

Abstract
Background
Statin treatment of hypercholesterolemia is accompanied also with depletion of the mevalonate intermediates, including farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) necessary for proper function of small GTPases. These include Ras proteins, prevalently mutated in pancreatic cancer. In our study, we evaluated the effect of three key intermediates of the mevalonate pathway on GFP-K-Ras protein localization and the gene expression profile in pancreatic cancer cells after exposure to individual statins.
Methods
These effects were tested on MiaPaCa-2 human pancreatic cancer cells carrying a K-Ras activating mutation (G12C) after exposure to individual statins (20 μM). The effect of statins (atorvastatin, lovastatin, simvastatin, fluvastatin, cerivastatin, rosuvastatin, and pitavastatin) and mevalonate intermediates on GFP-K-Ras protein translocation was analyzed using fluorescence microscopy. The changes in gene expression induced in MiaPaCa-2 cells treated with simvastatin, FPP, GGPP, and their combinations with simvastatin were examined by whole genome DNA microarray analysis.
Results
All tested statins efficiently inhibited K-Ras protein trafficking from cytoplasm to the cell membrane of the MiaPaCa-2 cells. The inhibitory effect of statins on GFP-K-Ras protein trafficking was partially prevented by addition of any of the mevalonate pathway’s intermediates tested. Expressions of genes involved in metabolic and signaling pathways modulated by simvastatin treatment was normalized by the concurrent addition of FPP or GGPP. K-Ras protein trafficking within the pancreatic cancer cells is effectively inhibited by the majority of statins; the inhibition is eliminated by isoprenoid intermediates of the mevalonate pathway.
Conclusions
Our data indicate that the anticancer effects of statins observed in numerous studies to a large extent are mediated through isoprenoid intermediates of the mevalonate pathway, as they influence expression of genes involved in multiple intracellular pathways.
Electronic supplementary material
The online version of this article (10.1186/s12944-017-0641-0) contains supplementary material, which is available to authorized users.
Keywords: Farnesyl pyrophosphate, Gene expression, Geranylgeranyl pyrophosphate, HMG-CoA reductase inhibitors, Isoprenoids, K-Ras oncogene, Mevalonate, Pancreatic cancer, Prenylation, Statins
Background
World-wide, statins are prescribed as hypocholesterolemic drugs preventing cardiovascular morbidity as well as mortality [1]. They lower cholesterol by inhibiting HMG-CoA reductase, the rate-limiting enzyme of the mevalonate pathway for de novo synthesis of cholesterol. However, the statins’ inhibitory action on HMG-CoA reductase also results in depletion of the downstream intermediate products of this pathway, such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which modify and target small GTPases, such as Ras proteins, to their site of action [2]. Farnesylation is also required for Ras protein anchoring in the plasma membrane, a compartment crucial for its proper signaling function [3]. Accordingly, the inhibition of prenylation (farnesylation and geranylgeranylation), became a plausible approach to modify tumor cell proliferation [4], but also seems to have therapeutic potential for cardiovascular and pulmonary medicine [5]. Importantly, in healthy cells, cholesterol and exogenous isoprenoids suppress HMG-CoA reductase via post-translational downregulation [6]. On the contrary, tumor cells are resistant to cholesterol-mediated suppression, although they remain sensitive to dietary, tumor-specific isoprenoid-mediated suppression of mevalonate synthesis [7]. The K-Ras signaling pathway is of special importance in pancreatic cancer, since >90% of these tumors bear activating mutations in K-Ras oncogene [8]. This oncogenic signaling pathway in pancreatic cancer is complex and passes through three major effector branches: Raf/Mek/Erk, PI3K/Pdk1/Akt and the Ral guanine nucleotide exchange factor pathway [9]. Because of this widespread effects, the K-Ras signaling pathway is a promising target in pancreatic cancer and various novel therapeutic approaches to suppress K-Ras signaling are currently under investigation. However, despite these comprehensive efforts, effective anti-Ras drugs have yet to reach the clinic [10].
Recently, we demonstrated substantial differences in the antitumor effects of individual statins used in clinical practice in an experimental model of human pancreatic cancer [11]. The effect of statins on the viability of pancreatic cells was, at least partially, caused by impairment of K-Ras protein trafficking, since all statins except for pravastatin efficiently inhibited GFP-K-Ras protein translocation from cytoplasm to the cell membrane [11].
Hence, the primary aim of the present study was to compare the effect of the mevalonate pathway intermediates on GFP-K-Ras protein dislocation in the pancreatic cancer cells treated with individual statins, and assess the role of these intermediates in the tumor-suppressive action of statins. The observations that addition of FPP or GGPP rescued the viability of cancer cells treated with statins [11] suggested that the effects of these compounds on gene expression changes were induced by statins. Thus, we also aimed to screen for differential gene expression of pancreatic cancer cells exposed to simvastatin, and to assess the gene expression profile after simultaneous exposure of these cells to FPP and GGPP in the whole genome expression array.
Methods
Materials
In each of the experiments, pure forms of the following statins were used: atorvastatin, lovastatin, simvastatin, fluvastatin, cerivastatin, rosuvastatin, and pitavastatin (all obtained from Alexis; San Diego, CA, USA); mevalonate, FPP, and GGPP were purchased from Sigma (St. Louis, MO, USA).
Cell cultures
The human pancreatic cancer cell line MiaPaCa-2 (ATCC, Manassas,VA, USA) carrying the activation mutation in the K-Ras oncogene (G12C) was maintained in the exponential phase of growth in a humidified atmosphere containing 5% CO2 at 37 °C, in DMEM medium supplemented with 10% fetal bovine serum. The final concentration of methanol, which was used for dissolving the experimental compounds, was below 1%. The cell line was authenticated at ATCC by STR profiling before distribution, and also re-authenticated by the end of the study by an external laboratory (Generi Biotech, Hradec Kralove, Czech Republic).
Transfection and imaging of GFP-K-Ras intracellular localization
MiaPaCa-2 cells were seeded in 6-well cell culture plates with sterile glass coverslips 5 h before transfection. Transfection with pEGFP-K-Ras plasmids, prepared as previously described [11], was carried out using FuGene® 6 (Roche, Basel, Switzerland) according to the manufacturer’s instructions. After 24 h of incubation, the medium was changed, and the tested compounds were added: statins to a final concentration of 20 μM, mevalonate to a final concentration of 600 μM, and FPP or GGPP to final concentrations of 17 μM. All experiments were performed in triplicates and repeated 4 times for all measurements.
The tested concentrations were chosen based on our previously published data [11], in which all the statins tested at the 20 μM concentration efficiently inhibited GFP-K-Ras protein accumulation in the plasma membrane, while FPP and GGPP at 17 μM substantially abrogated the tumor cell growth inhibitory effect of all statins tested in vitro. Simultaneously, mevalonate at the concentration of 600 μM completely eliminated the cytotoxic effect of all of the statins tested in vitro, whereas the equimolar concentration of mevalonate (17 μM) only had a partial effect [11].
After an additional 24 h, the cells were washed with phosphate buffered saline (PBS) and fixed with 4% formaldehyde in PBS for 20 min. Intracellular localization of GFP-K-Ras protein was visualized by fluorescence microscopy, using an Olympus IX-81 microscope and processing with Olympus xcellence RT software (Olympus, Tokyo, Japan).
DNA microarray analysis
The effects of 12 μM concentrations of simvastatin (corresponding to its IC50 value after 24 h [11]), and 17 μM FPP or GGPP on MiaPaCa-2 pancreatic cancer cell gene expression were investigated 24 h post-inoculation; the cells treated with vehicle (methanol <1%) served as control. Simvastatin was selected based on our previous study as the statin most efficiently inhibiting pancreatic cancer out of the clinically used statins [11].
The cells from two parallel cultures (10 cm2 culture dishes) were lysed in the stage of subconfluency using the RLT lysis buffer supplied in a RNeasy Mini Kit (Qiagen, USA). Total RNA was isolated by RNeasy Micro Kit (QIAGEN, USA) according to the procedure for animal cells. The quantity of RNA was measured by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies LLC, USA). The quality of the RNA was analyzed by an Agilent 2100 Bioanalyser (Agilent Technologies, CA, USA). Those RNA samples that had a RIN (RNA integrity number) above 9 were used for further analysis.
An Illumina HumanWG-6_V3 chip (Illumina, USA) was used for the microarray analysis following the standard protocol. Total RNA (150 ng) was amplified using an Illumina TotalPrep RNA Amplification Kit (Ambion, USA), and 1.5 μg of the amplified RNA was hybridized on the chips according to the manufacturer’s protocol. The analysis was performed in two biological replicates for the FPP, GGPP, and combination treatments, in four biological replicates for the simvastatin treatment, and in eight biological replicates for the control group.
The raw data was preprocessed using GenomeStudio software (version 1.9.0.24624; Illumina, CA, USA), with the beadarray [12] and the limma package [13] of the Bioconductor [14] within the R environment [15]: the transcription profiles were background corrected using a normal-exponential model, quantile normalized, and variance stabilized using base 2 logarithmic transformation. A moderated t-test was used to detect differentially expressed transcripts (within limma). Storey’s q-value (q < 0.05; [16]) and a minimally twofold change in expression intensity were required for significantly differentially transcribed genes. Batch effects were accounted for in the statistical model in limma, and eliminated for visualization purposes. The MIAME compliant data has been deposited to the ArrayExpress database (accession number E-MTAB-3263).
Gene set enrichment analysis (GSEA) was performed using the method of Tian et al. [17], and Fisher’s exact test on KEGG pathways [18]. We required statistical significance in both tests, with a false discovery rate of FDR < 0.001 in the more sensitive test of Tian, and FDR < 0.05 in Fisher’s exact test.
Quantitative real-time PCR
The genes for quantitative real-time PCR analysis were selected to validate DEG and their possible effect on KEGG pathways affected by simvastatin, FPP, and GGPP treatment. This analysis also served to validate changes detected by the microarray technology in general. The RNA derived from the same pool as for microarray experiments was used for quantitative real-time PCR analyses. Reverse transcription was performed with a QuantiTect Reverse Transcription Kit (QIAGEN Inc., USA). The RT-PCR was performed with a LightCycler 2.0 System using a LightCycler 480 DNA SYBR Green I Master kit (Roche Diagnostics, Germany), with the results analyzed by the LightCycler software. The resulting crossing point values were normalized using reference genes RPS9, TBP, and GAPDH. Relative fold changes of expression intensity in statin-treated against control samples were computed under assumption of perfect effectivity of PCR amplification. Statistical significance was estimated using Student’s t-test. All computations were performed within the R environment [15]. The list of amplicons/primers of target and housekeeping genes are provided in Additional file 1: Table S1.
Results
The effect of mevalonate, FPP, and GGPP on statin-mediated GFP-K-Ras protein translocation in the MiaPaCa-2 human pancreatic cancer cells
In our current study, we have confirmed the inhibitory effect of all clinically used statins on K-Ras protein accumulation in the cell plasma membrane (Fig. 1, column A). Pravastatin was not tested, since it failed to inhibit the GFP-K-Ras protein trafficking from the cytoplasm to the plasma membrane in our previous study [11]. This statin-mediated action was completely eliminated by an excess of mevalonate (Fig. 1, column B), indicating that this biologically important effect of statins is due to the inhibition of the mevalonate pathway.
Fig. 1.
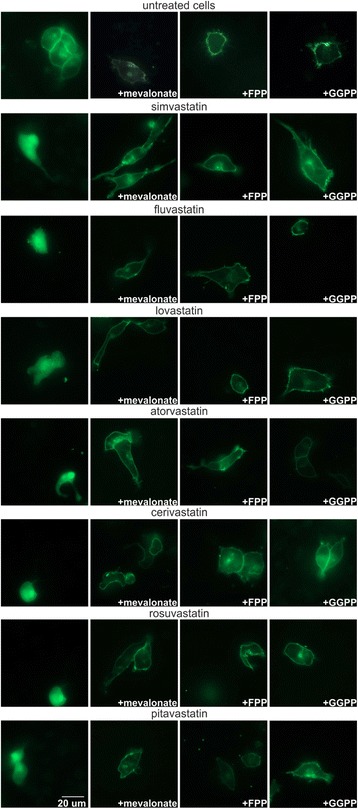
The effect of mevalonate (column B), FPP (column C), and GGPP (column D) on statin-mediated GFP-K-Ras protein translocation in the human pancreatic cancer cells. MiaPaCa-2 human pancreatic cancer cells expressing GFP-K-Ras were treated with statins (20 μM) together with mevalonate (600 μM), FPP (17 μM) or GGPP (17 μM), respectively, for 24 h. MiaPaCa-2 cells treated only with statins (column A) were used as the control of the statins’ effect on GFP-K-Ras protein localization. Simva = simvastatin; FPP = farnesyl pyrophosphate; GGPP = geranylgeranyl pyrophosphate
This observation was also corroborated by the same eliminating effects mediated by both FPP and GGPP even at much smaller concentrations (17 μM), although the trafficking of the GFP-K-Ras protein to the plasma membrane was not rescued in all of the treated cells (Fig. 1, columns C and D). We did not observe any apparent differences in these effects between both isoprenoid compounds, confirming the aforementioned statement that all of the tested statins affected the GFP-K-Ras protein translocation due to isoprenoids depletion caused by HMG-CoA reductase inhibition.
The effect of FPP and GGPP on simvastatin-mediated changes in the gene expression of MiaPaCa-2 human pancreatic cancer cells
Overall numbers of genes with significant changes in expression after simvastatin, FPP, or GGPP treatment of MiaPaCa-2 human pancreatic cancer cells are listed in Additional file 2: Table S2. There were about 200 genes affected by the simvastatin treatment. The changes in gene expression caused by simvastatin were eliminated by simultaneous addition of FPP with simvastatin, and the gene expression profile of the cells treated concurrently with simvastatin and FPP resembled control untreated cells (Fig. 2). In particular, we observed normalization of expression of KRAS, which is directly related to pancreatic cancer progression, and mevalonate pathway related genes (HMGCS1, MVD, MVK, and PDSS1). FPP per se influenced gene expression of the cells and the genes, whose expression was the most significantly affected by FPP treatment, were largely identical to those affected by the simvastatin treatment. We observed changes in the genes involved in DNA replication (POLA2, POLD4, PRIM1), RAS signaling related genes (RRAS, RHOB, RAB17), and genes of the mevalonate pathway (MVD, HMGCS1, HMGCR). However, the number of the genes affected by FPP was smaller and their change of expression weaker. GGPP alone did not affect gene expression of MiaPaCa-2 human pancreatic cancer cells; nevertheless, it eliminated almost all the changes in gene expression caused by simvastatin when the cells were treated simultaneously with GGPP and simvastatin (Table 1).
Fig. 2.
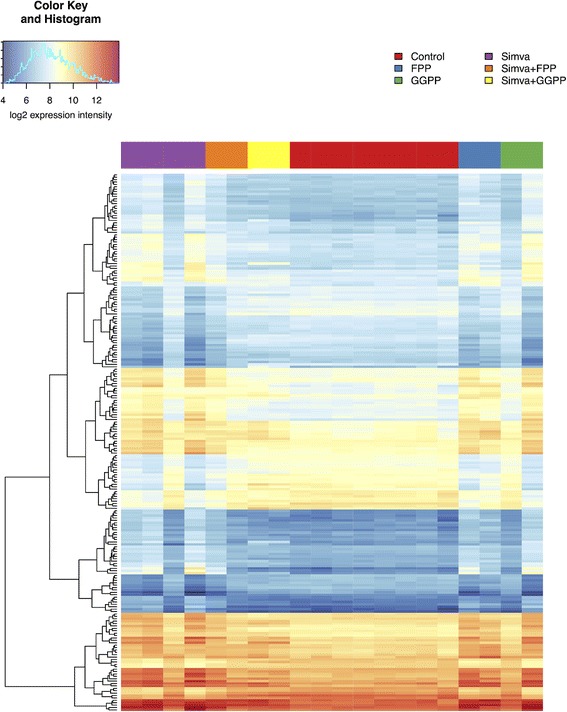
Heatmap illustrating log2-expression intensities of the genes that are differentially expressed after treatment of MiaPaCa-2 cells with simvastatin, FPP, GGPP, or their combinations. Only the differentially transcribed genes with at least two-fold change in expression intensity and q < 0.05 are shown. The list and sequence of the genes correspond to those stated in the Additional file 1: Table S4. Simva = simvastatin; FPP = farnesyl pyrophosphate; GGPP = geranylgeranyl pyrophosphate
Table 1.
Genes with significantly changed expression after simvastatin, FPP, and GGPP treatment of MiaPaCa-2 human pancreatic cancer cells
| Simva (S) | FPP (F) | Overlap (S) and (F) | Simva + FPP | GGPP | Simva + GGPP (SG) | Overlap (S) and (SG) | |
|---|---|---|---|---|---|---|---|
| Upregulated genes (No.) | 127 | 56 | 34 | 0 | 0 | 5 | 3 |
| Downregulated genes (No.) | 72 | 14 | 12 | 0 | 0 | 0 | 0 |
| Differentially expressed genes (No.) | 199 | 70 | 46 | 0 | 0 | 5 | 3 |
Number of genes differentially transcribed between samples treated with simvastatin (12 μM), FPP (17 μM) or GGPP (17 μM) compared to control samples of untreated cells, as well as for each combination of simvastatin and an inhibitor. Only the transcripts with a minimally two-fold change in expression intensity and q < 0.05 were counted. See Fig. 2 and Additional file 1: Table S4 for more details
Simva simvastatin, FPP farnesyl pyrophosphate, GGPP geranylgeranyl pyrophosphate
There were eight carcinogenesis-related KEGG pathways significantly affected by simvastatin treatment (Table 2, see Additional file 3: Table S3 for all affected pathways). Deregulation of these pathways was eliminated by simultaneous treatment of the cells with simvastatin and FPP or GGPP. Only two pathways remained deregulated, namely the DNA replication and cell cycle. Simultaneous treatment of the MiaPaCa-2 cells with simvastatin and GGPP eliminated the effect of simvastatin on DNA replication and cell cycle regulation more prominently than simvastatin in combination with FPP, accounting for better efficacy of GGPP on cancer cell viability [8]. The expression of a large number of genes implicated in DNA replication, cell cycle regulation, and mismatch repair were affected by FPP alone (Table 2). While only marginally significant after treatment by simvastatin, the genes involved in oxidative phosphorylation were significantly upregulated by treatment with FPP, simvastatin and FPP, and simvastatin and GGPP (Table 2, Fig. 3).
Table 2.
Signaling and metabolic pathways enriched by genes that change expression intensity after simvastatin, FPP, and GGPP treatment of MiaPaCa-2 human pancreatic cancer cells
| KEGG Path ID | Path name | Simva | FPP | Simva + FPP | GGPP | Simva + GGPP |
|---|---|---|---|---|---|---|
| hsa03030 | DNA replication | + | + | + | – | + |
| hsa04110 | Cell cycle | + | + | + | – | + |
| hsa03430 | Mismatch repair | + | + | – | – | – |
| hsa00100 | Steroid biosynthesis | + | – | – | – | – |
| hsa03440 | Homologous recombination | + | – | – | – | – |
| hsa04010 | MAPK signaling pathway | + | – | – | – | – |
| hsa00240 | Pyrimidine metabolism | + | – | – | – | – |
| hsa00190 | Oxidative phosphorylation | (−) | + | + | – | + |
Gene set enrichment analysis (GSEA) performed on KEGG signaling and metabolic pathways reveals the pathways enriched for genes that are differentially transcribed after simvastatin (12 μM) treatment, and the modulatory effects of FPP (17 μM) and GGPP (17 μM)
+ GSEA FDR < 0.001; − GSEA FDR > 0.001; (−) oxidative phosphorylation was only marginally significant in GSEA analysis by Tian with FDR < 0.05
Simva simvastatin, FPP farnesyl pyrophosphate, GGPP geranylgeranyl pyrophosphate, FDR false discovery rate
Fig. 3.
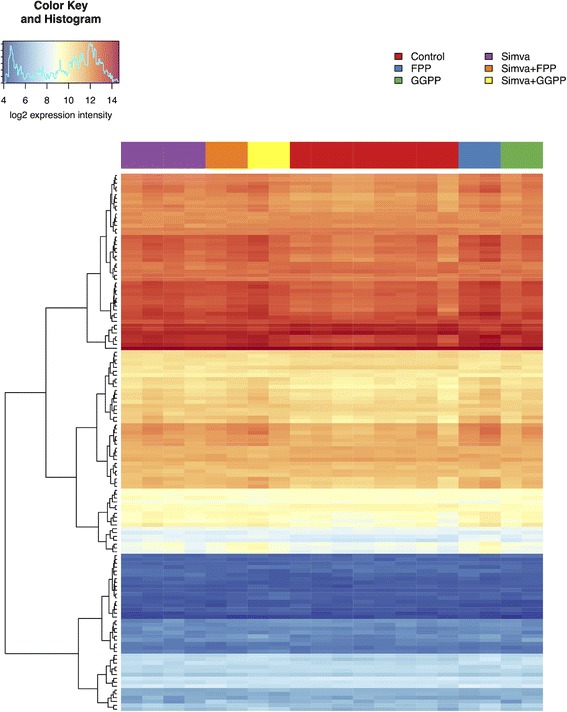
Heatmap illustrating log2-expression intensities of the genes that are involved in oxidative phosphorylation (KEGG pathway hsa00190). The list and sequence of the genes correspond to those stated in the Additional file 1: Table S4. Simva = simvastatin; FPP = farnesyl pyrophosphate; GGPP = geranylgeranyl pyrophosphate
Quantitative PCR analysis of selected genes revealed in most of the gene tested satisfactory agreement with the microarray data obtained, thus supporting their validity (Additional file 1: Table S4).
Discussion
Although prenylated proteins are estimated to constitute ~ 2% of the total mammalian proteome [19] (including key intracellular signaling proteins involved in carcinogenesis [20]), and many pre-clinical and clinical studies have implicated the role of protein prenylation in various human pathologies (such as certain cancers, cardiovascular, and/or neurodegenerative diseases), the therapeutic potential of isoprenoid synthesis inhibitors, including statins, still awaits to be explored in more detail [21]. In fact, various approaches to suppress K-Ras signaling are currently under investigation, although effective treatment is still far from the clinical application [10]. In this respect, it should be noted that statin use has been reported to significantly improve prognosis of pancreatic cancer in several [22–24], although not all clinical studies [25].
Consistent with this data, we previously observed that the inhibitory effects of statins on the viability of pancreatic cancer cells were partially prevented by concomitant addition of mevalonate, FPP, or GGPP, indicating the contribution of isoprenoids for growth and viability of pancreatic cancer cells [11]. Concurrently, Cafforio et al. have shown that the addition of mevalonate, GGPP, or FPP to cerivastatin-treated cells derived from human leukemia and myeloma resulted in complete recovery of both cell viability and proliferation; while no effect was observed by squalene supplementation [26]. However, other studies on the role of FPP and GGPP in the biology of various tumor-derived cells including cervical as well as head and neck squamous cell carcinomas [27], acute myeloid leukemia [28], or colon cancer [29] have produced different results. While in these studies FPP supplementation was shown to have had only a partial effect on statin-induced cancer cell apoptosis, that of GGPP was markedly higher. Even more so, in another in vitro study on human leukemia cells, inhibition of geranylgeranyl pyrophosphate synthase, leading to GGPP depletion with simultaneous FPP accumulation, led to enhanced statin-induced apoptosis of leukemia cells, while depletion of both isoprenoids resulted in the vanishing of the apoptotic effect [30] suggesting superiority of GGPP over FPP in the process of apoptosis in this particular model. A similar disparate effect was also demonstrated for colon cancer cells in a study by Agarwal et al., in which addition of GGPP prevented lovastatin-induced apoptosis, whereas FPP, even in high concentrations had no such effect [31]. These data apparently suggest different roles of both isoprenoids in carcinogenesis, and may also explain the discordant effects of these compounds on the gene expression profiles of the pancreatic cancer cells observed in our study.
We also demonstrated, that all the statins (except pravastatin) effectively eliminated the GFP-K-Ras protein trafficking to the cell membrane [11], suggesting that depletion of FPP caused by statin-mediated inhibition of the mevalonate pathway disables proper posttranslational modification and trafficking of the GFP-K-Ras protein. Our data extend previous observations by other researchers demonstrating the inhibitory effect of lovastatin on K-Ras protein translocation in mesothelioma [32] or NIH-3 T3 cells [33]. As proven in our current study, the localization of GFP-K-Ras protein has been effectively rescued with both FPP and GGPP supplementation, which is consistent with their rescue effect on the pancreatic cancer cell viability observed in our previous study [11]. The fact that both FPP and GGPP comparably rescued GFP-K-Ras protein trafficking to the cell plasma membrane (as suggested by Fig. 1) is likely to be the result of the cross-prenylation phenomenon [34]. The K-Ras protein preferentially undergoes farnesylation [34], but may also be geranylgeranylated, as demonstrated in cancer cells treated with farnesyltransferase inhibitors [35, 36].
Although both FPP and GGPP eliminated the block of GFP-K-Ras protein posttranslational modification caused by statins, the gene expression profiles after exposure of the simvastatin-treated cells to FPP and GGPP differed substantially. Genes whose expressions were most significantly affected by FPP treatment are largely identical with those affected by simvastatin treatment. This similarity was probably caused by the same inhibitory potential of FPP for HMG-CoA reductase, as reported for simvastatin [7, 37]. Nevertheless, this negative feedback was not observed for GGPP (Table 1), consistent with the fact that the excess of GGPP does not have an inhibitory effect on HMG-CoA reductase [38].
The observed data on gene expression in the pancreatic cancer cells treated with statin and isoprenoids might be explained by some complex regulatory activity, which FPP and GGPP exert on HMG-CoA reductase and other intracellular signaling pathways. These involve transcriptional (via modulation of sterol regulatory element-binding protein (SREBP) transcription factors), posttranscriptional (by accelerating protein acceleration, as described for statins), as well as posttranslational regulation [39]. In particular, changes could be seen in SREBP transcription factor regulated genes such as MVD, FDFT1, HMGCS1, and LSS, as well as RAS-related genes RRAS, RAB17, and RHOB. In addition, FPP and GGPP may directly (and specifically for each isoprenoid) influence the expression of Ras and Ras-related proteins [40]. Last but not least, both isoprenoids can directly modulate the activities of certain nuclear receptors. For instance, FPP can stimulate peroxisome proliferator-activated receptors (PPARs) [41], thyroid, estrogen, and glucocorticoid receptors [42], while GGPP itself has been shown to act as an antagonist of liver X receptor (LXR) [43]. Collectively, all this data demonstrates the multifaceted regulatory system exerted by isoprenoids. With a few exceptions, farnesylation and geranylgeranylation processes are highly substrate specific [19], further accounting for the observed changes in the gene expression profiles observed in our study.
We also observe an interesting effect of GGPP on those genes involved in the oxidative phosphorylation pathway. It is known that ubiquinone synthesis is blocked by the inhibitory action of statins [44], leading to the lack of ubiquinone in the mitochondria, with a consequent block of oxidative phosphorylation in the statin-treated cells. This, however, cannot be overcome by concomitant addition of GGPP or FPP, due to a lack of isopentenyl-PP (the FPP and GGPP upstream metabolites of the mevalonate pathway), which are also necessary for ubiquinone synthesis (Fig. 4). However, the results of the microarray analysis demonstrated that the process of the oxidative phosphorylation was only slightly affected by simvastatin, and was not affected by GGPP; however, the simultaneous treatment of cells with simvastatin and GGPP significantly changed the expression of genes encoding proteins of mitochondrial oxidative phosphorylation (Table 2, Fig. 3). In most of the cancer cell lines, the mitochondrial oxidative phosphorylation system is deficient, and the energy for growth (ATP production) is almost exclusively derived via glycolysis [45]. Whereas expressions of the genes involved in glycolysis were not affected in any of our experiments (data not shown), it seems that the anti-tumor effect of simvastatin on pancreatic cancer cells may also involve the inhibition of the mitochondrial oxidative phosphorylation, a phenomenon also described for simvastatin-treated prostate cancer cells [46].
Fig. 4.
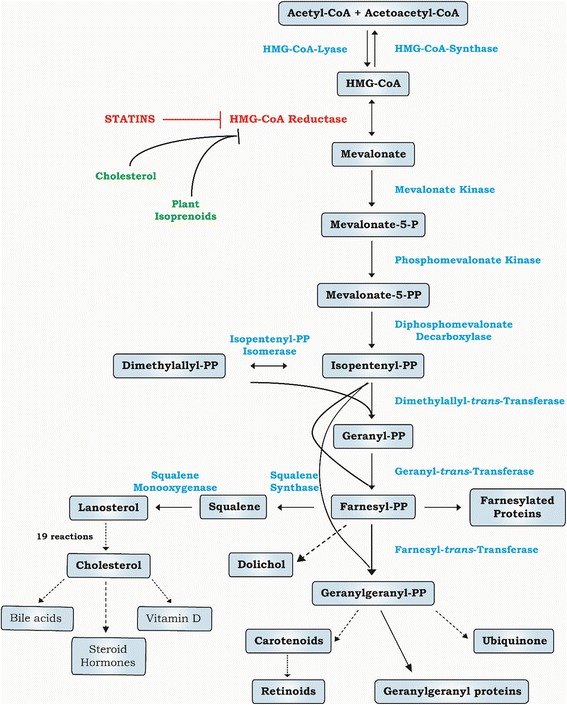
Scheme of the mevalonate pathway and isoprenoid synthesis. The mevalonate pathway (also known as the isoprenoid pathway) is an essential metabolic pathway in a human body producing a five-carbon building block called isopentenyl pyrophosphate (IPP). IPP is used to make fully functional biomolecules belonging into a large group of isoprenoids. Two intermediates in this pathway, FPP and GGPP, play also an immense role in post-translational modification of signaling proteins involved in a wide array of cellular functions including proliferation, and also carcinogenesis. The whole mevalonate pathway begins with acetyl-CoA, the rate limiting enzymatic step is catalyzed by HMG-CoA reductase (a therapeutic target of statins), whose inhibition leads to depletion signaling isoprenoids. Continuous and dashed lines represent one and multistep reaction, respectively. HMG-CoA reductase can be blocked by statins (in red)
One limitation of our study is high concentration of statins used. Since the clinical data on chemoadjuvant effects of clinically relevant doses of statins on pancreatic cancer are promising, it is apparent that further studies focused on determination of statins and isoprenoids in blood or even pancreatic cancer tissue might confirm our experimental data. In fact, such analytical methods are currently available [47] and are being used in our current studies [48]. Interestingly, gram doses of statins have already been used to treat cancer patients [49, 50], nevertheless it is certain that further clinical studies are needed to evaluate escalating doses of statins to define the therapeutic windows in oncology settings [51].
Conclusions
Our data indicate that the anticancer effects of statins observed in numerous studies to a large extent are mediated through isoprenoid intermediates of the mevalonate pathway, as they influence expression of genes involved in multiple intracellular pathways.
Additional files
List of primers used for quantitative real-time PCR analyses. Table S4. Quantitative RT PCR analysis of selected genes. (DOCX 18 kb)
Table S2. Differentially expressed genes after simvastatin, FPP, or GGPP treatment of MiaPaCa-2 human pancreatic cancer cells. (XLS 1507 kb)
Table S3. Gene set enrichment analysis on KEGG pathways affected by simvastatin, FPP, or GGPP treatment of MiaPaCa-2 human pancreatic cancer cells. (XLSX 22 kb)
Acknowledgements
NA
Funding
The research was supported by the European Social Fund CZ.1.07/2.3.00/30.0060, grants NT13112-4 and RVO-VFN64165/2017 given out by the Czech Ministry of Health, and the grant APVV-15-0217 given by the Slovak Research and Development Agency.
Availability of data and materials
The gene microarray data has been deposited to the ArrayExpress database (accession number E-MTAB-3263).
Abbreviations
- FPP
farnesyl pyrophosphate
- GFP
green fluorescent protein
- GGPP
geranylgeranyl pyrophosphate
- HMG-CoA
3-hydroxy-3-methylglutaryl-coenzyme A
- LXR
liver X receptor
- PPARs
peroxisome proliferator-activated receptors
- SREBP
sterol regulatory element-binding protein
Authors’ contributions
HG, SR and ZK performed all in vitro studies, JS, MK and HS performed all the bioinformatics analyses, WCD contributed to genetic analyses, TR and LV were responsible for study design and data interpretation. All authors contributed to writing of the whole manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
NA
Consent for publication
NA
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12944-017-0641-0) contains supplementary material, which is available to authorized users.
Contributor Information
Helena Gbelcová, Email: helena.gbelcova@fmed.uniba.sk.
Silvie Rimpelová, Email: Silvie.Rimpelova@vscht.cz.
Zdeněk Knejzlík, Email: Zdenek.Knejzlik@vscht.cz.
Michal Kolář, Email: kolarmi@img.cas.cz.
Hynek Strnad, Email: strnad@img.cas.cz.
Vanda Repiská, Email: vanda.repiska@fmed.uniba.sk.
Walter Cosimo D’Acunto, Email: waldacun@gmail.com.
Tomáš Ruml, Email: tomas.ruml@vscht.cz.
Libor Vítek, Email: vitek@cesnet.cz.
References
- 1.Lim S, Sakuma I, Quon MJ, Koh KK. Potentially important considerations in choosing specific statin treatments to reduce overall morbidity and mortality. Int J Cardiol. 2013;167(5):1696–1702. doi: 10.1016/j.ijcard.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 2.Ghaneh P, Neoptolemos J. A new approach to managing intraductal papillary mucinous pancreatic neoplasms. Gut. 2007;56(8):1041–1044. doi: 10.1136/gut.2006.113068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magee T, Marshall C. New insights into the interaction of Ras with the plasma membrane. Cell. 1999;98(1):9–12. doi: 10.1016/S0092-8674(00)80601-7. [DOI] [PubMed] [Google Scholar]
- 4.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–555. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 5.Yeganeh B, Wiechec E, Ande SR, Sharma P, Moghadam AR, Post M, et al. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol Ther. 2014;143(1):87–110. doi: 10.1016/j.pharmthera.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plemenitas A, Watson JA. Down-regulation of mammalian 3-hydroxy-3-methylglutaryl coenzyme a reductase activity with highly purified liposomal cholesterol. Eur J Biochem. 1999;266(2):317–326. doi: 10.1046/j.1432-1327.1999.00829.x. [DOI] [PubMed] [Google Scholar]
- 7.Mo H, Elson CE. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med (Maywood) 2004;229(7):567–585. doi: 10.1177/153537020422900701. [DOI] [PubMed] [Google Scholar]
- 8.Zeitouni D, Pylayeva-Gupta Y, Der CJ, Bryant KL. KRAS mutant pancreatic cancer: no lone path to an effective treatment. Cancers (Basel). 2016;8(4). doi:10.3390/cancers8040045. [DOI] [PMC free article] [PubMed]
- 9.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111(5):817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mann KM, Ying H, Juan J, Jenkins NA, Copeland NG. KRAS-related proteins in pancreatic cancer. Pharmacol Ther. 2016;168:29–42. doi: 10.1016/j.pharmthera.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Gbelcova H, Lenicek M, Zelenka J, Knejzlik Z, Dvorakova G, Zadinova M, et al. Differences in antitumor effects of various statins on human pancreatic cancer. Int J Cancer. 2008;122(6):1214–1221. doi: 10.1002/ijc.23242. [DOI] [PubMed] [Google Scholar]
- 12.Dunning MJ, Smith ML, Ritchie ME, Tavare S. Beadarray. R classes and methods for Illumina bead-based data. Bioinformatics. 2007;23(16):2183–2184. doi: 10.1093/bioinformatics/btm311. [DOI] [PubMed] [Google Scholar]
- 13.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21(9):2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 14.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.A language and environment for statistical computing [https://www.r-project.org/]. Accessed 16 Dec 2017.
- 16.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian L, Greenberg SA, Kong SW, Altschuler J, Kohane IS, Park PJ. Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci U S A. 2005;102(38):13544–13549. doi: 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen UT, Guo Z, Delon C, Wu Y, Deraeve C, Franzel B, et al. Analysis of the eukaryotic prenylome by isoprenoid affinity tagging. Nat Chem Biol. 2009;5(4):227–235. doi: 10.1038/nchembio.149. [DOI] [PubMed] [Google Scholar]
- 20.Caraglia M, Budillon A, Tagliaferri P, Marra M, Abbruzzese A, Caponigro F. Isoprenylation of intracellular proteins as a new target for the therapy of human neoplasms: preclinical and clinical implications. Curr Drug Targets. 2005;6(3):301–323. doi: 10.2174/1389450053765833. [DOI] [PubMed] [Google Scholar]
- 21.Park J, Matralis AN, Berghuis AM, Tsantrizos YS. Human isoprenoid synthase enzymes as therapeutic targets. Front Chem. 2014;2:50. doi: 10.3389/fchem.2014.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khurana V, Sheth A, Caldito G, Barkin JS. Statins reduce the risk of pancreatic cancer in humans: a case-control study of half a million veterans. Pancreas. 2007;34(2):260–265. doi: 10.1097/MPA.0b013e318030e963. [DOI] [PubMed] [Google Scholar]
- 23.Lee HS, Lee SH, Lee HJ, Chung MJ, Park JY, Park SW, et al. Statin use and its impact on survival in pancreatic cancer patients. Medicine (Baltimore) 2016;95(19):e3607. doi: 10.1097/MD.0000000000003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.H uang BZ, Chang JI, Li E, Xiang AH, Wu BU. Influence of statins and cholesterol on mortality among patients with pancreatic cancer. J Natl Cancer Inst. 2016;109(5). doi:10.1093/jnci/djw275. [DOI] [PubMed]
- 25.Cui X, Xie Y, Chen M, Li J, Liao X, Shen J, et al. Statin use and risk of pancreatic cancer: a meta-analysis. Cancer Causes Control. 2012;23(7):1099–1011. doi: 10.1007/s10552-012-9979-9. [DOI] [PubMed] [Google Scholar]
- 26.Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26(5):883–891. doi: 10.1093/carcin/bgi036. [DOI] [PubMed] [Google Scholar]
- 27.Dimitroulakos J, Marhin WH, Tokunaga J, Irish J, Gullane P, Penn LZ, et al. Microarray and biochemical analysis of lovastatin-induced apoptosis of squamous cell carcinomas. Neoplasia. 2002;4(4):337–346. doi: 10.1038/sj.neo.7900247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia Z, Tan MM, Wong WW, Dimitroulakos J, Minden MD, Penn LZ. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia. 2001;15(9):1398–1407. doi: 10.1038/sj.leu.2402196. [DOI] [PubMed] [Google Scholar]
- 29.Agarwal B, Rao CV, Bhendwal S, Ramey WR, Shirin H, Reddy BS, et al. Lovastatin augments sulindac-induced apoptosis in colon cancer cells and potentiates chemopreventive effects of sulindac. Gastroenterology. 1999;117(4):838–847. doi: 10.1016/S0016-5085(99)70342-2. [DOI] [PubMed] [Google Scholar]
- 30.Dudakovic A, Wiemer AJ, Lamb KM, Vonnahme LA, Dietz SE, et al. Inhibition of geranylgeranyl diphosphate synthase induces apoptosis through multiple mechanisms and displays synergy with inhibition of other isoprenoid biosynthetic enzymes. J Pharmacol Exp Ther. 2008;324(3):1028–1036. doi: 10.1124/jpet.107.132217. [DOI] [PubMed] [Google Scholar]
- 31.Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin Cancer Res. 1999;5(8):2223–2229. [PubMed] [Google Scholar]
- 32.Rubins JB, Greatens T, Kratzke RA, Tan AT, Polunovsky VA, Bitterman P. Lovastatin induces apoptosis in malignant mesothelioma cells. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1616–1622. doi: 10.1164/ajrccm.157.5.9709020. [DOI] [PubMed] [Google Scholar]
- 33.Chang MY, Jan MS, Won SJ, Liu HS. Ha-rasVal12 oncogene increases susceptibility of NIH/3T3 cells to lovastatin. Biochem Biophys Res Commun. 1998;248(1):62–68. doi: 10.1006/bbrc.1998.8911. [DOI] [PubMed] [Google Scholar]
- 34.Wang M, Casey PJ. Protein prenylation: unique fats make their mark on biology. Nat Rev Mol Cell Biol. 2016;17(2):110–122. doi: 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- 35.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez O, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272(22):14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 36.Rowell CA, Kowalczyk JJ, Lewis MD, Garcia AM. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem. 1997;272(22):14093–14097. doi: 10.1074/jbc.272.22.14093. [DOI] [PubMed] [Google Scholar]
- 37.Gardner RG, Hampton RY. A highly conserved signal controls degradation of 3-hydroxy-3-methylglutaryl-coenzyme a (HMG-CoA) reductase in eukaryotes. J Biol Chem. 1999;274(44):31671–31678. doi: 10.1074/jbc.274.44.31671. [DOI] [PubMed] [Google Scholar]
- 38.Correll CC, Ng L, Edwards PA. Identification of farnesol as the non-sterol derivative of mevalonic acid required for the accelerated degradation of 3-hydroxy-3-methylglutaryl-coenzyme a reductase. J Biol Chem. 1994;269(26):17390–17393. [PubMed] [Google Scholar]
- 39.Holstein SA, Hohl RJ. Isoprenoids: remarkable diversity of form and function. Lipids. 2004;39(4):293–309. doi: 10.1007/s11745-004-1233-3. [DOI] [PubMed] [Google Scholar]
- 40.Holstein SA, Wohlford-Lenane CL, Hohl RJ. Isoprenoids influence expression of Ras and Ras-related proteins. Biochemistry. 2002;41(46):13698–13704. doi: 10.1021/bi026251x. [DOI] [PubMed] [Google Scholar]
- 41.Liliom K, Tsukahara T, Tsukahara R, Zelman-Femiak M, Swiezewska E, Tigyi G. Farnesyl phosphates are endogenous ligands of lysophosphatidic acid receptors: inhibition of LPA GPCR and activation of PPARs. Biochim Biophys Acta. 2006;1761(12):1506–1514. doi: 10.1016/j.bbalip.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das S, Schapira M, Tomic-Canic M, Goyanka R, Cardozo T, Samuels HH. Farnesyl pyrophosphate is a novel transcriptional activator for a subset of nuclear hormone receptors. Mol Endocrinol. 2007;21(11):2672–2686. doi: 10.1210/me.2007-0080. [DOI] [PubMed] [Google Scholar]
- 43.Gan X, Kaplan R, Menke JG, MacNaul K, Chen Y, Sparrow CP, et al. Dual mechanisms of ABCA1 regulation by geranylgeranyl pyrophosphate. J Biol Chem. 2001;276(52):48702–48708. doi: 10.1074/jbc.M109402200. [DOI] [PubMed] [Google Scholar]
- 44.Maltese WA, Aprille JR. Relation of mevalonate synthesis to mitochondrial ubiquinone content and respiratory function in cultured neuroblastoma cells. J Biol Chem. 1985;260:11524–11529. [PubMed] [Google Scholar]
- 45.Pedersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- 46.Oliveira KA, Zecchin KG, Alberici LC, Castilho RF, Vercesi AE. Simvastatin inducing PC3 prostate cancer cell necrosis mediated by calcineurin and mitochondrial dysfunction. J Bioenerg Biomembr. 2008;40(4):307–314. doi: 10.1007/s10863-008-9155-9. [DOI] [PubMed] [Google Scholar]
- 47.Kosek V, Stranska M, Fenclova M, Ruml T, Vitek L, Hajslova J. High resolution mass spectrometry based method applicable for a wide range of 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase inhibitors in blood serum including intermediates and products of the cholesterol biosynthetic pathway. J Chrom A. 2017;1489:86–94. doi: 10.1016/j.chroma.2017.01.084. [DOI] [PubMed] [Google Scholar]
- 48.Gbelcova H, Rimpelova S, Ruml T, Fenclova M, Kosek V, Hajslova J, et al. Variability in statin-induced changes in gene expression profiles of pancreatic cancer. Sci Rep. 2017;7:44219. doi: 10.1038/srep44219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thibault A, Samid D, Tompkins AC, Figg WD, Cooper MR, Hohl RJ, et al. Phase I study of lovastatin, an inhibitor of the mevalonate pathway, in patients with cancer. Clin Cancer Res. 1996;2(3):483–491. [PubMed] [Google Scholar]
- 50.Kim WS, Kim MM, Choi HJ, Yoon SS, Lee MH, Park K, et al. Phase II study of high-dose lovastatin in patients with advanced gastric adenocarcinoma. Investig New Drugs. 2001;19(1):81–83. doi: 10.1023/A:1006481423298. [DOI] [PubMed] [Google Scholar]
- 51.de Wolf E, Abdullah MI, Jones SM, Menezes K, Moss DM, Drijfhout FP, et al. Dietary geranylgeraniol can limit the activity of pitavastatin as a potential treatment for drugresistant ovarian cancer. Sci Rep. 2017;7:5410. doi: 10.1038/s41598-017-05595-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of primers used for quantitative real-time PCR analyses. Table S4. Quantitative RT PCR analysis of selected genes. (DOCX 18 kb)
Table S2. Differentially expressed genes after simvastatin, FPP, or GGPP treatment of MiaPaCa-2 human pancreatic cancer cells. (XLS 1507 kb)
Table S3. Gene set enrichment analysis on KEGG pathways affected by simvastatin, FPP, or GGPP treatment of MiaPaCa-2 human pancreatic cancer cells. (XLSX 22 kb)
Data Availability Statement
The gene microarray data has been deposited to the ArrayExpress database (accession number E-MTAB-3263).