correlation test. All statistical analyses were performed using GraphPad Prism software (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA). Two-sided P<0.05 was considered to indicate a statistically significant difference. All results were repeated three times.
Results
Expression of PD-1 in infiltrating CD8+ T lymphocytes is associated with the level of PGE2 in lung cancer tissue homogenate
It was revealed that the normal functions of T lymphocytes were suppressed in numerous types of solid tumors, resulting in reduced antitumor immunity and evasion of host immune surveillance (16,17). As an inflammatory factor, PD-1 exhibited marked immunosuppressive effects in a number of different types of tumors (18,19). In order to determine the change in the number of PD-1+ T cells during the progression of lung cancer, the levels of PD-1 expression in infiltrating CD4+ T cells and CD8+ T cells was determined in patients with lung cancer at four disease stages: Stage I (n=20), II (n=25), III (n=17) and IV (n=13).
Following isolation of tumor infiltrating lymphocytes from patient tissues, FITC-conjugated anti-CD4 and PE-conjugated anti-CD8 were used to label CD4+ and CD8+ T cells, respectively. As presented in Fig. 1A and B, the percentage of CD4+ T cells was not significantly altered in the four lung cancer groups. By contrast, the proportion of CD8+ T cells gradually decreased along with lung cancer development. Subsequently, the level of PD-1 expression in sorted CD4+ and CD8+ T cells was analyzed.
Figure 1.

Differential distribution of infiltrating CD4+ and CD8+ T cells in patients with lung cancer at various stages. (A) The gating strategy used in flow cytometric analysis. (B) Percentage of infiltrating CD4+ and CD8+ T lymphocytes isolated from lung cancer tissues. (C) The expression level of PD-1 in sorted CD4+ and CD8+ T cells as determined by quantitative polymerase chain reaction method. The data is presented as the mean ± standard deviation. *P<0.05. CD, cluster of differentiation; FSC, forward scatter; PD-1, programmed cell death 1; SSC, side scatter.
The PD-1 expression level in sorted CD8+ T cells was significantly increased in stages III and IV compared with stages I and II of disease (Fig. 1C). By contrast, there were no marked changes between the stages of disease in sorted CD4+ T cells (Fig. 1C). These results revealed that a major feature of the immune microenvironment in lung cancer tissues was the inhibition of CD8+ T cells rather than CD4+ T cells, and this immunosuppression may be mediated by the increased level of PD-1 expression on CD8+ T cells.
As a key pro-inflammatory factor, PGE2 is highly expressed in numerous types of solid tumors, including liver, prostate and lung cancer, mediating tumor proliferation and metastasis by secreting into the tumor microenvironment (7–9). To clarify whether PGE2 secreted by lung cancer cells directly affected PD-1 expression, the concentration of PGE2 in lung cancer tissue homogenate was detected using the ELISA method in the present study. As presented in Fig. 2A, the level of PGE2 expression was at similarly low levels at stages I and II of disease, but expression was significantly increased at stage III compared with stage I. Similarly, there was a marked increase in the level of PGE2 expression at stage IV compared with stage III. Considering that the pattern of changes in the levels of PGE2 was similar to the changes observed in the level of PD-1 expression in CD8+ T cells, the association between PGE2 and PD-1 expression was further analyzed. As presented in Fig. 2B, the level of PGE2 produced by lung cancer cells was positively correlated with the level of PD-1 expression in CD8+ T cells. This finding indicated that PGE2 may induce the inhibition of CD8+ T cells by promoting the level of PD-1 in the lung cancer microenvironment.
Figure 2.

Association between PGE2 and PD-1 level. (A) The concentration of PGE2 in lung cancer tissue homogenate was determined by ELISA. Data is presented as the mean ± standard deviation. *P<0.05. (B) The expression level of PD-1 in sorted CD8+ T cells was positively associated with the concentration of PGE2 in cancer tissues. Each data point represents an individual subject. PGE2, prostaglandin E2; PD-1, programmed cell death 1; CD, cluster of differentiation.
Expression level of PD-1 was regulated by PGE2 via EP2- and EP4-associated signaling pathways
The cellular effects of PGE2 are mediated by a family of G-protein-coupled receptors designated EP1, −2, −3 and −4. Of the four EP receptor subtypes, EP1 and EP3 generally elicit excitatory actions, whereas EP2 and EP4 elicit inhibitory actions on cellular function (20–22).
In the present study, sorted CD8+ T cells were stimulated by PGE2 in vitro, and the level of PD-1 expression was demonstrated to be upregulated in a time- and dose-dependent manner (Fig. 3A and B). In the present study, it was indicated that the levels of mRNA expression of EP2 and EP4 were all increased following PGE2 treatment, whereas there was no statically significant change in the production of EP1 or EP3 following treatment with 0.5 µM PGE2 for 6 h (Fig. 4A).
Figure 3.
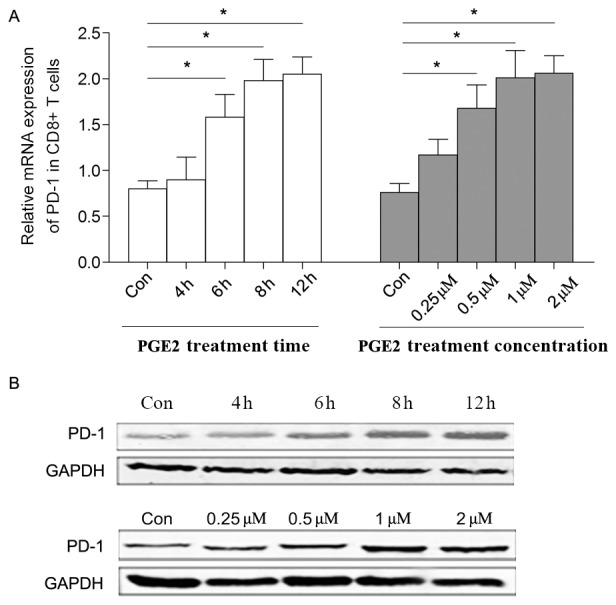
Effect of PGE2 on the level of PD-1 expression. PGE2 was added into the culture of sorted CD8+ T cells and a dose- and time-dependent effect of PGE2 on the PD-1 expression level was confirmed by (A) quantitative polymerase chain reaction and (B) western blot analysis. Data is presented as the mean ± standard deviation. *P<0.05. Con, control group with no PGE2 added; PGE2, prostaglandin E2; PD-1, programmed cell death 1; CD, cluster of differentiation.
Figure 4.

Differential effects of PGE2-associated pathways on the level of PD-1 expression. The culture medium was supplemented with 0.5 µM PGE2, and the sorted CD8+ T cells were stimulated for 6 h. (A) Potential receptors (EP1, EP2, EP3 and EP4) that may be associated with PGE2 stimulation in vitro were primarily screened using quantitative polymerase chain reaction analysis, which revealed that EP1 and EP3 levels were not significantly affected by PGE2 treatment. EP2 antagonist ONO-AE1-259-01 (5 nM) and EP4 antagonist GW627368 (100 nM) were used to inhibit the corresponding pathways in sorted CD8+ T cells for an additional 6 h. The level of PD-1 expression was subsequently detected by (B) quantitative polymerase chain reaction and (C) western blot analysis. Data is presented as the mean ± standard deviation. *P<0.05. The control group was treated with PGE2, but not with an antagonist. Con, control group; PGE2, prostaglandin E2; PD-1, programmed cell death-1; CD, cluster of differentiation.
To investigate the specific pathways through which PGE2 mediates its effects, the medium was supplemented with EP2 and EP4 antagonists for an additional 6 h to block the corresponding signaling pathways. The results demonstrated that the level of PD-1 expression was closely associated with the PGE2/EP2 and PGE2/EP4 signaling pathways (Fig. 4B and C). Therefore, the level of PD-1 expression in CD8+ T cells may be regulated by PGE2 via the EP2 and EP4 signaling pathways.
Notably, although there was no association between PD-1 expression in CD4+ T cells and the level of PGE2, it was indicated that the activation of PGE2 signaling may also increase the level of PD-1 expression in CD4+ T cells in vitro (Fig. 5). Therefore, complex regulatory mechanisms for PD-1 expression may exist in CD4+ T cells in vivo, which may involve other cytokines or signaling transduction pathways.
Figure 5.
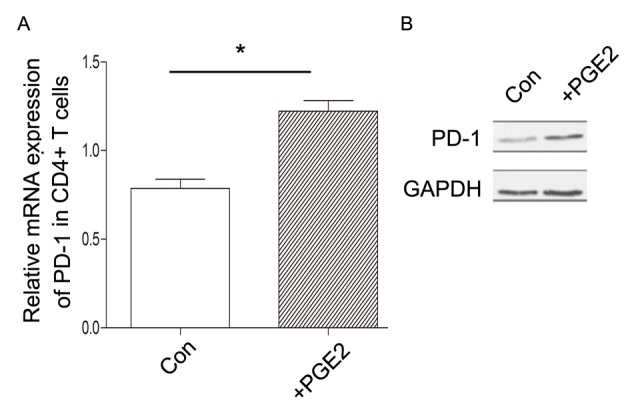
Effect of PGE2 on PD-1 expression in CD4+ T cells. The sorted CD4+ T cells were stimulated with 0.5 µM PGE2 for 6 h. The level of PD-1 expression in CD4+ T cells was detected by (A) quantitative polymerase chain reaction and (B) western blot analysis. Data is presented as the mean ± standard deviation. *P<0.05. Con, control group without PGE2 treatment; PGE2, prostaglandin E2; PD-1, programmed cell death-1; CD, cluster of differentiation.
Discussion
Cancer immunotherapy targeting the inflammatory microenvironment is a promising strategy in numerous types of solid tumors, including lung cancer. As a key factor widely expressed in the immune system and malignant tumor cells, PD-1 facilitates an inhibitory immune response. PD-L1 on lung cancer cells was revealed to be able to increase the apoptosis of antigen-specific T cells and to inhibit the activation of CD4+ and CD8+ T cells via PD-1, resulting in reduced antitumor immunity and evasion of host immune surveillance (23,24). In the present study, the level of PD-1 expression in infiltrating CD4+ and CD8+ T cells in lung cancer was investigated. Although the percentage of CD8+ T cells gradually decreased as the disease stage increased, there was an increased ratio of PD-1 expression in this subset of T cells.
Considering the immunosuppressive signaling induced by PD-1, it was hypothesized that the decreased percentage of CD8+ T cells was potentially mediated by high PD-1 expression. Conversely, there were no marked changes in the percentage of CD4+ T cells between different stages of disease, which may be due to the presence of various CD4+ T subsets which would lead to different and even opposite immune effects. Regulatory T cells (CD4+ CD25+) and other Th2-type T cells always act as immune inhibitors, whereas Th1-type T cells are able to enhance immune responses (25,26). Therefore, the PD-1 level of each different CD4+ T cell subset requires analyzing in order to determine its association with lung cancer development in future studies.
To investigate how lung cancer cells regulate the level of PD-1 expression, the present study focused on the inflammatory cytokine, PGE2, which is highly expressed in lung cancer tissues (27,28). It was revealed that the level of PGE2 expression in the lung cancer tissue homogenates was positively correlated with the level of PD-1 expression in CD8+ T cells. To clarify the potential mechanism, PGE2-associated signaling pathways were analyzed in the present study.
The signaling initiated from PGE2 is primarily mediated by its four receptors, EP1, EP2, EP3 and EP4. The role of EP1 is reported to be associated with intracellular calcium concentration, and promoter analysis of the EP2 and EP4 genes indicated the presence of several consensus sequences associated with inflammatory stimuli, including interleukin-6, nuclear factor-κB and activator protein 2 (29,30). A previous study revealed that EP4 is able to mediate PGE2-induced migration of A549 lung cancer cells (31). EP3 is distinct as it has multiple isoforms generated by alternative mRNA splicing, and EP3 has also been found to be crucial for tumor stroma formation and tumor growth (32,33). The functions of various PGE2 receptors in regulating the level of PD-1 expression were distinguished.
By treating the sorted T cells with PGE2 and the antagonists of PGE2 receptors in vitro in the present study, it was observed that the activation of EP2- and EP4-signaling was able to promote the level of PD-1 expression. Treatment with PGE2 was able to markedly promote PD-1 expression in CD4+ and CD8+ T cells in vitro. However, the level of PD-1 expression in CD4+ T cells was not correlated with the concentration of PGE2 in the tissue homogenates. Since CD4+ T cells contain multiple subsets with differential functions, the present study considered that more complex regulatory mechanisms may exist in order to regulate PD-1 expression in various CD4+ T subsets in vivo, which may be associated with the synergistic effect between PGE2 and other cytokines, as well as the crosstalk between CD4+ T cells and lung cancer cells via direct contact.
In conclusion, the results of the present study revealed that the level of PD-1 expression in the infiltrating CD8+ T cells of patients with lung cancer at various disease phases was positively regulated by PGE2 via the EP2- and EP4-associated signaling pathways. More thorough studies should be performed to reveal the characteristics of various T-cell subsets in the tumor microenvironment in order to be able to reverse immune tolerance and improve the immunotherapy of lung cancer.
Acknowledgements
The present study was supported by the Innovation Project from the Department of Education of Guangdong (grant no. 2014KTSCX043), the Norman Bethune Program of Jilin University (grant no. 2015328), and the Natural Science Foundation of Guangdong (grant no. 2015A030313264).
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Sun S, Schiller JH, Spinola M, Minna JD. New molecularly targeted therapies for lung cancer. J Clin Invest. 2007;117:2740–2750. doi: 10.1172/JCI31809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rolfo C, Sortino G, Smits E, Passiglia F, Bronte G, Castiglia M, Russo A, Santos ES, Janssens A, Pauwels P, Raez L. Immunotherapy: Is a minor god yet in the pantheon of treatments for lung cancer? Expert Rev Anticancer Ther. 2014;14:1173–1187. doi: 10.1586/14737140.2014.952287. [DOI] [PubMed] [Google Scholar]
- 4.Fazilleau N, Mark L, McHeyzer-Willams LJ, McHeyzer-Williams MG. Follicular helper T cells: Lineage and location. Immunity. 2009;30:324–335. doi: 10.1016/j.immuni.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006;66:7741–7747. doi: 10.1158/0008-5472.CAN-05-3766. [DOI] [PubMed] [Google Scholar]
- 6.Lee-Chang C, Bodogai M, Martin-Montalvo A, Wejksza K, Sanghvi M, Moaddel R, de Cabo R, Biragyn A. Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. J Immunol. 2013;191:4141–4151. doi: 10.4049/jimmunol.1300606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Callaghan G, Houston A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br J Pharmacol. 2015;172:5239–5250. doi: 10.1111/bph.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–293. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 9.Baratelli F, Lin Y, Zhu L, Yang SC, Heuzé-Vourc'h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005;175:1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 10.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 11.Park JJ, Omiya R, Matsumura Y, Sakoda Y, Kuramasu A, Augustine MM, Yao S, Tsushima F, Narazaki H, Anand S, et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood. 2010;116:1291–1298. doi: 10.1182/blood-2010-01-265975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 13.Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, Dominguez CX, Rosenberg DW, Kaech SM. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21:327–334. doi: 10.1038/nm.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rami-Porta R, Bolejack V, Giroux DJ, Chansky K, Crowley J, Asamura H, Goldstraw P, International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee, Advisory Board Members and Participating Institutions The IASLC lung cancer staging project: The new database to inform the eighth edition of the TNM classification of lung cancer. J Thorac Oncol. 2014;9:1618–1624. doi: 10.1097/JTO.0000000000000334. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, Lim KH, Weber A, Chow P, Chung A, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66:342–351. doi: 10.1136/gutjnl-2015-310814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, Fridman WH. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene. 2010;29:1093–1102. doi: 10.1038/onc.2009.416. [DOI] [PubMed] [Google Scholar]
- 18.Lin Z, Chen X, Li Z, Luo Y, Fang Z, Xu B, Han M. PD-1 antibody monotherapy for malignant melanoma: A systematic review and meta-analysis. PLoS One. 2016;11:e0160485. doi: 10.1371/journal.pone.0160485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Bao Z, Zhang X, Li F, Lai T, Cao C, Chen Z, Li W, Shen H, Ying S. Effectiveness and safety of PD-1/PD-L1 inhibitors in the treatment of solid tumors: A systematic review and meta-analysis. Oncotarget. 2017;8:59901–59914. doi: 10.18632/oncotarget.18316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang L, Yamagata N, Yadav R, Brandon S, Courtney RL, Morrow JD, Shyr Y, Boothby M, Joyce S, Carbone DP, Breyer RM. Cancer-associated immunodeficiency and dendritic cell abnormalities mediated by the prostaglandin EP2 receptor. J Clin Invest. 2003;111:727–735. doi: 10.1172/JCI16492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI200113640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von der Emde L, Goltz D, Latz S, Müller SC, Kristiansen G, Ellinger J, Syring I. Prostaglandin receptors EP1-4 as a potential marker for clinical outcome in urothelial bladder cancer. Am J Cancer Res. 2014;4:952–962. [PMC free article] [PubMed] [Google Scholar]
- 23.Han L, Liu F, Li R, Li Z, Chen X, Zhou Z, Zhang X, Hu T, Zhang Y, Young K, et al. Role of programmed death ligands in effective T-cell interactions in extranodal natural killer/T-cell lymphoma. Oncol Lett. 2014;8:1461–1469. doi: 10.3892/ol.2014.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology. 2012;1:1223–1225. doi: 10.4161/onci.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakaguchi S, Vignali DA, Rudensky AY, Niec RE, Waldmann H. The plasticity and stability of regulatory T cells. Nat Rev Immunol. 2013;13:461–467. doi: 10.1038/nri3464. [DOI] [PubMed] [Google Scholar]
- 26.Yun X, Shang Y, Li M. Effect of Lactobacillus salivarius on Th1/Th2 cytokines and the number of spleen CD4+ CD25+ Foxp3+ Treg in asthma Balb/c mouse. Int J Clin Exp Pathol. 2015;8:7661–7674. [PMC free article] [PubMed] [Google Scholar]
- 27.Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, Shen J, Li Y, Hu J, Meng Q, et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/β-catenin signalling pathway. Mol Immunol. 2017;90:197–210. doi: 10.1016/j.molimm.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Fan Y, Wang Y, Wang K. Prostaglandin E2 stimulates normal bronchial epithelial cell growth through induction of c-Jun and PDK1, a kinase implicated in oncogenesis. Respir Res. 2015;16:149. doi: 10.1186/s12931-015-0309-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quan Y, Jiang J, Dingledine R. EP2 receptor signaling pathways regulate classical activation of microglia. J Biol Chem. 2013;288:9293–9302. doi: 10.1074/jbc.M113.455816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho JS, Han IH, Lee HR, Lee HM. Prostaglandin E2 Induces IL-6 and IL-8 Production by the EP Receptors/Akt/NF-κB pathways in nasal polyp-derived fibroblasts. Allergy Asthma Immunol Res. 2014;6:449–457. doi: 10.4168/aair.2014.6.5.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JI, Lakshmikanthan V, Frilot N, Daaka Y. Prostaglandin E2 promotes lung cancer cell migration via EP4-betaArrestin1-c-Src signalsome. Mol Cancer Res. 2010;8:569–577. doi: 10.1158/1541-7786.MCR-09-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Israel DD, Regan JW. EP(3) prostanoid receptor isoforms utilize distinct mechanisms to regulate ERK 1/2 activation. Biochim Biophys Acta. 2009;1791:238–245. doi: 10.1016/j.bbalip.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amano H, Hayashi I, Endo H, Kitasato H, Yamashina S, Maruyama T, Kobayashi M, Satoh K, Narita M, Sugimoto Y, et al. Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J Exp Med. 2003;197:221–232. doi: 10.1084/jem.20021408. [DOI] [PMC free article] [PubMed] [Google Scholar]