

ABSTRACT
ZipA is essential for cell division in Escherichia coli, acting early in the process to anchor polymers of FtsZ to the cytoplasmic membrane. Along with FtsA, FtsZ and ZipA form a proto-ring at midcell that recruits additional proteins to eventually build the division septum. Cells carrying the thermosensitive zipA1 allele divide fairly normally at 30°C in rich medium but cease dividing at temperatures above 34°C, forming long filaments. In a search for suppressors of the zipA1 allele, we found that deletions of specific genes involved in amino acid biosynthesis could partially rescue cell growth and division at 34°C or 37°C but not at 42°C. Notably, although a diverse group of amino acid biosynthesis gene deletions could partially rescue the growth of zipA1 cells at 34°C, only deletions of genes related to the biosynthesis of threonine, glycine, serine, and methionine could rescue growth at 37°C. Adding exogenous pyridoxal 5-phosphate (PLP), a cofactor for many of the enzymes affected by this study, partially suppressed zipA1 mutant thermosensitivity. For many of the deletions, PLP had an additive rescuing effect on the zipA1 mutant. Moreover, added PLP partially suppressed the thermosensitivity of ftsQ and ftsK mutants and weakly suppressed an ftsI mutant, but it failed to suppress ftsA or ftsZ thermosensitive mutants. Along with the ability of a deletion of metC to partially suppress the ftsK mutant, our results suggest that perturbations of amino acid metabolic pathways, particularly those that redirect the flow of carbon away from the synthesis of threonine, glycine, or methionine, are able to partially rescue some cell division defects.
IMPORTANCE Cell division of bacteria, such as Escherichia coli, is essential for their successful colonization. It is becoming increasingly clear that nutritional status and central metabolism can affect bacterial size and shape; for example, a metabolic enzyme (OpgH) can moonlight as a regulator of FtsZ, an essential cell division protein. Here, we demonstrate a link between amino acid metabolism and ZipA, another essential cell division protein that binds directly to FtsZ and tethers it to the cytoplasmic membrane. Our evidence suggests that altering flux through the methionine-threonine-glycine-serine pathways and supplementing with the enzyme cofactor pyridoxal-5-phosphate can partially compensate for an otherwise lethal defect in ZipA, as well as several other cell division proteins.
KEYWORDS: Escherichia coli, FtsZ, ZipA, amino acid biosynthesis, cell division
INTRODUCTION
To divide, Escherichia coli cells use a number of essential proteins to form a cytokinetic ring at midcell and synthesize a cell division septum (1). Three of these, FtsZ, FtsA, and ZipA, are required for assembly of the initial ring structure, called the proto-ring (2). The tubulin-like FtsZ protein forms dynamic treadmilling polymers that are attached to the inner surface of the cytoplasmic membrane by FtsA and ZipA (3). Together, these three proteins form several mobile complexes that recruit additional cell division proteins, including enzymes that synthesize septal peptidoglycan, which build the division septum (3–6).
FtsZ can still localize to the ring structure in the absence of either FtsA or ZipA, but the rings are not able to proceed in division, indicating that FtsA and ZipA have partially overlapping functions (7–10). Notably, the essential function of ZipA can be completely bypassed in certain cases, including in mutants of FtsA or FtsL, or when FtsN is overproduced, suggesting that ZipA is not an indispensable structural component of the septal ring but perhaps part of a checkpoint mechanism (11–15). FtsA is attached to the membrane with an amphipathic helix (16), theoretically allowing it to bind reversibly, while ZipA is bound permanently to the membrane through an N-terminal transmembrane domain (17). ZipA interacts with FtsZ through a C-terminal FtsZ-binding domain (18, 19); its two functional domains are connected by an intrinsically disordered peptide linker (20). ZipA enhances the bundling of FtsZ protofilaments in vitro (17, 21). Consistent with this idea, a single amino acid substitution in FtsZ that increases self-bundling can also bypass the need for ZipA (22).
Studies of essential bacterial cell division genes have been facilitated by conditional mutants, particularly thermosensitive (ts) mutants. Such mutants can divide at lower temperatures, such as 30°C, but fail to divide at nonpermissive temperatures, such as 42°C, conveniently allowing the rapid inactivation of cell division by a simple temperature shift. There is only one known ts mutant of zipA, called zipA1, which consists of one missense mutation in the N-terminal transmembrane domain and three missense mutations in the FtsZ binding domain (9). Cells harboring the zipA1 allele divide fairly normally at 30°C in rich medium, although not as well as wild-type (WT) cells, but rapidly cease division when shifted from 30°C to 42°C, forming long filaments with multiple nucleoids (9).
In this study, we isolated and characterized thermoresistant suppressors of the zipA1 allele that are not in other cell division genes but are instead in genes involved in amino acid metabolism. In particular, we find that inactivating genes involved in methionine-threonine-glycine biosynthesis or adding pyridoxal-5-phosphate cofactor suppresses the thermosensitivity of the zipA1 mutant and some, but not all, thermosensitive cell division mutants. Our evidence supports a novel connection between amino acid biosynthesis and cell division.
RESULTS
The original zipA1 mutant in W3110 exhibits medium-dependent growth and division defects at the permissive temperature that are partially suppressed by added Casamino Acids, glycine, or l-threonine.
Cells of the wild-type E. coli parent strain W3110 and its zipA1 mutant derivative WM2991 grew and divided normally at the permissive temperature of 30°C, although some WM2991 cells were slightly longer (Fig. 1A and G), probably because of residual defects of the zipA1 allele at this temperature. However, when we grew these two strains in minimal medium (either morpholinepropanesulfonic acid [MOPS] or M9), we were surprised to find that both grew poorly (Fig. 1M). Although W3110 cells remained short under these growth conditions (Fig. 1B and C), WM2991 cells became highly filamentous (Fig. 1H and I), indicating that any small cell division defect seen in LB was greatly exacerbated in minimal medium.
FIG 1.
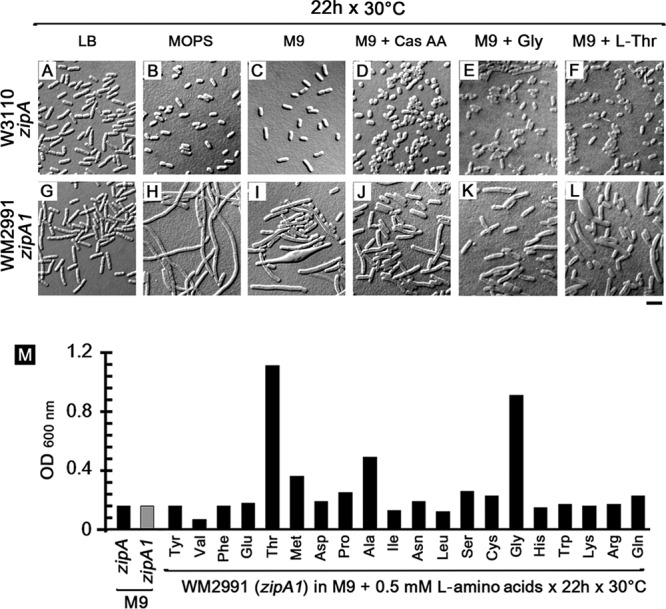
The zipA1 mutant in W3110 displays a medium-dependent cell division defect at the permissive temperature. Wild-type E. coli W3110 exhibited normal cell morphology when grown at 30°C in LB (A), MOPS (B), M9 (C), M9 plus 1% Casamino Acids (Cas AA) (D), M9 plus 0.5 mM glycine (E), or M9 plus 0.5 mM l-threonine (F). The thermosensitive zipA1 mutant (WM2991) was slightly elongated in LB (G), filamented in MOPS (H), and filamented with some misshapen cells in M9 (I), and these defects were partially suppressed when M9 was supplemented with 1% Casamino Acids (J), glycine (K), or l-threonine (L). (M) The bar graph represents the growth of W3110 (wild-type zipA) and WM2991 (zipA1 allele) in M9 and the effects of different l-amino acids on the growth of WM2991 after 22 h at 30°C. Scale bar, 4 μm.
These medium-dependent defects in cell growth and division prompted us to ask whether adding nutrients back to M9 medium could suppress these defects. We found that the addition of Casamino Acids partially suppressed the filamentation of WM2991 cells (Fig. 1J). We then added back individual amino acids to WM2991 cells grown in M9 and found that only glycine (Fig. 1K) or l-threonine (Fig. 1L) could significantly suppress the cell division and growth defects (Fig. 1M). Even though WM2991 and the W3110 parent exhibited similar growth defects in M9, the fact that cell division defects in the zipA1 mutant were medium dependent and specifically suppressed by glycine or l-threonine prompted us to consider a possible link between cell division and the glycine-threonine biosynthesis pathway.
To determine whether the observed medium-dependent effects of the zipA1 allele on growth and division were specific to the W3110 strain background, we used a linked nupC::Tn10 marker to transduce the zipA1 allele by P1 transduction into MG1655, creating WM5337. Cells of WM5337 were moderately filamentous at 30°C in both M9 (see Fig. S1D in the supplemental material) and in LB at 30°C (Fig. S1E), presumably because of residual thermosensitivity of the zipA1 allele. As expected, WM5337 cells were severely filamentous in LB at the nonpermissive temperature of 42°C (Fig. S1F), and they grew well in a spot assay in LB only at 30°C (Fig. S1G) but not at 42°C (Fig. S1H). A plasmid expressing wild-type zipA was able to complement WM5337 for growth at 42°C (Fig. S1H), while the same plasmid expressing the zipA1 gene failed to complement. Conversely, when we converted WM2991 (W3110 zipA1) to zipA+ by transduction creating WM5322, the filamentation phenotype either in M9 (Fig. S1A) or LB (Fig. S1B and C) was abolished. We conclude that zipA1 confers a thermosensitive cell division phenotype on MG1655 as well as W3110.
However, we also found that when the zipA1 allele was introduced into the MG1655 background (WM5337), the strain grew more robustly in M9 compared to the equivalent W3110 derivative, WM2991. This, along with the lack of a strong medium-dependent cell division phenotype in WM5337, led us to consider that W3110 itself is auxotrophic under nutrient deprivation conditions. To test this more definitively, we measured the cell densities of W3110, WM2991, MG1655, and WM5337 grown in parallel at 30°C for 24 h in LB, MOPS, and M9. Even in rich LB medium, MG1655 and its zipA1 mutant derivative WM5337 grew slightly better than W3110 and its zipA1 mutant derivative WM2991 (Fig. S2A). However, these strain-background growth differences were much more evident in MOPS (Fig. S2B) and, at least for W3110, in M9 (Fig. S2C). The growth defects of W3110 and WM2991 were greatly suppressed when glycine or l-threonine was added to MOPS (Fig. S2D and E) or M9 (Fig. S2G and H) but not when l-methionine was added (Fig. S2F and I), indicating that W3110 itself is starved for glycine or l-threonine. Although the reasons for this are not known, we chose to use the MG1655 background for all further studies of the zipA1 allele to eliminate confounding effects on cell division and growth caused by the W3110 background.
Blocking l-threonine biosynthesis partially suppresses zipA1 mutant growth and division defects.
Prior to our realization that the zipA1 mutant strain WM2991 (and its parent W3110) seemed to be auxotrophic for threonine and glycine and thus differed in important ways from MG1655 derivatives, we asked whether l-threonine had any effect on the zipA1 allele in the MG1655 strain background by deleting genes required for threonine biosynthesis (thrA, thrB, and thrC) (Fig. S3). Although we could not test effects in M9 medium because cells cannot grow without added l-threonine, we decided to explore whether there were any effects of these deletions on the zipA1 mutant in rich medium. In LB broth at the permissive temperature (30°C), we found that the zipA1 strain and all three thr mutant derivatives of the zipA1 strain displayed normal growth and morphology (data not shown). In LB at 42°C, cells of the zipA1 parent (Fig. 2A) and the zipA1 ΔthrA double mutant (Fig. 2B) grew to high density but were very filamentous, as expected. However, we noticed many more short cells in the zipA1 ΔthrB (Fig. 2C) and zipA1 ΔthrC (Fig. 2D) double mutants than in the zipA1 and zipA1 ΔthrA mutants, which were mostly filaments (Fig. 2A and B). Despite this hint of suppression in broth at 42°C, none of the double mutants formed colonies on LB plates in spot assays at 42°C (Fig. 2H), indicating that the ΔthrB and ΔthrC mutants could not completely bypass ZipA at the most nonpermissive temperature.
FIG 2.
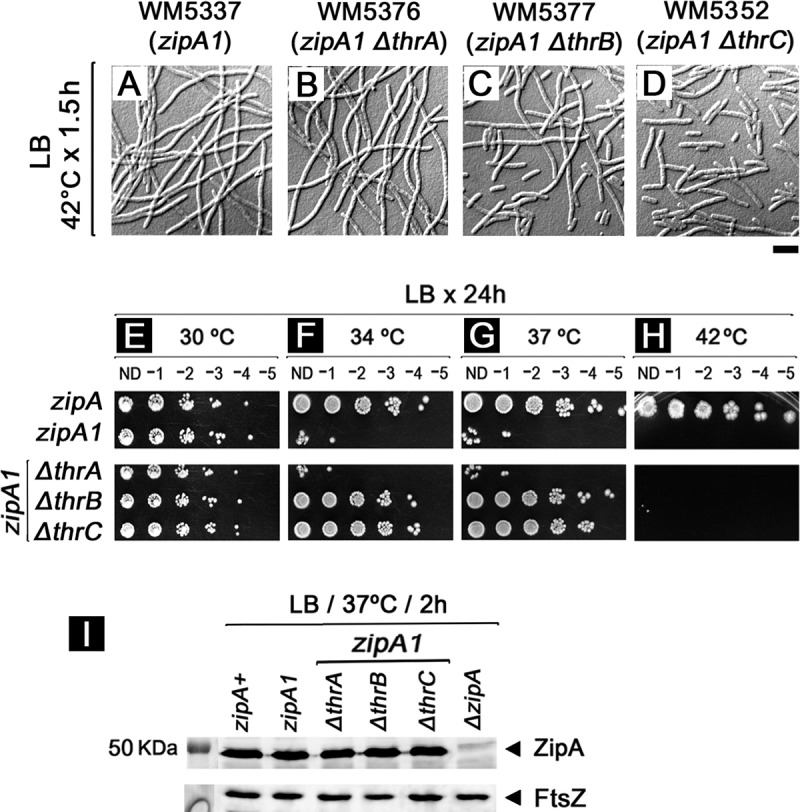
Defects in l-threonine biosynthesis partially suppress zipA1 mutant thermosensitivity. (A to D) WM5337 and mutants lacking thrA, thrB, or thrC were grown for 1.5 h at 42°C in LB. (E to H) Viability was measured by spotting assay on LB at 30°C (E), 34°C (F), 37°C (G), or 42°C (H) for 24 h. ND, no dilution. (I) The levels of ZipA and FtsZ protein in indicated cells grown at 37°C for 2 h were determined by Western blotting using antibody against ZipA or FtsZ.
Because they showed some suppression of filamentation at 42°C, we then tested whether the ΔthrB or ΔthrC mutation might suppress the zipA1 mutant at potentially less restrictive temperatures (34 and 37°C). Although both temperatures were still lethal for the zipA1 parent strain, they allowed partial suppression of zipA1 mutant thermosensitivity in the ΔthrB and ΔthrC derivatives (Fig. 2F and G). The rates of exponential growth of the wild-type parent and the partially suppressed zipA1 ΔthrB and zipA1 ΔthrC derivatives at 37°C were similar to those of nonsuppressed zipA1 and zipA1 ΔthrA mutant strains, although the zipA1 and zipA1 ΔthrA mutant strains reached lower cell densities, as expected (Fig. S4). At the permissive temperature of 30°C, no differences in viability (Fig. 2E) or cellular ZipA protein levels (Fig. 2I) were detected among the zipA+, zipA1, or the ΔthrA, ΔthrB, or ΔthrC derivatives of zipA1. Together, these results suggest that deletion mutations in thrB and thrC, the last two steps of l-threonine biosynthesis from aspartate after the synthesis of l-homoserine, can partially suppress the thermosensitivity of the zipA1 allele.
Deletions of other genes directly or indirectly involved in l-threonine or glycine metabolism also partially suppress cell zipA1 mutant thermosensitivity.
Based on the unexpected effects on suppressing the thermosensitivity of the zipA1 strain caused by deleting genes involved in the l-threonine metabolism, we asked whether other genes encoding enzymes in related amino acid biosynthesis pathways (Fig. S3) had similar effects. First, using the zipA+ or zipA1 MG1655 derivative, we deleted glyA, encoding serine hydroxymethyltransferase (SHMT), which catalyzes the interconversion of serine and glycine (23, 24); ltaE, encoding low-specificity l-threonine aldolase, which converts l-threonine or l-allo-threonine to glycine (23–25); tdcB, encoding a catabolic threonine dehydratase, the first step in l-threonine and l-serine degradation (23, 26); or tdh, encoding threonine dehydrogenase, which catalyzes the conversion of l-threonine to l-2-amino-3-oxobutanoate, yielding either aminoacetone or conversion to glycine (27). In the zipA+ (MG1655) background, none of the single deletions affected cell division significantly (data not shown), but in a zipA1 (MG1655) background, mutants lacking glyA, ltaE, or tdcB, but not those lacking tdh, partially suppressed cell filamentation at 37°C (Fig. 3B, C, and E; compare with Fig. 3A and D), similar to the effects of thrB and thrC deletions.
FIG 3.
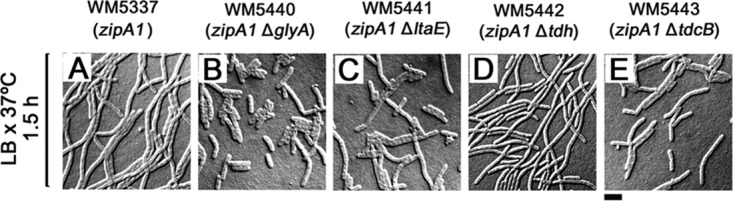
Deletion of glyA, ltaE, and tdcB but not tdh partially suppresses zipA1 mutant thermosensitivity. The effects of suppressing zipA1 mutant thermosensitivity were determined by growing WM5337 (A) and zipA1 mutants lacking glyA (B), ltaE (C), tdh (D), or tdcB (E) in LB broth for 1.5 h at 37°C. Scale bar, 4 μm.
Next, based on the observation that except for thrB, all the deleted genes that partially suppressed zipA1 mutant thermosensitivity required pyridoxal-5-phosphate (PLP) as a cofactor (Fig. S3), we asked whether deleting other PLP-dependent genes near or far from the glycine-serine-threonine pathway could suppress the thermosensitivity of the zipA1 allele. Using the zipA1 MG1655 derivative (WM5337) as a host, we generated 19 new single-deletion mutants in amino acid biosynthesis genes. Including our original mutants, we could now test a total of 15 PLP-dependent and 11 non-PLP dependent genes (Table 1).
TABLE 1.
Capacity of single-deletion mutants to partially suppress thermosensitivity of the zipA1 allele
| Strain | Relevant genotype | PLP dependence | Suppression of thermosensitivity by tempa |
|||
|---|---|---|---|---|---|---|
| 30°C | 34°C | 37°C | 42°C | |||
| WM5163 | MG1655 | + | + | + | + | |
| WM5337 | MG1655 zipA1 ΔnupC::tet | + | − | − | − | |
| WM5376 | WM5337 ΔthrA::kan | No | + | − | − | − |
| WM5377 | WM5337 ΔthrB::kan | No | + | + | + | − |
| WM5352 | WM5337 ΔthrC::kan | Yes | + | + | + | − |
| WM5354 | WM5337 ΔglyA::kan | Yes | + | + | +/− | − |
| WM5378 | WM5337 Δtdh::kan | No | + | + | − | − |
| WM5379 | WM5337 ΔtdcB::kan | Yes | + | + | +/− | − |
| WM5473 | WM5337 ΔilvA::kan | Yes | + | + | +/− | − |
| WM5698 | WM5337 Δkbl::kan | Yes | + | + | +/− | − |
| WM5433 | WM5337 ΔltaE::kan | Yes | + | + | +/− | − |
| WM5695 | WM5337 ΔtrpA::kan | Yes | + | − | − | − |
| WM5696 | WM5337 ΔtrpB::kan | Yes | + | − | − | − |
| WM5388 | WM5337 ΔmetA::kan | No | + | − | − | − |
| WM5389 | WM5337 ΔmetB::kan | Yes | + | + | + | − |
| WM5390 | WM5337 ΔmetC::kan | Yes | + | + | + | − |
| WM5430 | WM5337 ΔmetE::kan | No | + | − | − | − |
| WM5431 | WM5337 ΔmetH::kan | No | + | +/− | − | − |
| WM5432 | WM5337 ΔmmuM::kan | No | + | +/− | − | − |
| WM5475 | WM5337 ΔmetL::kan | No | + | − | − | − |
| WM5477 | WM5337 ΔlysC::kan | No | + | + | + | − |
| WM5693 | WM5337 ΔargD::kan | Yes | + | +/− | − | − |
| WM5694 | WM5337 ΔlysA::kan | Yes | + | − | − | − |
| WM5697 | WM5337 ΔmalY::kan | Yes | + | − | − | − |
| WM5705 | WM5337 ΔaspC::kan | Yes | + | +/− | − | − |
| WM5706 | WM5337 ΔhisC::kan | Yes | + | +/− | − | − |
| WM5707 | WM5337 ΔhisD::kan | No | + | +/− | − | − |
| WM5708 | WM5337 ΔasnB::kan | No | + | +/− | − | − |
To determine the effects of single deletions on suppression of the thermosensitivity of the zipA1 allele, spot assays were carried out in LB at 30, 34, 37, and 42°C by using all the mutants generated in a zipA1 background. All 26 single-deletion mutants grew well at 30°C (Table 1, +, and Fig. 4P), with spots out to the 10−4 dilution, although the ΔglyA and ΔltaE mutant spots were weaker. At 34°C, essentially no viability was observed for the parental zipA1 strain and mutants lacking thrA, trpA, trpB, metA, metE, metL, lysA, and malY (Table 1, −, and Fig. 4Q), partial viability was observed for mutants lacking metH, mmuM, argD, aspC, hisC, hisD, and asnB (Table 1, +/−, and Fig. 4Q), and growth equivalent to that at 30°C was observed in mutants lacking thrB, thrC, glyA, tdcB, tdh, ilvA, kbl, ltaE, metB, metC, and lysC (Table 1, +, and Fig. 4Q).
FIG 4.

Deletion of many genes involved in amino acid biosynthesis partially suppresses zipA1 mutant thermosensitivity. To determine the effects of suppressing zipA1 mutant thermosensitivity, MG1655 (A), WM5337 (MG1655 zipA1) (B), and zipA1 mutants lacking thrA (C), thrB (D), thrC (E), glyA (F), tdcB (G), tdh (H), ilvA (I), kbl (J), ltaE (K), metB (L), metC (M), lysC (N), or argD (O) were grown in LB broth for 2 h at 37°C, at which time cell density was measured by optical density and cell morphology examined by phase-contrast microscopy. Scale bar, 4 μm. Spot assays were used to measure cell viabilities in LB at 30°C (P), 34°C (Q), 37°C (R), and 42°C (S). ND, no dilution.
The less permissive temperature of 37°C caused a general reduction in viability compared with 34°C for most of the mutants. Mutants that previously grew well at 34°C showed partial viability at 37°C, and those with partial viability at 34°C were generally unable to survive at 37°C (Table 1). Only mutants lacking thrB, thrC, metB, metC, and lysC were fully viable either at 34 or 37°C (Table 1 and Fig. 4R). However, despite the suppressing effects on thermosensitivity observed on plates at 37°C, cells of most of the mutants were filamentous when grown in LB broth at the same temperature (Fig. 4C to O), suggesting that the filaments could divide enough to form colonies under these conditions. As expected, no growth was observed on spot plates at 42°C for any of the double mutants (Table 1 and Fig. 4S). These data support the idea that inactivation of some genes involved in amino acid biosynthesis can partially suppress the thermosensitivity of the zipA1 allele.
We then asked whether combinations of some of these deletions had an additive effect on suppression. Deletions of metB, glyA, ltaE, tdcB, or tdh were individually introduced by P1 transduction into a zipA1 background lacking metC, which exhibits partial suppression of zipA1 mutant thermosensitivity (Fig. 4). No differences in the viability of the parental strain and the double-deletion mutants were detected in a comparison of spot assays on LB at 30, 34, 37, or 42°C (data not shown). Therefore, at least in the case of the metC deletion, combining it with deletions that individually can partially suppress zipA1 thermosensitivity suggests that the effects are not additive. The possibility remains that combining more than two deletions, or deletions in other pathways, is additive.
Effects on other cell division genes.
We next asked whether these gene deletions had a general stimulatory effect on cell division versus a more specific effect on zipA1. To test this, we chose deletions of thrC or metC, because both could suppress zipA1 strongly at 37°C and were not located upstream of other amino acid genes where they could potentially have indirect effects. The ΔthrC::kan and ΔmetC::kan alleles were introduced by P1 transduction into ftsA12, ftsZ84, ftsK44, ftsQ1, and ftsI23 thermosensitive cell division mutants, either in an MG1655 background (the first three) or in an MC4100 background (the last two). The double mutants were grown in LB at 37°C for 2 h and checked for cell filamentation. None of the combined mutant cells divided significantly better than the single mutants based on a qualitative examination of multiple micrographs (Fig. 5A to O).
FIG 5.

Effects of pyridoxal-5-phosphate and deletions of thrC or metC in suppressing thermosensitivity of fts cell division mutants. Thermosensitive mutants ftsA12, ftsZ84, fts123, ftsQ1, and ftsK44 (A, D, G, J, and M, respectively) and single deletions of thrC (B, E, H, K, and N) or metC (C, F, I, L, and O) generated in each fts strain background were grown for 1.5 h in LB at the semipermissive temperature of 37°C and examined by microscopy (A to O). After transferring the ftsQ1 and ftsI23 alleles to the MG1655 strain background, fts mutant cells with or without ΔmetC or ΔthrC were spotted on LB or LB with no added NaCl at four different temperatures (P). Five fts mutants were tested for the ability of 1 mM PLP to suppress their thermosensitivity after 22 h of incubation at the temperatures indicated; the ftsQ1 and ftsI23 derivatives shown here are in MC4100, while the others are in MG1655 (Q). To rule out effects due to strain background, derivatives of ftsQ1 or ftsI23 mutants with or without ΔmetC or ΔthrC in either the MC4100 strain background or the MG1655 background were compared for the ability of 1 mM PLP to rescue viability after 20 h of incubation at 42°C (R). ND, no dilution. Scale bar, 4 μm.
To obtain a more complete picture of how well the ΔmetC or ΔthrC mutant might suppress cell division defects, we tested their effects on the viability of fts mutants in spot assays on LB plates at several temperatures. To ensure that any effects on viability were not a result of differences between the MG1655 and MC4100 backgrounds, the ftsQ1 and ftsI23 mutants were introduced into the MG1655 background harboring the other fts mutants. We tested all of the MG1655-derived fts mutants at 30°C, 34°C, 37°C, or 42°C on LB or LB with no added NaCl, which is a more nonpermissive condition for many fts mutants that might allow the detection of more subtle suppression effects. As expected, the ftsZ84, ftsA12, and ftsQ1 mutants were thermosensitive at intermediate temperatures on LB with no salt, making it possible to detect small suppression effects by the ΔmetC or ΔthrC mutation. Notably, the ΔmetC mutation suppressed the ftsK44 mutant quite strongly (Fig. 5P). The ΔthrC mutation also suppressed the ftsZ84 mutant, but very weakly. The levels of FtsZ in the zipA1 strain or in the zipA1 strain with a ΔthrA, ΔthrB, or ΔthrC mutation were unchanged (Fig. 2I), ruling out the possibility that the suppression of zipA1 was due to enhanced expression of the ftsZ or zipA1 mutant gene, at least in those deletion strains. The ability of the ftsK44 mutant to be suppressed by the ΔmetC mutation suggests that components of the cell division machinery other than ZipA can be affected by amino acid metabolism.
Exogenous PLP partially suppresses zipA1 mutant thermosensitivity.
Although we observed partial suppression of zipA1 mutant thermosensitivity by deleting either PLP or non-PLP dependent genes (Table 1 and Fig. 4), we nevertheless were interested to know whether PLP plays a common role in that thermosensitivity. PLP serves as a cofactor for a large number of essential enzymes which catalyze more than 140 distinct reactions, including important steps in amino acid metabolism (28, 29).
We determined the effect of PLP on zipA1 mutant viability in spot assays on LB plates by supplementing with 0.25 or 1 mM PLP and growth at 30, 34, 37, and 42°C for 20 h. We added 1 mM thiamine hydrochloride (vitamin B1) to LB under the same conditions as a negative control. At 30°C, all cells grew well in the presence of either 0.25 or 1 mM PLP (Fig. 6A), although as before, ΔglyA and ΔltaE cell spots were weaker. Strikingly, adding 1 mM PLP greatly suppressed the thermosensitivity of the parental zipA1 strain at 34°C (Fig. 6B) and 37°C (Fig. 6C) and even had a weak suppressing effect at 42°C. Moreover, 1 mM PLP improved the growth of several double mutants at 34°C and 37°C, and with the exception of the glyA zipA1 mutant, permitted partial to full growth rescue even at 42°C (Fig. 6D). The lower (0.25 mM) concentration of PLP was unable to support viability at 42°C except for the ΔthrB zipA1 and ΔthrC zipA1 mutants, which were viable 42°C with either 0.25 or 1 mM PLP.
FIG 6.

Exogenous pyridoxal-5-phosphate partially suppresses zipA1 mutant thermosensitivity and has an additive effect with many of the gene deletions. To determine the effect of pyridoxal-5-phosphate (PLP) on suppressing zipA1 mutant thermosensitivity, cells used for the spot assay in Fig. 3 were also spotted on LB plates supplemented with 0.25 mM or 1 mM PLP and incubated for 20 h at 30°C (A), 34°C (B), 37°C (C), and 42°C (D). ND, no dilution.
Added PLP can partially suppress other cell division mutants.
Based on these unexpected results, we wanted to know if this ability of PLP to rescue the zipA1 mutant would translate to the suppression of other thermosensitive cell division mutants. Strikingly, the ftsK44 and ftsQ1 mutants were able to grow at 42°C when LB was supplemented with 1 mM PLP (Fig. 5Q), with the ftsK44 mutant being more strongly suppressed. The ftsI23 mutant was very weakly suppressed at 42°C. In contrast, the growth of the ftsA12 and ftsZ84 mutants at 42°C was not rescued (Fig. 5Q), suggesting that the inactivation of the ftsA12 and ftsZ84 mutants at 42°C may be more complete than that of the other fts mutants, or perhaps that PLP affects a later stage of cell division than the proto-ring. We confirmed that the MC4100 strain background of the ftsQ1 and ftsI23 mutants shown in Fig. 5Q was not a significant factor in suppression by PLP, as MG1655 derivatives of these two mutants showed similar PLP suppression profiles (Fig. 5R). Moreover, the presence or absence of a ΔmetC or ΔthrC mutation in the ftsQ1 or ftsI23 mutant strain did not appreciably affect the ability of PLP to suppress thermosensitivity.
Overall, these data support a role of PLP in partially suppressing the thermosensitivity of the parental zipA1 mutant. Such suppressing effects were more evident when zipA1 was combined with single deletions of genes involved in amino acid biosynthesis. The ability of PLP to partially suppress the thermosensitivity of the ftsK44 and ftsQ1 mutants indicates that this effect is not exclusive to the zipA1 allele.
DISCUSSION
In this study, we initially found that a thermosensitive zipA (zipA1) strain in W3110 (WM2991) exhibited a growth and division defect in minimal medium at the permissive temperature. This was not surprising, as zipA1 cells were reported to have a mild cell division defect at 30°C when grown in LB (9). However, it was somewhat surprising that the zipA1 mutant defect persisted under slow-growth conditions in minimal medium, because some cell division defects are suppressed at lower growth rates. For example, ΔftsN mutants that are partially suppressed by mutations in ftsB or ftsL divide better in minimal medium than in rich medium (12). We were further surprised to find that the parent W3110 strain had a similar growth defect in minimal medium, although the cells did not form filaments like the zipA1 mutant derivatives. Further examination of this phenomenon indicated that both W3110 and its zipA1 derivative WM2991 behaved as if they were starved for l-threonine or glycine, as the addition of either of these two amino acids could rescue the growth defect. Although we do not understand the reason for this unexpected auxotrophic phenotype, we wanted to work in a strain background that did not have these defects, so we moved the zipA1 allele into MG1655 (WM5337). This strain grew and divided fairly normally in M9 and LB at 30°C and formed the expected filaments at 42°C. We also found that WM5337 formed filaments and was not viable at temperatures as low as 34°C, which allowed us to look for suppressors of the zipA1 allele that could survive at this and other temperatures lower than 42°C.
As our initial studies with WM2991 were focused on glycine and l-threonine, we made deletions in genes involved in glycine and threonine biosynthesis to see if limiting the cells for these amino acids might exacerbate thermosensitivity of the zipA1 mutant. The fact that we found the opposite effect was clearly serendipitous. We then deleted other genes in the methionine-threonine-glycine-serine pathways and found that most, but not all, also partially suppressed zipA1 mutant thermosensitivity at 34° and 37°C but not at the most nonpermissive temperature of 42°C. This partial suppression was not a general effect on all stages of cell division, as two deletions that rescued the zipA1 mutant (ΔthrC and ΔmetC) failed to significantly rescue cell division in ftsZ, ftsA, ftsQ, and ftsI mutants at the nonpermissive temperature. However, we were surprised to find that ΔmetC could suppress the ftsK44 mutants quite efficiently. Little is known about the defect in the ftsK44 mutant, but this ability of the ΔmetC (but not ΔthrC) mutation to suppress its thermosensitivity suggests that other aspects of cell division may be sensitive to perturbations in amino acid metabolism.
One curious aspect of the suppression was the ability of the ΔthrB and ΔthrC mutations but not the ΔthrA mutation, to suppress zipA1 thermosensitivity. One mechanism for this specificity might involve an increase in l-homoserine levels. l-Aspartate is converted into l-aspartate-4-semialdehyde (ASA) by the three aspartate kinase (AK) genes thrA (AK-I), metL (AK-II), and lysC (AK-III) and the aspartate semialdehyde dehydrogenase gene (asd). ASA is subsequently converted into l-homoserine by homoserine dehydrogenases encoded by thrA or metL. Interestingly, deletion of either thrA or metL failed to suppress zipA1 thermosensitivity. In the absence of thrA, less l-homoserine should be synthesized, whereas in the absence of either thrB or thrC, which encode the enzymes needed to convert l-homoserine to l-threonine, cellular l-homoserine levels should increase.
Our work also revealed a role for PLP in partial rescue of the zipA1 allele's phenotypic defects. Of the 17 genes we deleted that were in pathways within or related to threonine metabolism, eight of these were capable of partially suppressing zipA1 thermosensitivity in LB (Table 1). Of these eight, seven (thrC, glyA, ltaE, tdcB, metB, metC, and ilvA) are known to require PLP as a cofactor; only thrB is PLP independent. We therefore initially hypothesized that the zipA1 allele might perturb PLP homeostasis, which was supported by the ability of extra PLP to rescue the growth and division of the zipA1 mutant at 37°C in LB. To test this hypothesis, we deleted a number of other amino acid biosynthesis genes, both PLP dependent and PLP independent. In addition to the ΔthrB mutation, we found that deletions of other PLP-independent amino acid biosynthesis genes in related pathways, including tdh, metH, mmuM, and lysC, could partially suppress thermosensitivity of the zipA1 allele to various degrees. Any one of these deletions might indirectly perturb PLP homeostasis because they affect the threonine-glycine-serine and methionine pathways and thus could affect PLP-dependent enzymes indirectly. Moreover, deletions of amino acid biosynthesis genes, such as ΔhisD and ΔasnB, in seemingly unrelated pathways were able to weakly suppress the zipA1 allele at 34°C but are not PLP dependent. The reason why the PLP-independent ΔthrB mutation can suppress zipA1 thermosensitivity is unclear, but perhaps the accumulation of l-homoserine as described above might alter flux through various pathways (see below), indirectly causing increased demand for PLP. ThrB can also synthesize l-4-phosphohydroxythreonine, a precursor of PLP, via an alternative pathway that has the potential to compete with serine biosynthesis (30), so potential diversion of metabolites into the serine biosynthesis pathway because of the absence of thrB might play a role in the effects.
The relationship between central metabolism and cell shape/division in bacteria has emerged in recent years, with several notable examples of metabolic enzymes moonlighting as cell division regulators (31), including direct interaction between glucolipid enzymes or NAD(H) oxidoreductases and FtsZ (32–35). Amino acid biosynthesis enzymes have not yet been implicated in directly regulating cell division proteins, although a deficiency of shikimate kinase 1 (encoded by aroK), part of the aromatic amino acid pathway, can partially suppress the thermosensitivity of the ftsZ84 mutant by an unknown mechanism (36). In addition, a deficiency of S-adenosylmethionine (SAM) has been shown to inhibit cell division in E. coli (37). However, the present study does not support the idea of SAM involvement in the suppression of the zipA1 phenotype, particularly as blocking several steps in the synthesis of l-methionine helps to rescue division in zipA1 cells (and in the case of ΔmetC, even ftsK44 cells). Another potential mechanism of suppression might be slower growth, which could compensate for a deficiency in cell division. Such a mechanism may be why Δmin ΔslmA mutants can divide in M9 medium but not LB (38), or, as mentioned earlier, why growth in minimal medium can enhance the cell division of ftsN mutants in certain cases (12). However, we think this possibility is unlikely, because the suppressed strains all had colony sizes similar to the nonsuppressed strains and grew as fast as or faster than the nonsuppressed strains in LB broth when switched from 30°C to 37°C.
By analogy to the role of pyruvate in regulating cell division in Bacillus subtilis (39), it is possible that the zipA1 allele can perturb PLP homeostasis if PLP itself is a regulator of ZipA. For example, ZipA may require PLP for its proper function in cell division, and the ZipA1 mutant protein may be defective in utilizing PLP. This defect would be relieved, and ZipA function partially restored, either by directly providing extra PLP to the growth medium, or by shunting amino acid biosynthesis pathways away from PLP-intensive enzymes, such as those in the threonine-methionine-glycine pathways. Such shunting could be accomplished by rerouting biosynthesis by blocking specific PLP-intensive steps. Consistent with this idea, a modest deficiency of pyridoxine 5′ oxidase (PdxH), a key enzyme that converts pyridoxine 5′-phosphate to PLP, was reported to inhibit E. coli cell division (40). However, because deletions of genes encoding PLP-independent enzymes suppress the zipA1 mutant, and added PLP can partially rescue later cell division defects of ftsK44 and ftsQ1 mutants, any model in which PLP regulates ZipA is likely too simplistic. Although it is known that ZipA is required for recruiting later cell division proteins (10), it is also not clear which step(s) of cell division is being rescued by the suppressing factors. Future dissection of these pathways and their relationships with cell division proteins should provide more mechanistic insights into the relationship between PLP, amino acid biosynthesis, and cell division.
MATERIALS AND METHODS
Bacterial strains, plasmids, growth media, and chemicals.
The Escherichia coli strains used are listed in Table 2. Plasmid pDSW210 (a pBR322 derivative with a weakened trp-lac promoter and a gfp gene) was used to clone the zipA or zipA1 mutant genes by EcoRI/SalI digestion. Cells were grown at 30, 34, 37, and 42°C in either Luria-Bertani (LB) broth with 0.5% NaCl, LB with no NaCl added, MOPS-glucose, or M9-glucose (41). For experiments in minimal medium, cells were first inoculated in LB and incubated for 16 h at 30°C, after which cells were pelleted, washed twice, resuspended in minimal medium, and diluted to 1:100 or 1:1,000 in M9 or MOPS. For all growth assays in liquid medium, cells were incubated with shaking in a BioTek Synergy MX microplate reader for the times indicated in Fig. 1M, 4A to O, and S2 and S4 in the supplemental material prior to measuring the cell optical density at 600 nm (OD600). For microscopic analysis, 10 μl of culture was spread onto an agarose-covered glass microscope slide and topped with a glass coverslip.
TABLE 2.
E. coli strains used in this studya
| Strain | Description | Source |
|---|---|---|
| W3110 | rpoSam rph-1 INV (rrnD-rrnE) | Lab collection |
| WM1074 | Wild-type MG1655 F− λ− ilvG rfb50 rph-1 lacU169 | Lab collection |
| MC4100 | F− [araD139]B/r Δ(argF-lac)169 λ− e14− flhD5301 Δ(fruK-yeiR)725 (fruA25) relA1 rpsL150(Strr) rbsR22 Δ(fimB-fimE)632(::IS1) deoC1 | Lab collection |
| WM2991 | W3110 zipA1 (M44V, T211L, Y229C, and N281S) | |
| WM5322 | WM2991 zipA+ ΔnupC::Tn10 | zipA+, nupC::Tn10 × WM2991 |
| WM5337 | MG1655 zipA1 ΔnupC::Tn10 | zipA1, nupC::Tn10 × MG1655 |
| WM5339 | MG1655 nupC::Tn10 | zipA+, nupC::Tn10 × MG1655 |
| WM5372 | WM5339 ΔthrA::kan | ΔthrA, JW0001-1a × WM5339 |
| WM5376 | WM5337 ΔthrA::kan | ΔthrA, JW0001-1 × WM5337 |
| WM5373 | WM5339 ΔthrB::kan | ΔthrB, JW0002-3 × WM5339 |
| WM5377 | WM5337 ΔthrB::kan | ΔthrB, JW0002-3 × WM5337 |
| WM5351 | WM5339 ΔthrC::kan | ΔthrC, JW0003-2 × WM5339 |
| WM5352 | WM5337 ΔthrC::kan | ΔthrC, JW0003-2 × WM5337 |
| WM5369 | WM5339 ΔglyA::kan | ΔglyA, JW2535-1 × WM5339 |
| WM5354 | WM5337 ΔglyA::kan | ΔglyA, JW2535-1 × WM5337 |
| WM5429 | WM5339 ΔltaE::kan | ΔltaE, JW0854-1 × WM5339 |
| WM5433 | WM5337 ΔltaE::kan | ΔltaE, JW0854-1 × WM5337 |
| WM5374 | WM5339 Δtdh::kan | Δtdh, JW3591-4 × WM5339 |
| WM5378 | WM5337 Δtdh::kan | Δtdh, JW3591-4 × WM5337 |
| WM5375 | WM5339 ΔtdcB::kan | ΔtdcB, JW3088-2 × WM5339 |
| WM5379 | WM5337 ΔtdcB::kan | ΔtdcB, JW3088-2 × WM5337 |
| WM5385 | WM5339 ΔmetA::kan | ΔmetA, JW3973-1 × WM5339 |
| WM5388 | WM5337 ΔmetA::kan | ΔmetA, JW3973-1 × WM5337 |
| WM5386 | WM5339 ΔmetB::kan | ΔmetB, JW3910-1 × WM5339 |
| WM5389 | WM5337 ΔmetB::kan | ΔmetB, JW3910-1 × WM5337 |
| WM5387 | WM5339 ΔmetC::kan | ΔmetC, JW2975-1 × WM5339 |
| WM5390 | WM5337 ΔmetC::kan | ΔmetC, JW2975-1 × WM5337 |
| WM5426 | WM5339 ΔmetE::kan | ΔmetE, JW3805-1 × WM5339 |
| WM5430 | WM5337 ΔmetE::kan | ΔmetE, JW3805-1 × WM5337 |
| WM5474 | WM5339 ΔmetL::kan | ΔmetL, JW3911-1 × WM5339 |
| WM5475 | WM5337 ΔmetL::kan | ΔmetL, JW3911-1 × WM5337 |
| WM5427 | WM5339 ΔmetH::kan | ΔmetH, JW3979-1 × WM5339 |
| WM5431 | WM5337 ΔmetH::kan | ΔmetH, JW3979-1 × WM5337 |
| WM5428 | WM5339 ΔmmuM::kan | ΔmmuM, JW0253-1 × WM5339 |
| WM5432 | WM5337 ΔmmuM::kan | ΔmmuM, JW0253-1 × WM5337 |
| WM5472 | WM5339 ΔilvA::kan | ΔilvA, JW3745-2 × WM5339 |
| WM5473 | WM5337 ΔilvA::kan | ΔilvA, JW3745-2 × WM5337 |
| WM5476 | WM5339 ΔlysC::kan | ΔlysC, JW3984-1 × WM5339 |
| WM5477 | WM5337 ΔlysC::kan | ΔlysC, JW3984-1 × WM5337 |
| WM5693 | WM5337 ΔargD::kan | ΔargD, JW3322-1 × WM5337 |
| WM5694 | WM5337 ΔlysA::kan | ΔlysA, JW2806-1 × WM5337 |
| WM5695 | WM5337 ΔtrpA::kan | ΔtrpA, JW1252-1 × WM5337 |
| WM5696 | WM5337 ΔtrpB::kan | ΔtrpB, JW1253-1 × WM5337 |
| WM5697 | WM5337 ΔmalY::kan | ΔmalY, JW1614-1 × WM5337 |
| WM5698 | WM5337 Δkbl::kan | Δkbl, JW3592-2 × WM5337 |
| WM5705 | WM5337 ΔaspC::kan | ΔaspC, JW0911-1 × WM5337 |
| WM5706 | WM5337 ΔhisC::kan | ΔhisC, JW2003-1 × WM5337 |
| WM5707 | WM5337 ΔhisD::kan | ΔhisD, JW2002-1 × WM5337 |
| WM5708 | WM5337 ΔasnB::kan | ΔasnB, JW0660-2 × WM5337 |
| WM5440 | WM5337 ΔglyA::frt | WM5354 cured by pCP20 |
| WM5441 | WM5337 ΔltaE::frt | WM5433 cured by pCP20 |
| WM5442 | WM5337 Δtdh::frt | WM5378 cured by pCP20 |
| WM5443 | WM5337 ΔtdcB::frt | WM5379 cured by pCP20 |
| WM5444 | WM5337 ΔmetC::frt | WM5390 cured by pCP20 |
| WM5462 | WM5337 ΔltaE::frt ΔtdcB::kan | ΔtdcB, JW3088-2 × WM5441 |
| WM5463 | WM5337 ΔltaE::frt ΔmetC::kan | ΔtdcB, JW3088-2 × WM5444 |
| WM5464 | WM5337 ΔglyA::frt ΔltaE::kan | ΔltaE, JW0854-1 × WM5440 |
| WM5485 | WM1125 (ftsZ84) ΔthrC::kan in WM1074 | ΔthrC, JW0003-2 × WM1125 |
| WM5486 | WM1125 (ftsZ84) ΔmetC::kan in WM1074 | ΔmetC, JW2975-1 × WM1125 |
| WM5487 | WM3993 (ftsI23) ΔthrC::kan in MC4100 | ΔthrC, JW0003-2 × WM3993 |
| WM5488 | WM3993 (ftsI23) ΔmetC::kan in MC4100 | ΔmetC, JW2975-1 × WM3993 |
| WM5489 | WM3994 (ftsQ1) ΔthrC::kan in MC4100 | ΔthrC, JW0003-2 × WM3994 |
| WM5490 | WM3994 (ftsQ1) ΔmetC::kan in MC4100 | ΔmetC, JW2975-1 × WM3994 |
| WM5491 | WM2101 (ftsK44) ΔthrC::kan in WM1074 | ΔthrC, JW0003-2 × WM2101 |
| WM5492 | WM2101 (ftsK44) ΔmetC::kan in WM1074 | ΔmetC, JW2975-1 × WM2101 |
| WM4649 | WM3993 (ftsI23) in WM1074 | ftsI23 leuO::Tn10 × WM1074 |
| WM4661 | WM3994 (ftsQ1) in WM1074 | ftsQ1 leuO::Tn10 × WM1074 |
| WM5817 | WM4649 (ftsI23) ΔthrC::kan in WM1074 | ΔthrC, JW0003-2 × WM4649 |
| WM5818 | WM4649 (ftsI23) ΔmetC::kan in WM1074 | ΔmetC, JW2975-1 × WM4649 |
| WM5819 | WM4661 (ftsQ1) ΔthrC::kan in WM1074 | ΔthrC, JW0003-2 × WM4661 |
| WM5820 | WM4661 (ftsQ1) ΔmetC::kan in WM1074 | ΔmetC, JW2975-1 × WM4661 |
Phage P1 lysates were made from the indicated JW strains in the KEIO collection and used as donors in transductions.
Where noted, M9 or MOPS medium was supplemented with one of the following compounds (Sigma-Aldrich): Casamino Acids, l-tyrosine, l-valine, l-phenylalanine, l-glutamic acid, l-threonine, l-methionine, l-aspartic acid, l-proline, l-alanine, l-isoleucine, l-asparagine, l-leucine, l-serine, l-cysteine, glycine, l-histidine, l-tryptophan, l-lysine, l-arginine, l-glutamic acid, or pyridoxal 5-phosphate. Kanamycin (50 μg · ml−1), ampicillin (50 μg · ml−1), chloramphenicol (20 μg · ml−1), or tetracycline (10 μg · ml−1) was added to the plates or broth as needed. For spotting assays, an overnight culture was diluted 250-fold, followed by growth at 30°C for 2 h. Five microliters of 10-fold serial dilutions was spotted onto prewarmed plates and incubated at selected temperatures.
Strain construction.
Single-deletion mutant strains listed in Table 1 were generated by growing P1 phage on strains from the KEIO collection (42) and then transducing the kanamycin resistance (Kanr) markers into the appropriate recipient. For the construction of double mutants, the Kanr cassette was removed by using plasmid pCP20 (43); the normal plasmid curing protocol was modified by using 30°C instead of higher temperatures to avoid thermoinactivating the zipA1 allele. The presence of each mutation was confirmed by diagnostic PCR. The ftsQ1 and ftsI23 mutants were transferred from the MC4100 background to the MG1655 background (WM1074) by P1 transduction using the linked leuO::Tn10 marker present in the MC4100 strains, followed by a screen for thermosensitivity. The ΔmetC::kan and ΔthrC::kan alleles were introduced into these strains by transduction and confirmed by PCR, as described above.
Immunoblotting.
Equivalent OD600 units of cell lysate were separated on a 12% SDS polyacrylamide gel and transferred to a nitrocellulose membrane. Immunoblotting was performed with standard procedures using a 1:10,000 dilution of polyclonal ZipA or FtsZ primary antibody (14). Secondary horseradish peroxidase-conjugated goat anti-rabbit and chemiluminescent substrate were applied prior to developing and imaging.
Supplementary Material
ACKNOWLEDGMENTS
We thank Marcin Krupka, Steven Distelhorst, and Kara Schoenemann for helpful discussions, Jiqiang Ling for generously sharing his Keio strain collection, and Kevin Morano for the use of his microplate reader.
This work was supported by NIH award GM61074 to W.M.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00535-17.
REFERENCES
- 1.Haeusser DP, Margolin W. 2016. Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol 14:305–319. doi: 10.1038/nrmicro.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rico AI, Krupka M, Vicente M. 2013. In the beginning, Escherichia coli assembled the proto-ring: an initial phase of division. J Biol Chem 288:20830–20836. doi: 10.1074/jbc.R113.479519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang X, Lyu Z, Miguel A, McQuillen R, Huang KC, Xiao J. 2017. GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355:744–747. doi: 10.1126/science.aak9995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowlett VW, Margolin W. 2014. 3D-SIM super-resolution of FtsZ and Its membrane tethers in Escherichia coli cells. Biophys J 107:L17–L20. doi: 10.1016/j.bpj.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisson-Filho AW, Hsu Y-P, Squyres GR, Kuru E, Wu F, Jukes C, Sun Y, Dekker C, Holden S, VanNieuwenhze MS, Brun YV, Garner EC. 2017. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355:739–743. doi: 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egan AJ, Vollmer W. 2013. The physiology of bacterial cell division. Ann N Y Acad Sci 1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- 7.Hale CA, de Boer PA. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88:175–185. doi: 10.1016/S0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- 8.Addinall SG, Bi E, Lutkenhaus J. 1996. FtsZ ring formation in fts mutants. J Bacteriol 178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pichoff S, Lutkenhaus J. 2002. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J 21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hale CA, de Boer PAJ. 2002. ZipA is required for recruitment of FtsK, FtsQ, FtsL, and FtsN to the septal ring in Escherichia coli. J Bacteriol 184:2552–2556. doi: 10.1128/JB.184.9.2552-2556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pichoff S, Du S, Lutkenhaus J. 2015. The bypass of ZipA by overexpression of FtsN requires a previously unknown conserved FtsN motif essential for FtsA-FtsN interaction supporting a model in which FtsA monomers recruit late cell division proteins to the Z ring. Mol Microbiol 95:971–987. doi: 10.1111/mmi.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu B, Persons L, Lee L, de Boer PA. 2015. Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol Microbiol 95:945–970. doi: 10.1111/mmi.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsang M-J, Bernhardt TG. 2015. A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol Microbiol 95:925–944. doi: 10.1111/mmi.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geissler B, Elraheb D, Margolin W. 2003. A gain-of-function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci U S A 100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pichoff S, Shen B, Sullivan B, Lutkenhaus J. 2012. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA's self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol 83:151–167. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pichoff S, Lutkenhaus J. 2005. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol 55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- 17.Hale CA, Rhee AC, de Boer PA. 2000. ZipA-induced bundling of FtsZ polymers mediated by an interaction between C-terminal domains. J Bacteriol 182:5153–5166. doi: 10.1128/JB.182.18.5153-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosyak L, Zhang Y, Glasfeld E, Haney S, Stahl M, Seehra J, Somers WS. 2000. The bacterial cell-division protein ZipA and its interaction with an FtsZ fragment revealed by X-ray crystallography. EMBO J 19:3179–3191. doi: 10.1093/emboj/19.13.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moy FJ, Glasfeld E, Mosyak L, Powers R. 2000. Solution structure of ZipA, a crucial component of Escherichia coli cell division. Biochemistry 39:9146–9156. doi: 10.1021/bi0009690. [DOI] [PubMed] [Google Scholar]
- 20.Ohashi T, Hale CA, de Boer PA, Erickson HP. 2002. Structural evidence that the P/Q domain of ZipA is an unstructured, flexible tether between the membrane and the C-terminal FtsZ-binding domain. J Bacteriol 184:4313–4315. doi: 10.1128/JB.184.15.4313-4315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raychaudhuri D. 1999. ZipA is a MAP-Tau homolog and is essential for structural integrity of the cytokinetic FtsZ ring during bacterial cell division. EMBO J 18:2372–2383. doi: 10.1093/emboj/18.9.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haeusser DP, Rowlett VW, Margolin W. 2015. A mutation in Escherichia coli ftsZ bypasses the requirement for the essential division gene zipA and confers resistance to FtsZ assembly inhibitors by stabilizing protofilament bundling. Mol Microbiol 97:988–1005. doi: 10.1111/mmi.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neidhardt FC, Curtiss R, Ingraham JL, Lin ECC, Kow KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE. 1996. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed ASM Press, Washington, DC. [Google Scholar]
- 24.Ogawa H, Gomi T, Fujioka M. 2000. Serine hydroxymethyltransferase and threonine aldolase: are they identical? Int J Biochem Cell Biol 32:289–301. doi: 10.1016/S1357-2725(99)00113-2. [DOI] [PubMed] [Google Scholar]
- 25.Liu JQ, Dairi T, Itoh N, Kataoka M, Shimizu S, Yamada H. 1998. Gene cloning, biochemical characterization and physiological role of a thermostable low-specificity l-threonine aldolase from Escherichia coli. Eur J Biochem 255:220–226. doi: 10.1046/j.1432-1327.1998.2550220.x. [DOI] [PubMed] [Google Scholar]
- 26.Goss TJ, Schweizer HP, Datta P. 1988. Molecular characterization of the tdc operon of Escherichia coli K-12. J Bacteriol 170:5352–5359. doi: 10.1128/jb.170.11.5352-5359.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boylan SA, Dekker EE. 1981. l-Threonine dehydrogenase. Purification and properties of the homogeneous enzyme from Escherichia coli K-12. J Biol Chem 256:1809–1815. [PubMed] [Google Scholar]
- 28.Mooney S, Leuendorf J-E, Hendrickson C, Hellmann H. 2009. Vitamin B6: a long known compound of surprising complexity. Molecules 14:329–351. doi: 10.3390/molecules14010329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenberg J, Ischebeck T, Commichau FM. 2017. Vitamin B6 metabolism in microbes and approaches for fermentative production. Biotechnol Adv 35:31–40. doi: 10.1016/j.biotechadv.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Kershner JP, Novikov Y, Shoemaker RK, Copley SD. 2010. Three serendipitous pathways in E. coli can bypass a block in pyridoxal-5′-phosphate synthesis. Mol Syst Biol 6:436. doi: 10.1038/msb.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperber AM, Herman JK. 2017. Metabolism shapes the cell. J Bacteriol 199:e0039-17. doi: 10.1128/JB.00039-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radhakrishnan SK, Pritchard S, Viollier PH. 2010. Coupling prokaryotic cell fate and division control with a bifunctional and oscillating oxidoreductase homolog. Dev Cell 18:90–101. doi: 10.1016/j.devcel.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 33.Beaufay F, Coppine J, Mayard A, Laloux G, De Bolle X, Hallez R. 2015. A NAD-dependent glutamate dehydrogenase coordinates metabolism with cell division in Caulobacter crescentus. EMBO J 34:1786–1800. doi: 10.15252/embj.201490730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weart RB, Lee AH, Chien AC, Haeusser DP, Hill NS, Levin PA. 2007. A metabolic sensor governing cell size in bacteria. Cell 130:335–347. doi: 10.1016/j.cell.2007.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill NS, Buske PJ, Shi Y, Levin PA. 2013. A moonlighting enzyme links Escherichia coli cell size with central metabolism. PLoS Genet 9:e1003663. doi: 10.1371/journal.pgen.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vinella D, Gagny B, Joseleau-Petit D, D'Ari R, Cashel M. 1996. Mecillinam resistance in Escherichia coli is conferred by loss of a second activity of the AroK protein. J Bacteriol 178:3818–3828. doi: 10.1128/jb.178.13.3818-3828.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman EB, Budman LI, Chan EC, Greene RC, Lin RT, Woldringh CL, D'Ari R. 1998. Lack of S-adenosylmethionine results in a cell division defect in Escherichia coli. J Bacteriol 180:3614–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernhardt TG, de Boer PA. 2005. SlmA, a nucleoid-associated, FtsZ-binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol Cell 18:555–564. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monahan LG, Hajduk IV, Blaber SP, Charles IG, Harry EJ. 2014. Coordinating bacterial cell division with nutrient availability: a role for glycolysis. mBio 5:e00935=14. doi: 10.1128/mBio.00935-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lam HM, Winkler ME. 1992. Characterization of the complex pdxH-tyrS operon of Escherichia coli K-12 and pleiotropic phenotypes caused by pdxH insertion mutations. J Bacteriol 174:6033–6045. doi: 10.1128/jb.174.19.6033-6045.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neidhardt FC, Bloch PL, Smith DF. 1974. Culture medium for enterobacteria. J Bacteriol 119:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.