

Abstract
Background
Knowledge on the capacity of Australian ticks to carry Borrelia species is currently limited or missing. To evaluate the potential of ticks to carry bacterial pathogens and their DNA, it is imperative to have a robust workflow that maximises recovery of bacterial DNA within ticks in order to enable accurate identification. By exploiting the bilateral anatomical symmetry of ticks, we were able to directly compare two DNA extraction methods for 16S rRNA gene diversity profiling and pathogen detection. We aimed to assess which combination of DNA extraction and 16S rRNA hypervariable region enables identification of the greatest bacterial diversity, whilst minimising bias, and providing the greatest capacity for the identification of Borrelia spp.
Results
We collected Australian endemic ticks (Bothriocroton undatum), isolated DNA from equal tick halves using two commercial DNA extraction methods and sequenced samples using V1-V3 and V3-V4 16S rRNA gene diversity profiling assays. Two distinct Borrelia spp. operational taxonomic units (OTUs) were detected using the V1-V3 16S rRNA hypervariable region and matching Borrelia spp. sequences were obtained using a conventional nested-PCR. The tick 16S rRNA gene diversity profile was dominated by Rickettsia spp. (98–99%), while the remaining OTUs belonged to Proteobacteria (51–81%), Actinobacteria (6–30%) and Firmicutes (2–7%). Multiple comparisons tests demonstrated biases in each of the DNA extraction kits towards different bacterial taxa.
Conclusions
Two distinct Borrelia species belonging to the reptile-associated Borrelia group were identified. Our results show that the method of DNA extraction can promote bias in the final microbiota identified. We determined an optimal DNA extraction method and 16S rRNA gene diversity profile assay that maximises detection of Borrelia species.
Electronic supplementary material
The online version of this article (doi: 10.1186/s13071-017-2579-5) contains supplementary material, which is available to authorized users.
Keywords: Bothriocroton undatum, Borrelia, Goanna tick, DNA extraction, MiSeq, Illumina, NGS, Ixodidae, Coxiella burnetii
Background
Ticks are known to have the ability to transmit bacterial, viral and protozoal agents during blood feeding episodes, making them one of the most important arthropod vectors parasitising reptilian, avian and mammalian species, including humans [1–3]. In Australia, Ixodes holocyclus and Bothriocroton hydrosauri can carry and transmit Rickettsia australis and Rickettsia honei, respectively, causing rickettsiosis in humans [4–7]. More recently, a novel species of Candidatus Borrelia tachyglossi was identified in Bothriocroton concolor and I. holocyclus ticks collected from the short-beaked echidna (Tachyglossi aculeatus) [8, 9]. Borrelia species and their associated human illnesses such as Lyme disease, have been a highly debated and controversial topic for over 25 years in Australia [10–12]. To bring a new line of evidence to the debate on Lyme disease and other human tick-borne diseases, a complete map of bacteria in Australian ticks is needed [9, 13, 14]. Lyme disease agents [spirochetes Borrelia burgdorferi (sensu lato)] are not known to be transmitted locally in Australia, but Lyme-like disease caused by unknown pathogens are considered to exist [10–12, 15, 16]. Baseline data and a consensus of potentially pathogenic bacteria in Australian ticks are imperative to initiate inquiry into the existence of the bacterial causality of human disease. To evaluate the potential of ticks to carry bacterial pathogens, specifically bacterial DNA, it is imperative to obtain a high quality and quantity of bacterial DNA in order to enable unbiased tick surveys.
Next-generation sequencing (NGS) technologies have seen a shift in tick microbial studies from targeting individual bacterial species, to sequencing whole tick assemblages [17, 18]. However, a universal consensus on the optimal DNA extraction protocol for hard tick species is still lacking: a significant issue when the hard chitinous exoskeleton and multiple life stages of ixodid ticks are considered [4, 6, 7, 9, 19, 20]. DNA extraction is a critical component of microbiota studies, due to its influence on the abundance and diversity of bacterial species identified [21–23]. Determining the bias of DNA extraction methods towards or away from certain bacterial groups is confounded by variability introduced before the DNA is even extracted, due to the heterogeneity of the individual tick, and their small size [18, 24, 25]. Thus, there is a need to determine the bias of DNA extraction methods on a homogenous tick population.
The aim of this study was to assess which combination of DNA extraction method with 16S ribosomal (rRNA) hypervariable region through NGS sequencing gave the greatest bacterial diversity and enabled the identification of Borrelia spp. and Rickettsia spp. To directly compare pre-sequencing methodologies, we exploited the symmetrical body plan of ticks [26]. Our approach involved longitudinally bisecting tick samples (Bothriocroton undatum) and processing each assumed identical half with a different DNA extraction method. Our workflow allowed us to determine and associate microbiome variability with the method and assay applied. We were then able to confirm our workflow using conventional PCR techniques targeting Borrelia spp. and Rickettsia spp.
Methods
Collection and identification of ticks
A total of 25 ticks (Tick1–25) were collected from a wild lace monitor (Varanus varius) in Terrey Hills, New South Wales, Australia in December 2016 (Additional file 1: Table S1). The lace monitor was brought to the attention of the veterinary surgeon by a member of the public, and was euthanized by the registered veterinarian due to suspected rat poisoning and emaciation. Ticks were collected opportunistically post-mortem, immediately submerged in 70% (w/v) ethanol and submitted to the Sydney School of Veterinary Sciences (SSVS), The University of Sydney. Ticks were stored at -20 °C until morphological identification and DNA isolation. Ticks were morphologically identified with the aid of a stereomicroscope (Olympus, Macquarie Park, Australia), using dichotomous keys and character matrices [26, 27].
Bisecting ticks for comparison of DNA extraction methods
Prior to DNA isolation, individual ticks were surface sterilised [28]; 1 min submerged in 3% hydrogen peroxide (H2O2), 30 s submerged in 70% (w/v) ethanol and 2 min submerged in phosphate buffered saline (PBS, pH = 7.4).
Tick1 to Tick6 (n = 6) were chosen based on level of engorgement and size (fully engorged, uniformly ~1 cm in width). Each tick was longitudinally bisected using a sterile disposable scalpel blade (Fig. 1) and each half was subjected to a different DNA extraction method. Method 1 exploited a mechanical maceration protocol where the first half of each tick was placed in a 1.5 ml bead-beater with ceramic beads and homogenised using a high-speed benchtop homogeniser FastPrep-24 (MP Biomedicals, Seven Hills, Australia) for 40 s at 6.0 m/s, followed by extraction using the ISOLATE Fecal DNA Kit (Bioline, Eveleigh, Australia) as per the manufacturer’s instructions. Method 2 consisted of enzymatic cell lysis followed by the ISOLATE II Genomic DNA Kit (Bioline) as per the manufacturer’s instructions, with overnight (16 h) digestion with Proteinase K. To monitor DNA isolation efficiency, 5 μl of DNA Extraction Control 670 (Bioline) was included in each sample, regardless of the DNA extraction method applied. DNA quality and quantity was further confirmed using a Qubit dsDNA HS assay on the Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, North Ryde, Australia). DNA was stored at -20 °C until PCR amplification. DNA Extraction Control 670 (Bioline) served to validate the success of the extraction step and was amplified as per the manufacturer’s protocol and instructions.
Fig. 1.
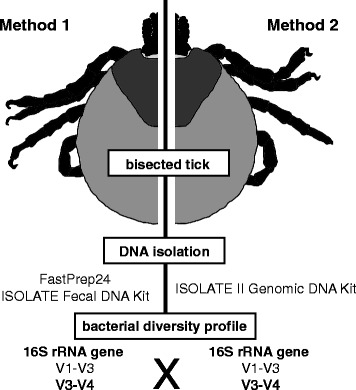
Summary of a workflow taking advantage of the symmetrical body plan of ticks. All tick samples were longitudinally bisected, each half being subjected to DNA extraction by either the ISOLATE Fecal DNA kit (Method 1) or the ISOLATE II Genomic DNA Kit (Method 2). All samples were sequenced using gene diversity profiling assays, targeting both the V1-V3 and V3-V4 16S rRNA hypervariable regions
Tick7 to Tick25 (n = 19) were surface sterilised, longitudinally bisected and both halves were together subjected to Method 1 for DNA extraction and processed as described above.
Amplification of the tick’s cytochrome c oxidase subunit 1
A 658 nucleotide (nt) fragment of the cytochrome c oxidase subunit 1 (cox1) gene was amplified using PCR with forward primer S0725 (F1) (5′-TAC TCT ACT AAT CAT AAA GAC ATT GG-3′) and reverse primer S0726 (R1) (5′-CCT CCT CCT GAA GGG TCA AAA AAT GA-3′) (primers as published in O. Kushimo thesis ‘The tick genus Amblyomma in Africa: Phylogeny and multilocus DNA barcoding’ from Georgia Southern University, 2013; primers cited as M. Montagna, unpublished data). MyTaq™ Red Mix (Bioline) was used for 30 μl reactions with 2 μl of DNA template in a T100 Thermal Cycler (BioRad, Gladesville, Australia) as previously described [29]. PCR products were bi-directionally sequenced by Macrogen Ltd. (Seoul, South Korea). Sequences were assembled and aligned using CLC Main Workbench 6.9.1 (CLC bio, Denmark). The evolutionary history of tick cox1 gene sequences was inferred using the Minimum Evolution (ME) method, with model selection based on maximum likelihood in MEGA7 (v.7.0.14) [30]. Bootstrap support was calculated as a percentage of replicate trees in which the associated taxa clustered together as implemented in MEGA7.
Next-generation sequencing of bacterial 16S rRNA gene amplicons
Tick DNA samples (Tick1–6) were subjected to 16S rRNA gene diversity profiling assays at the Australian Genome Research Facility (Brisbane, Australia). Sequencing of the V1-V3 and V3-V4 16S rRNA hypervariable regions was performed on the Illumina Miseq (300-nt pair-end) using the following assay; 16S (V1-V3): 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) with 519R (5′-GWA TTA CCG CGG CKG CTG-3′) and 16S (V3-V4) 341F (5′-CCT AYG GGR BGC ASC AG-3′) with 806R (5′-GGA CTA CNN GGG TAT CTA AT-3′). Paired-end reads were assembled using PEAR (version 0.9.5). Primers were identified and trimmed. Trimmed sequences were processed using Quantitative Insights into Microbial Ecology (QIIME 1.8/1.9.1), USEARCH (v.8.0.1623) and UPARSE software. Sequences were quality filtered and full length duplicate sequences were removed and sorted by abundance using USEARCH. Singletons or unique reads in the data set were discarded. Sequences were clustered followed by chimera filtering using the “rdp gold” database as a reference. To obtain the number of reads in each OTU, reads were mapped back to OTUs with a minimum identity of 97%. Using QIIME, taxonomy was assigned using the Greengenes database 5 (version 13 8, Aug 2013). Unassigned OTUs were excluded as well as any belonging to mitochondria and chloroplasts.
Multivariate statistical analysis of tick microbiome
Multivariate analytical procedures were used to investigate patterns of variation in the composition of bacterial assemblages among tick samples. The microbiome abundance matrix with taxonomy of each OTU and sample associated factors were imported for multivariate statistical analysis in PRIMER v.7 [31]. Data from V1-V3 and V3-V4 16S rRNA hypervariable regions were analysed separately. Each sample was associated with the following factors: method and tick number. The data matrix of OTU abundances (or chosen taxonomical level abundance) was fourth-root-transformed. A reasonably severe transformation was appropriate in order to reduce the contribution of the most abundant taxonomical categories (OTU, species, genera, etc.). Variation in the structure of bacteria within the ticks was examined using Bray-Curtis resemblance measures. An analysis of similarities (ANOSIM [32]) was used to test the null hypothesis of no differences among the communities of defined groupings (significance level, P = 0.05). Non-metric multidimensional scaling (nMDS) ordination [33] was undertaken to visualize and explore the patterns of community similarities amongst all samples. The goodness-of-fit of the resulting two dimensional nMDS plot was measured using Kruskal’s stress formula I [33]. Visualisation was enhanced by marking different factors (e.g. age and parasite status) superimposed on the ordination plots as symbols and numbers to help evaluate their potential effects on bacterial assemblage structure.
Diversity indices including S, total number of species (OTUs); N, species richness (Margalef); H′, Shannon diversity index with logs to the base e; and 1-λ’, Simpson’s diversity index, were calculated for the OTU tables in PRIMER v.7 [31]. Shannon diversity indices were evaluated using t-tests in Microsoft Excel (2016) to compare diversity estimates between V1-V3 and V3-V4 16S rRNA hypervariable regions, and Method 1 and Method 2. Richness was obtained by calculating the number of different taxonomic families and genera found within V1-V3 and V3-V4 16S rRNA hypervariable regions, where a threshold of 30 reads was applied. Venn diagrams were constructed in RStudio® (version 1.0.143 RStudio, Inc.) using the ‘VennDiagram’ package [34].
Abundance at different taxonomic levels and bar reads representing uncultured or unknown bacteria were calculated using Microsoft Excel (2016). Abundance measures for each phylum, class, order, family and genus between each assay (Method 1 + V1-V3, Method 1 + V3-V4, Method 2 + V1-V3, Method 2 + V3-V4) were used to further determine the presence of bias. A Tukey’s multiple comparisons test was conducted in GraphPad Prism 7.03 (GraphPad Software Inc. 2017), comparing each measure of bacterial abundance between each of the four combinations.
Detection of Borrelia spp. and Rickettsia spp. using conventional nested-PCR, and Coxiella burnetii using conventional PCR
Detection of Borrelia spp. spirochetes in ticks was performed by two independent conventional nested-PCR assays (16S rRNA gene, 23S rRNA gene). A 16S rRNA gene nested-PCR amplifying a ~1250-nt fragment was performed on all tick samples (Tick1–25) at SSVS using previously published primers [9]. The 16S rRNA gene of Borrelia spirochetes was amplified in a reaction volume of 30 μl, containing 15 μl of MyTaq Red™ Mix (Bioline), 10 pmol of each primer (Bor-16F (S0778), 5′-TGC GTC TTA AGC ATG CAA GT-3′ / Bor-1360R (S0779), 5′-GTA CAA GGC CCG AGA ACG TA-3′ for the first round; Bor-27F (S0780), 5′-CAT GCA AGT CAA ACG GAA TG-3′ / Bor-1232R (S0781), 5′-ACT GTT TCG CTT CGC TTT GT-3′ for the second round [9]), template (2 μl of purified DNA for the first round and 1 μl aliquot of the first PCR product in the second round), and PCR-grade water. The PCR was run on a T100 Thermal Cycler (Bio-Rad), including initial denaturation at 95 °C for 3 min, followed by 35 cycles of 95 °C for 15 s, 55 °C for 15 s, 72 °C for 30 s, and a final elongation for 5 min at 72 °C for both primary and secondary reactions. Each run included a blank negative control with sterile PCR-grade water. A positive control (16S rRNA gene amplification) was excluded to minimise contamination. A 23S rRNA gene nested-PCR amplifying a 222-nt fragment was performed on Tick1–6 at the Biology Centre of the Academy of Science of the Czech Republic [35]. The 23S rRNA gene of Borrelia spirochetes was amplified in a reaction volume of 25 μl, containing 12.5 μl of FastStart PCR MasterMix (Roche, Praha, Czech Republic), 10 pmol of each primer (Bor-1, 5′-AGA AGT GCT GGA GTC GA-3′ / Bor-2, 5′-TAG TGC TCT ACC TCT ATT AA-3′ for the first round; Bor-3, 5′-GCG AAA GCG AGT CTT AAA AGG-3′ / Bor-4, 5′-ACT AAA ATA AGG CTG AAC TTA AAT-3′ for the second round [35]), template (4 μl of purified DNA for the first round, 1 μl of aliquot for the first PCR product in the second round), and PCR-grade water. The PCR was run on an Applied Biosystems 2720 Thermal Cycler (ThermoFisher Scientific), including initial denaturation at 95 °C for 10 min followed by 40 cycles of 95 °C for 30 s, 53 °C for 30 s, 72 °C for 30 s, and a final elongation for 7 min at 72 °C. The amplification program for the second round was the same, except for the annealing temperature which was 58 °C.
A diagnostic conventional nested-PCR assay targeting the gltA gene of Rickettsia spp. was applied to Tick1–6 as previously described and adopted [29, 36]. A ~650-nt fragment of gltA was amplified in a final reaction volume of 30 μl, containing 15 μl MyTaq Red™ Mix (Bioline), 10 pmol of each primer (gltA-F1 (S0659), 5′-GCA AGT ATT GGT GAG GAT GTA ATC-3′ / gltA-R1 (S0660), 5′-CTG CGG CAC GTG GGT CAT AG-3′ for the first round; gltA-F2 (S0661), 5′-GCG ACA TCG AGG ATA TGA CAT-3′ / gltA-R2 (S0662) 5′-GGA ATA TTC TCA GAA CTA CCG-3′, for the second round), template (2 μl of purified DNA for the first round and 1 μl aliquot of the first PCR product in the second round), and PCR-grade water. The PCR was run on a T100 Thermal Cycler (Bio-Rad), including initial denaturation at 95 °C for 3 min, followed by 35 cycles of 95 °C for 15 s, 55 °C for 15 s, and a final elongation for 5 min at 72 °C for both primary and secondary reactions. Each run included a blank negative control with sterile PCR-grade water.
Coxiella burnetii was detected using a conventional PCR targeting IS1111-repetitive elements with IS1111aF (S0797) (5′-GTC TTA AGG TGG GCT GCG TG-3′) and IS1111aR (S0798): (5′-CCC CGA ATC TCA TTG ATC AGC-3′) [37]. MyTaq™ Red Mix (Bioline) was used in 30 μl reactions with 2 μl of DNA template in a T100 Thermal Cycler (BioRad). The cycling conditions were as follows: initial denaturation at 95 °C for 3 min, followed by 34 cycles of 95 °C for 15 s, 55 °C for 15 s, 72 °C for 15 s, and a final elongation for 5 min at 72 °C. The positive control for C. burnetii was DNA extracted from the Q Fever vaccine (Q-VAX®) (kindly provided by Jacqui Norris). Sterile PCR-grade water was used as a blank negative control.
PCR products were visualized on 2% (w/v) agarose gel with GelRed™ (Biotium, Inc., Fremont, CA, USA). Products from the 16S rRNA gene assay (Borrelia) and gltA gene assay (Rickettsia) were directly and bi-directionally sequenced using amplification primers at Macrogen Ltd. (Seoul, South Korea). Sequences were assembled and chromatographs visually inspected and compared to related sequences using CLC Main Workbench 6.9.1 (CLC bio, Aarhus, Denmark). The evolutionary history of Borrelia spp. 16S rRNA gene sequences was inferred using the Minimum Evolution (ME) method, with model selection based on maximum likelihood in MEGA7 (v.7.0.14) [30]. Bootstrap support was calculated as a percentage of replicate trees in which the associated taxa clustered together as implemented in MEGA7.
Quantification of bacteria using real-time PCR
Bacterial load was determined using a universal bacterial real-time PCR assay targeting the 16S rRNA gene [38]. The 10 times dilution of Escherichia coli strain ATCC 11775 (kindly gifted by Denise Wigney) suspended cells was used to construct a standard curve. A set of generic bacterial primers (S0775) (5′-TCC TAC GGG AGG CAG CAG T-3′) / (S0776) (5′-GGA CTA CCA GGG TAT CTA ATC CTG TT-3′) and probe (S0777) (5′-CGT ATT ACC GCG GCT GCT GGC AC-3′) labelled with FAM-BHQ1 was used to quantify the amount of bacterial DNA present within each tick sample from Method 1 and Method 2. SensiFAST™ Probe No-ROX Kit (Bioline) was used according to the manufacturer’s instructions. The PCR included 2 μl of DNA template and was run on the CFX96 Touch Real-Time PCR (Bio-Rad), and copy number analysed using the corresponding CFX Manager 3.1 (Bio-Rad) software. Real-time PCR cycling conditions included an initial denaturation step at 95 °C for 3 min, followed by 35 cycles of 95 °C for 10 s and 60 °C for 30 s. An extraction control real-time PCR was performed for each sample to monitor DNA isolation efficiency and PCR inhibitors as described previously. A t-test using Microsoft Excel (2016) was used to calculate significant differences between the average numbers of bacterial cells found using each extraction kit.
Results
The goanna tick (Bothriocroton undatum) from the lace monitor (Varanus varius)
All ticks (n = 25) collected from the lace monitor (Varanus varius) were morphologically identified as females from the species B. undatum. Amplification and DNA sequences of cox1 mtDNA from the DNA of six tick specimens demonstrated close identities with the cox1 sequences of B. undatum (98.9%, 650/658, JN863728). All eight nucleotide substitutions were synonymous, revealing 100% (219/219) identity with the cox1 amino acid sequence of B. undatum (JN863728). Multiple sequence alignment with related Bothriocroton spp. sequences at either nucleotide or amino acid sequence confirmed conspecificity with B. undatum (Fig. 2).
Fig. 2.
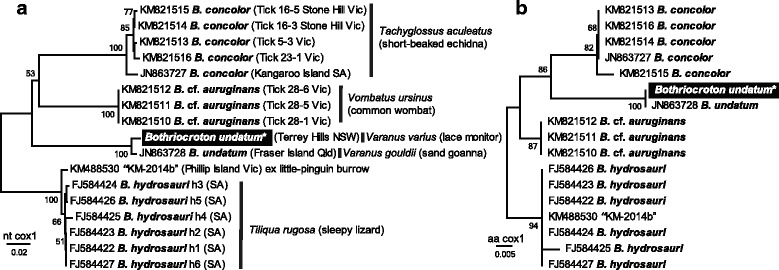
Phylogenetic position of the Goanna tick (Bothriocroton undatum). Ticks were collected from a wild lace monitor (Varanus varius) from Sydney, Australia. a The evolutionary history based on cox1 nucleotide (nt) sequences was inferred using the Minimum Evolution (ME) method with Kimura-2 parameter model and percent bootstrap support percentages (200 replicates) shown next to the branches was calculated in MEGA7. There were a total of 521 positions in the final dataset. b The evolutionary history based on cox1 amino acid (aa) sequences was inferred using the ME method, with bootstrap support percentages test (200 replicates) shown next to the branches. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of aa substitutions per site. There were a total of 173 positions in the final dataset. Accession number with tick species name, tick identifier and locality in Australia is followed by host name. *All B. undatum ticks (n = 6, Tick1 to Tick6) had identical cox1 sequences
The real-time PCR assay estimated the average number of bacterial cells for Method 1 and Method 2 to be 48,139 and 148,660 per entire tick, respectively (Tick 1–6). The results of the t-test demonstrated a significant increase (paired t-test, t (5) = 8.936, two-tailed P = 0.0003) in the number of bacterial cells detected in DNA extracted using Method 2 compared to Method 1. DNA extraction control was amplified for examined ticks, confirming successful and uninhibited DNA isolation. The Qubit dsDNA HS assay found the average quantity of DNA for Method 1 and Method 2 to be 21 ng/μl and 51 ng/μl, respectively (Additional file 1: Table S2).
The greater tick microbiome diversity using a combination of 16S rRNA assays
Ticks (n = 6, Tick1–6) were longitudinally bisected. DNA from each half was isolated with either Method 1 or Method 2, and a microbiome profile was generated using V1-V3 (n = 12) and V3-V4 (n = 12) 16S rRNA gene diversity profiling assays (Figs. 1 and 3, Additional file 1: Table S3). The V1-V3 16S rRNA gene diversity profiling assays yielded 1,112,938 raw reads that were quality filtered into 834,157 (min. 19,420; max. 114,048; n = 12) high quality reads (excluding singletons) and clustered into 126 bacterial OTUs. The V3-V4 16S rRNA gene diversity profiling assays yielded 1,895,826 raw reads that were quality filtered into 1,602,905 (min. 40,128; max. 225,303; n = 12) high quality reads (excluding singletons) and clustered into 124 bacterial OTUs. Both V1-V3 and V3-V4 16S rRNA gene diversity profiles using either method (Fig. 1) were dominated by OTUs belonging to the genus Rickettsia (98–99%). Excluding the dominating OTU (OTU_1; Rickettsia), reads belonging to the phyla Proteobacteria (51–81%), Actinobacteria (6–30%) and Firmicutes (2–7%) dominated both hypervariable regions (Fig. 3a). 16S rRNA gene diversity profiles from both Method 1 and Method 2 were dominated by Alphaproteobacteria (49–53%) and Gammaproteobacteria (39–63%) respectively (Fig. 3a). OTUs assigned to Borrelia spp. accounted for ~7% of bacterial reads between the two hypervariable regions, and Coxiella spp. accounted for 1.2% of bacterial reads. Shannon diversity indices (H′) demonstrated no significant difference in the level of bacterial diversity between V1-V3 and V3-V4 16S rRNA gene diversity profiling assays (paired t-test, t (5) = 1.4561, P > 0.05; paired t-test t (5) = 0.6848, P > 0.05), or between Method 1 and Method 2 (paired t-test, t (5) = 0.1419, P > 0.05; paired t-test t (5) = 1.3524, P > 0.05) (Table 1). A large proportion of bacterial genera and classes were unique to either the V1-V3 or V3-V4 16S rRNA gene diversity profiling assays (Fig. 3b). Multivariable analysis via non-metric MDS (nMDS) showed groupings of samples based on the method of DNA isolation used, regardless of the 16S rRNA gene diversity profiling assay used (Fig. 4a, b). Plotting vectors of the bacterial genera revealed alignment of samples with nMDS distribution with Borrelia and Coxiella (Fig. 4a, b). Focusing on the bacterial genus and class level of the microbiome, ANOSIM showed a significant difference in the bacterial community composition between those recovered using Method 1 compared to Method 2 (Fig. 4). Using a histogram of permutations, the observed R was outside the permutation distribution of the test statistics R under the null hypothesis from OTUs to class, and from OTU to order for both V1-V3 and V3-V4 16S rRNA gene diversity profiling assays, respectively (Fig. 4c, d). There were no significant differences between the individual ticks’ microbiomes at any taxonomical level, with the observed R close to the permutated distribution (Fig. 4c, d). Tukey’s multiple comparisons tests found significant levels of bias between Method 1 and Method 2 at all taxonomic levels, with the exception of phyla (Fig. 3a). Although not significant (Tukey’s multiple comparisons tests, P > 0.05), Method 1 found ~0.3–4.8% more reads representing Borrelia spp. at both V1-V3 and V3-V4 16S rRNA hypervariable regions than Method 2.
Fig. 3.
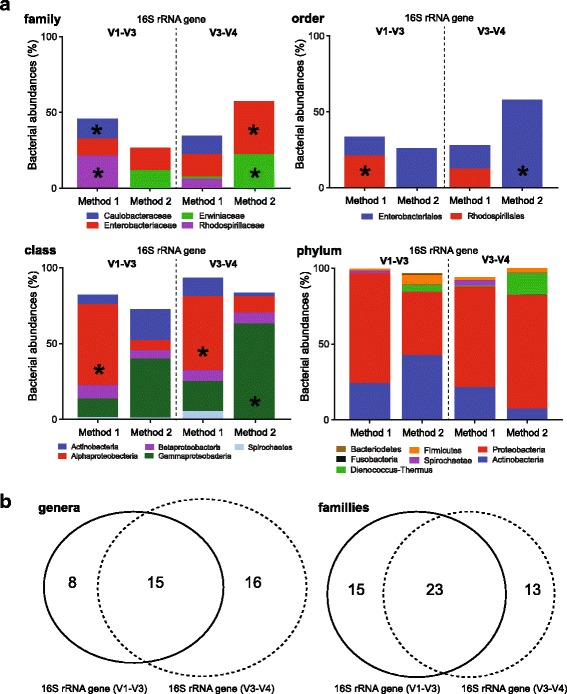
Bacterial diversity profiles of the Goanna tick (Bothriocroton undatum). Bacterial abundance and bias at family, order, class and phylum levels were retrieved from a combination of different workflows that included V1-V3 or V3-V4 16S rRNA gene diversity profiles assays, and either Method 1 or Method 2 for the Goanna tick (Bothriocroton undatum). a Graphs with bacterial families and orders show only those with significant bias between the workflows. The graph depicting bacterial classes demonstrates the five most dominant bacterial classes found in each of the workflows, including spirochetes (Borrelia spp.). The graph of bacterial phyla shows no bias found between any of the workflows. Bias between bacterial abundances at different taxonomical levels was evaluated using Tukey’s multiple comparisons test, (P < 0.05) and is indicated with ‘*’. b Venn diagrams representing the number of taxonomic families and genera identified within each 16S rRNA hypervariable region as a measure of richness. A threshold of 30 reads and 97% identification was applied
Table 1.
Bacterial diversity measures for the Goanna tick Bothriocroton undatum
| S | N | H′ | 1-λ’ | |
|---|---|---|---|---|
| V1-V3 16S rRNA gene | ||||
| Method 1 | 57 | 12.16 | 0.1303 | 0.03108 |
| Method 2 | 79 | 16.94 | 0.122 | 0.02785 |
| Method 1 + 2 | 126 | 27.14 | 0.133 | 0.02947 |
| V3-V4 16S rRNA gene | ||||
| Method 1 | 82 | 17.59 | 0.133 | 0.03071 |
| Method 2 | 87 | 18.67 | 0.1479 | 0.03601 |
| Method 1 + 2 | 124 | 26.71 | 0.148 | 0.03337 |
Abbreviations: S total number of species (OTUs), N species richness (Margalef), H′, Shannon index with logs to the base e, 1-λ’ Simpson index
Fig. 4.
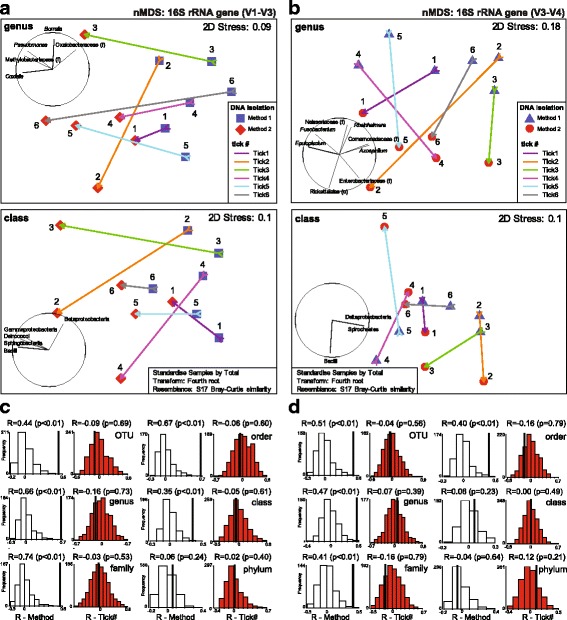
Multivariate analysis of the bacterial profiles of the Goanna tick (Bothriocroton undatum). Non-metric MDS ordination (nMDS) plots for bacterial abundance and analysis of similarity (ANOSIM). The ordinations are from abundances at the genus or class level of (a) 124 OTUs (V1-V3 16S rRNA gene diversity profiling assay) and (b) 126 OTUs (V3-V4 16S rRNA gene diversity profiling assay), with resulting stress values of (a) 0.09 (genus), 0.10 (class) and (b) 0.18 (genus), 0.10 (class). Superimposed are vector plots for taxa (correlation > 0.8) displaying observed responses to the gradient [direction reflects the Pearson correlation of transformed abundances, length represents the multiple correlation coefficient from the linear regression on the ordination points (circle is a correlation = 1)]. Individual halved ticks are joined by solid lines to identify the community differences using nMDS. Histograms of permutated distributions of the test statistics R (up to 999 permutations, ANOSIM; null hypothesis - is no significant difference among communities, P < 0.05) with observed R (bold) at different taxonomic level evaluated for DNA isolation method (transparent) and tick (red) (c, d)
Method 1 with V1-V3 16S rRNA gene diversity profiling assay demonstrates capacity for Borrelia detection and identification
The V1-V3 and V3-V4 16S rRNA gene diversity profiling assays revealed two Borrelia OTUs (V1-V3: OTU_51 and OTU_146), and one Borrelia OTU (V3-V4: OTU_26), respectively (Fig. 5a). Using Method 1, two ticks (33%, 2/6, Tick2 and Tick3) were positive for Borrelia, with the V3-V4 16S rRNA gene diversity profiling assay recovering identical OTU_26 (240-nt) in both ticks. The V1-V3 16S rRNA gene diversity profiling assay was able to detect OTUs in both Tick2 and Tick3 (V1-V3: OTU_51 and OTU_146, pairwise 96.7%, 8/240 identity). Method 2 only recovered Borrelia in Tick3 (V1-V3: OTU_51), matching the OTU using DNA extracted with Method 1. Using Method 2, Tick2 did not have any detectable Borrelia sequences, although it did have the lowest sequence depth of the examined ticks (20,237 sequence raw reads, Fig. 5a).
Fig. 5.

Molecular identification of reptile-associated Borrelia species in the Goanna tick (Bothriocroton undatum) from Sydney, Australia. a Summary table showing the V1-V3 and V3-V4 16S rRNA gene diversity profiling assay reads for Borrelia OTUs within each tick sample (Tick 1–6) recovered using either Method 1 or Method 2. The table includes the total number of high quality reads, the proportion of Borrelia reads, as well as the library identifier (JS2948-JS2970). b, c Multiple sequence alignment of the overlapping Borrelia OTU region of the V1-V3 (b) and V3-V4 (c) 16S rRNA gene sequence, with Borrelia sp. 16S rRNA gene sequence amplified from Tick2 and Tick3. Identical residues with the top reference are indicated by dots. Both Borrelia spp. Tick2 and Tick3 16S rRNA gene sequences are indistinguishable within the V3-V4 16S rRNA hypervariable region
Detection of two new reptile-associated Borrelia spp. in Bothriocroton undatum from Australia
Two tick samples (33%, 2/6, Tick2 and Tick3) analysed with the 16S rRNA gene diversity profiling assays yielded ~1250-nt Borrelia 16S rRNA gene specific amplification products using DNA obtained with both Method 1 and Method 2. DNA sequencing revealed an unambiguous 16S rRNA gene (1167-nt) sequence that was identical between amplicons from Method 1 and Method 2. The two 16S rRNA gene sequences (Tick2 and Tick3) were 98.5% (17/1167) identical to each other. When comparing data from the V1-V3 16S rRNA gene diversity profiling assay and the conventional PCR, 16S rRNA gene sequences obtained from Tick2 and Tick3 demonstrated 100% identity with OTU_146 (194/194) and OTU_51 (194/194), respectively (Fig. 5b). OTU_26 from the V3-V4 16S rRNA gene diversity profiling assay was 100% identical (240/240) with both Tick2 and Tick3 sequences obtained through conventional PCR (Fig. 5c).
The nested 23S rRNA gene PCR performed on Tick1–6 revealed the presence of Borrelia spirochetes in Tick2 (from DNA isolated using both Method 1 and Method 2, 222-nt product), while the remaining five ticks were PCR negative.
We subsequently inquired into the presence of Borrelia spp. in the remaining B. undatum ticks (Tick7–25). DNA was isolated using Method1 and subjected to the ~1250-nt Borrelia 16S rRNA gene specific conventional nested-PCR assay. A single DNA from Tick14 resulted in positive PCR amplification and the 16S rRNA gene sequence was 100% identical to Borrelia sp. Tick3 (1167/1167) 16S rRNA gene sequence.
In summary, three tick samples (12%, 3/25, Tick2, Tick3 and Tick14) yielded two distinct ~1250-nt Borrelia spp. 16S rRNA gene specific amplification products. We tentatively named these two Borrelia sequences Borrelia sp. Tick2 and Borrelia sp. Tick3/Tick14.
Multiple sequence alignment and phylogenetic analysis of 16S rRNA gene sequences from representative Borrelia spp. with Tick2 and Tick3/Tick14 sequences revealed 98.5% (18/1167) and 98.2% (21/1167) identity with Borrelia sp. TA2 (AB529427) within a monophyletic clade (Fig. 6). The phylogenetic analysis clearly (> 95% bootstrap support) resolved monophyly of Lyme disease Borrelia, Relapsing fever Borrelia spp. and the monotreme-associated “Candidatus B. tachyglossi” (Fig. 6). The recognised sequences and B. turcica within the reptile-associated Borrelia species form three paraphyletic clades according to their tick host: snake, turtle and lizard (Fig. 6). The Borrelia spp. Tick2 and Tick3/Tick14, with Borrelia sp. TA2 (AB529427), form the lizard clade of the reptile-associated Borrelia species.
Fig. 6.
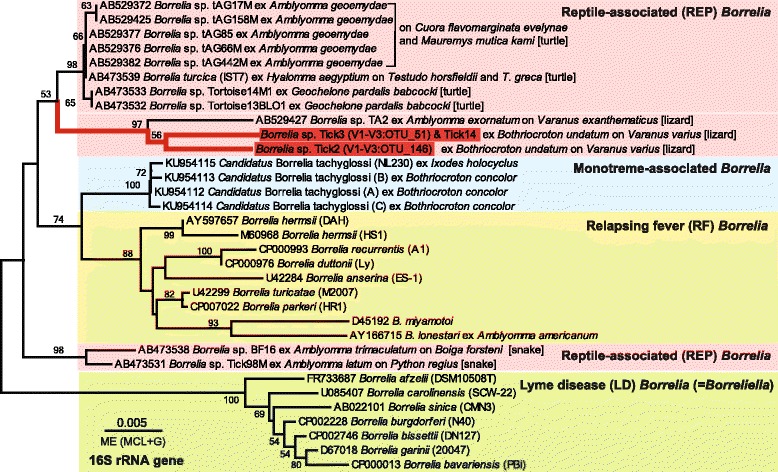
The evolutionary history of the reptile associated Borrelia 16S rRNA gene sequences. The tree was inferred using the Minimum Evolution (ME) method, with evolutionary distances computed using the Tajima-Nei method (TN), and the rate variation among sites modelled with a gamma distribution (+G, shape parameter = 0.28) in MEGA7. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The analysis involved 33 nucleotide sequences. All positions containing gaps and missing data were eliminated. There were a total of 1075 positions in the final dataset. Accession numbers and species name and strain are depicted on the right. Colour coded major clades within Borrelia species include the monophyletic Lyme disease Borrelia (=Borreliella), the monophyletic Relapsing fever Borrelia, the monotreme-associated Borrelia, and the polyphyletic reptile-associated Borrelia, including turtle, lizard and snake groups
Detection of Rickettsia cf. tamurae in Bothriocroton undatum from Australia
All tick samples (6/6, Tick1–6) yielded ~650-nt Rickettsia gltA specific amplification products. DNA sequencing revealed unambiguous gltA sequences (606 nt), which were identical across all six ticks (Tick1–6). Multiple sequence alignment and phylogenetic analysis of gltA sequences from representative Rickettsia spp. revealed a high identity (99.7%, 2/606) to Rickettsia tamurae type strain AT-1 (AF394896). High identity (> 99%) at the gltA gene is indicative of species identity, therefore we tentatively consider our recovered species as Rickettsia cf. tamurae (Fig. 7). Negative controls remained negative throughout the diagnostic assay.
Fig. 7.
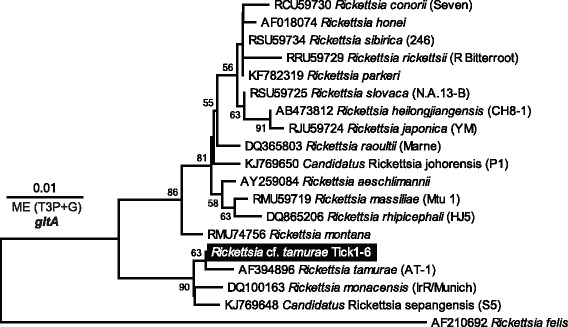
Phylogenetic tree of Rickettsia cf. tamurae in the Goanna tick (Bothriocroton undatum) from Sydney, Australia. The tree based on gltA gene sequences was inferred using the Minimum Evolution (ME) method with Tamura 3-parameter (T3P) distanced with gamma distribution (+G, shape parameter = 0.31) and calculated in MEGA7. Bootstrap support test (500 replicates) percentages are shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. There were a total of 606 positions in the final dataset
Absence of Coxiella burnetii in Bothriocroton undatum from Australia
The Coxiella burnetii species specific PCR assay was negative for the six ticks examined (Tick1–6). The positive control for C. burnetii yielded a positive amplicon of expected 294 nt size. Negative controls remained negative throughout the diagnostic assay.
Discussion
The study developed a methodology that resulted in the largest coverage of the tick microbiome, maximising the capacity to detect multiple Borrelia spp. within B. undatum, an endemic Australian tick species. The direct comparison of two DNA extraction protocols, each subjected to two 16S rRNA gene diversity profiling assays, demonstrated bias associated with routine DNA extraction methods. The approach has enabled us to objectively propose an optimised workflow for the detection of Borrelia and Rickettsia species in ticks.
Analysis of the 16S rRNA gene can be used to identify bacterial communities [39, 40]. The 16S rRNA gene contains nine hypervariable regions: V1-V9 [39, 41]. Each region, however, shows a different capacity to define bacterial diversity, therefore no single region is able to differentiate all microbiota within the community of a tick [39, 42]. Our results are in line with these findings, as the V1-V3 16S rRNA gene diversity profiling assay identified a larger number of families, while the V3-V4 16S rRNA gene diversity profiling assay identified a larger number of genera. Diversity analyses showed no significant differences in the level of bacterial diversity between V1-V3 and V3-V4 16S rRNA hypervariable regions, or Method 1 and Method 2, despite an increasing trend in the diversity of V3-V4 16S rRNA hypervariable region and Method 2. Traditionally, the V3-V4 16S rRNA hypervariable region is preferred because diversity estimates for bacterial communities are often the highest [18]. Nevertheless, sequencing errors potentially result in an overestimation of the microbiome, increased diversity and bacterial taxa identification [18, 43]. With a single region being unable to identify all microbiota in a community, a wider span of the microbiome is achieved when multiple 16S rRNA hypervariable regions are combined [18, 39, 44].
The design of the current study included obtaining near identical subsamples of each tick, providing us with the ability to demonstrate the effect of DNA isolation method as a significant source of variability. Previous research suggests that the type of sample collected, and DNA extraction protocol implemented during microbiome studies, can introduce variability and bias on the resulting microbiota [18, 45]. It is imperative to use a standardised DNA extraction approach to demonstrate microbiome associations with biotic factors. In fact, infections of B. burgdorferi in Ixodes spp. ticks were found to be significantly higher when sourced from woodlands as opposed to pastures, demonstrating the geographical effect on the presence of pathogenic bacteria in ticks [46]. Furthermore, the location and sex of Ixodes spp. has been shown to impact on the final microbiota identified, providing additional strength to our approach [25].
We demonstrated the presence of bacterial detection bias between two different approaches to DNA extraction (Method 1 and Method 2). These results conflict with those of Cruaud et al. [47] who argued that different DNA extraction methods lead to similar estimation of microbial diversity. Similarly, Rubin et al. [22] suggested that different DNA extraction protocols will only influence the ability, or lack thereof, of the microbiota to be sequenced, rather than impacting on the community recovered. Our results are in agreement with Vishnivetskaya et al. [45] who demonstrated that different DNA extraction kits can impact on the final interpretation of the microbiome. Evaluation of bias should be a prerequisite for any microbiome study because of the above noted controversies, which show the strength of the effect of DNA isolation approach on the subsequent microbiome sequenced.
For over 25 years there has been an ongoing debate on the presence of Lyme disease agents [Borrelia burgdorferi (s.l.)] in Australia, fuelled by findings of Borrelia spp. of unknown pathogenicity within Australian ticks [8–10, 15, 16, 48]. The current study demonstrates two more distinct Borrelia species of unknown pathogenic potential, found in 12% (3/25) of tick samples and tentatively named Borrelia sp. Tick2 and Borrelia sp. Tick3/Tick14. Recently, an Australian endemic “Candidatus B. tachyglossi” was identified in a closely related tick species, B. concolor [9]. Australian ticks are not free from Borrelia spp., yet the impacts of their presence on human and animal health remains unknown. Adopting a validated approach that has demonstrated the capacity to detect and differentiate Borrelia species should be applied across Australian ticks. Our results allow us to conclude that Method 1 combined with the V1-V3 16S rRNA hypervariable region results in the greatest capacity for Borrelia spp. detection.
Borrelia sp. Tick2 and Borrelia sp. Tick3/Tick14 both had a 98.2% similarity with Borrelia sp. TA2, a species previously identified within Amblyomma geomydae in Japan [49]. Takano et al. [49] found Borrelia sp. TA2 to be closely related to B. turcica, a reptile-associated (REP) Borrelia species. Because Borrelia sp. TA2, Borrelia sp. Tick2 and Borrelia sp. Tick3/Tick14 are monophyletic and were all sourced from hosts of the reptile genus Varanus, we believe that we have found two more REP Borrelia species that contribute to the ‘lizard’ clade.
In comparison to Borrelia spp., there is strong evidence for the presence of potentially pathogenic Rickettsia spp. within Australian ticks [4–7, 13]. Our study is in agreement with previous research, as our samples contained a high proportion (98–99%) of Rickettsia cf. tamurae (12/12, 100%). Rickettsia cf. tamurae is a member of the Spotted Fever Group Rickettsia, previously of unknown pathogenicity in humans [50]. However, in 2011 Imaoka et al. [51] described one of the first cases of human rickettsiosis in Japan caused by R. tamurae strain AT-1. In 2015, Kho et al. [52] further identified R. tamurae within Amblyomma ticks sourced from wild Python molurus in Malaysia. The current study has now identified R. tamurae within B. undatum, an endemic species of Australian tick. However, as our samples were only 99.7% similar to R. tamurae strain AT-1, is it possible that we have identified a new R. tamurae strain. Furthermore, when combined with previous research, our findings suggest the potential of this pathogenic bacteria to be distributed worldwide [49, 51, 52].
Conclusions
The aim of the current study was to assess which combination of DNA extraction method with 16S rRNA gene diversity profiling assay would give the greatest bacterial diversity, least amount of bias and greatest capacity for identification of Borrelia spp. and Rickettsia spp. We were able to establish an approach that satisfies all three aims, as well as demonstrate the presence of Borrelia spp. DNA in tick extracts. We have developed a molecular workflow capable of targeting Borrelia and Rickettsia spp., although we have limited insight on the origin of the bacteria identified and their pathogenic potential for human or animals.
Additional files
Summary of tick samples used. Table S2. Summary of Qubit DNA concentrations and qPCR bacterial numbers estimates. Table S3. Summary of DNA sequencing of bacterial 16S rRNA gene. (XLSX 18 kb)
Acknowledgements
The authors wish to acknowledge and thank Jacqui Norris and Denise Wigney for kindly providing the Coxiella burnetii positive control and culture of Escherichia coli strain ATCC 11775. We thank David Emery for discussions and encouragements. The authors acknowledge the volunteers and registered veterinarian, Natasha Pesce, who assisted in the admission and euthanasia of the wild lace monitor, as well as the collection of tick samples from Terrey Hills Animal Hospital.
Funding
The study was supported by the Dugdale Guy Peele Bequest, University of Sydney (JŠ). The work of JLP and JP was partly supported by Sydney School of Veterinary Science, University of Sydney Honours support funds. Study was in part supported by the Czech Science Foundation grants (17–27393S to RŠ and 17–27386S to OH). The funding bodies had no role in the design, collection, analysis, and interpretation of the data and in writing the manuscript.
Availability of data and materials
The tick exoskeletons were donated to the Australian National Insect Collection (ANIC) in Canberra, Australia under database numbers ANIC 48 006553 to ANIC 48 006558. Nucleotide sequence data from this study are available in the GenBank (National Center for Biotechnology Information, NCBI) database under accession numbers MG004673–MG004678 (Rickettsia gltA), MG004679–MG004680 and MG452684 (Borrelia 16S rRNA gene), and MG004681–MG004686 (Bothriocroton cox1) and the SRA database under BioProject accession number: PRJNA411926 (https://www.ncbi.nlm.nih.gov/bioproject/411926). BIOM, PRIMER and associated files are available at LabArchives (10.6070/H4W094FD).
Authors’ contributions
JLP and JŠ participated in the study design. JLP, SC and JP performed DNA isolation. JLP collected material, performed the conventional PCR, analysed the data and wrote the draft. RŠ and OH performed testing and provided scientific insight. NEDC performed the real-time PCR. JLP, NEDC and JŠ contributed to the draft. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable. Sample submissions were provided by the veterinary practitioner and approval was not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi: 10.1186/s13071-017-2579-5) contains supplementary material, which is available to authorized users.
Contributor Information
Jessica L. Panetta, Email: jpan7959@uni.sydney.edu.au
Radek Šíma, Email: radeksima@gmail.com.
Nichola E. D. Calvani, Email: nichola.calvani@sydney.edu.au
Ondřej Hajdušek, Email: hajdus@paru.cas.cz.
Shona Chandra, Email: scha8008@uni.sydney.edu.au.
Jessica Panuccio, Email: jpan7775@uni.sydney.edu.au.
Jan Šlapeta, Email: jan.slapeta@sydney.edu.au.
References
- 1.Jongejan F, Uilenberg G. The global importance of ticks. Parasitology. 2004;129(07):S3. doi: 10.1017/S0031182004005967. [DOI] [PubMed] [Google Scholar]
- 2.Cook MJ. Lyme borreliosis: a review of data on transmission time after tick attachment. Int J Gen Med. 2015;8:1–8. doi: 10.2147/IJGM.S73791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Fuente J, Antunes S, Bonnet S, Cabezas-Cruz A, Domingos AG, Estrada-Peña A, et al. Tick-pathogen interactions and vector competence: identification of molecular drivers for tick-borne diseases. Front Cell Infect Microbiol. 2017;7:114. doi: 10.3389/fcimb.2017.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Unsworth NB, Stenos J, Graves S, Faa AG, Cox E, Dyer JR, et al. Flinders Island spotted fever Rickettsioses caused by “marmionii” strain of Rickettsi honei. Eastern Australia Emerging Infect Dis. 2007;13(4):566–573. doi: 10.3201/eid1304.050087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Unsworth NB, Stenos J, McGregor AR, Dyer JR, Graves SR. Not only ‘Flinders Island’ spotted fever. Pathology. 2005;37(3):242–245. doi: 10.1080/00313020500099247. [DOI] [PubMed] [Google Scholar]
- 6.Vilcins IM, Old JM, Deane E. Molecular detection of Rickettsia, Coxiella and Rickettsiella DNA in three native Australian tick species. Exp Appl Acarol. 2009;49(3):229–242. doi: 10.1007/s10493-009-9260-4. [DOI] [PubMed] [Google Scholar]
- 7.Whiley H, Custance G, Graves S, Stenos J, Taylor M, Ross K, Gardner MG. Rickettsia detected in the reptile tick Bothriocroton hydrosauri from the lizard Tiliqua rugosa in South Australia. Pathogens. 2016;5(2):5(2). doi: 10.3390/pathogens5020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loh SM, Gillett A, Ryan U, Irwin P, Oskam C. Molecular characterization of ‘Candidatus Borrelia tachyglossi’ (family Spirochaetaceae) in echidna ticks, Bothriocroton concolor. Int J Syst Evol Microbiol. 2017;67(4):1075–1080. doi: 10.1099/ijsem.0.001929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loh SM, Gofton AW, Lo N, Gillett A, Ryan UM, Irwin PJ, Oskam CL. Novel Borrelia species detected in echidna ticks, Bothriocroton concolor, in Australia. Parasit Vectors. 2016;9(1):339. doi: 10.1186/s13071-016-1627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayne PJ. Investigation of Borrelia burgdorferi genotypes in Australia obtained from erythema migrans tissue. Clin Cosmet Investig Dermatol. 2012;5:69–78. doi: 10.2147/CCID.S31913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piesman J, Stone BF. Vector competence of the Australian paralysis tick, Ixodes holocyclus, for the lyme disease spirochete Borrelia burgdorferi. Int J Parasitol. 1991;21(1):109–111. doi: 10.1016/0020-7519(91)90127-S. [DOI] [PubMed] [Google Scholar]
- 12.Stewart A, Glass J, Patel A, Watt G, Cripps A, Clancy R. Lyme arthritis in the Hunter Valley. Med J Aust. 1982;1:139. doi: 10.5694/j.1326-5377.1982.tb132207.x. [DOI] [PubMed] [Google Scholar]
- 13.Graves S, Stenos J. Rickettsioses in Australia. Ann N Y Acad Sci. 2009;1166:151–155. doi: 10.1111/j.1749-6632.2009.04530.x. [DOI] [PubMed] [Google Scholar]
- 14.Graves SR, Stenos J. Tick-borne infectious diseases in Australia. Med J Aust. 2017;206(7):320–324. doi: 10.5694/mja17.00090. [DOI] [PubMed] [Google Scholar]
- 15.Mayne P, Song S, Shao R, Burke J, Wang Y, Roberts T. Evidence for Ixodes holocyclus (Acarina: Ixodidae) as a vector for human Lyme borreliosis infection in Australia. J Insect Sci. 2014;14:271. doi: 10.1093/jisesa/ieu133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chalada MJ, Stenos J, Bradbury RS. Is there a Lyme-like disease in Australia? Summary of the findings to date. One Health. 2016;2:42–54. doi: 10.1016/j.onehlt.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khoo JJ, Chen F, Kho KL, Ahmad Shanizza AI, Lim FS, Tan KK, et al. Bacterial community in Haemaphysalis ticks of domesticated animals from the Orang Asli communities in Malaysia. Ticks Tick Borne Dis. 2016;7(5):929–937. doi: 10.1016/j.ttbdis.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 18.Sperling JL, Silva-Brandao KL, Brandao MM, Lloyd VK, Dang S, Davis CS, et al. Comparison of bacterial 16S rRNA variable regions for microbiome surveys of ticks. Ticks Tick Borne Dis. 2017;8(4):453–461. doi: 10.1016/j.ttbdis.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Gofton AW, Doggett S, Ratchford A, Oskam CL, Paparini A, Ryan U, Irwin P. Bacterial profiling reveals novel “Ca. Neoehrlichia”, Ehrlichia, and Anaplasma species in Australian human-biting ticks. PLoS One. 2015;10(12):e0145449. doi: 10.1371/journal.pone.0145449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ammazzalorso AD, Zolnik CP, Daniels TJ, Kolokotronis SO. To beat or not to beat a tick: comparison of DNA extraction methods for ticks (Ixodes scapularis) PeerJ. 2015;3:e1147. doi: 10.7717/peerj.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill CA, Gutierrez JA. A method for extraction and analysis of high quality genomic DNA from ixodid ticks. Med Vet Entomol. 2003;17:224–7. [DOI] [PubMed]
- 22.Rubin BE, Sanders JG, Hampton-Marcell J, Owens SM, Gilbert JA, Moreau CS. DNA extraction protocols cause differences in 16S rRNA amplicon sequencing efficiency but not in community profile composition or structure. Microbiol Open. 2014;3(6):910–921. doi: 10.1002/mbo3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Willner D, Daly J, Whiley D, Grimwood K, Wainwright CE, Hugenholtz P. Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One. 2012;7(4):e34605. doi: 10.1371/journal.pone.0034605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rynkiewicz EC, Hemmerich C, Rusch DB, Fuqua C, Clay K. Concordance of bacterial communities of two tick species and blood of their shared rodent host. Mol Ecol. 2015;24(10):2566–2579. doi: 10.1111/mec.13187. [DOI] [PubMed] [Google Scholar]
- 25.Van Treuren W, Ponnusamy L, Brinkerhoff RJ, Gonzalez A, Parobek CM, Juliano JJ, et al. Variation in the microbiota of Ixodes ticks with regard to geography, species, and sex. Appl Environ Microbiol. 2015;81(18):6200–6209. doi: 10.1128/AEM.01562-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barker SC, Walker AR. Ticks of Australia. The species that infest domestic animals and humans. Zootaxa. 2014;3816:1–144. [DOI] [PubMed]
- 27.Roberts FHS. Australian ticks. Commonwealth Scientific and Industrial Research Organization: Canberra; 1970. [Google Scholar]
- 28.Swei A, Kwan JY. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2017;11(3):813–816. doi: 10.1038/ismej.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Šlapeta Š, Šlapeta J. Molecular identity of cat fleas (Ctenocephalides felis) from cats in Georgia, USA carrying Bartonella clarridgeiae, Bartonella henselae and Rickettsia sp. RF2125. Vet Parasitol (Amst) 2016;3(4):36–40. doi: 10.1016/j.vprsr.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 30.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke K, Gorley R. PRIMER v7: user manual/tutorial: PRIMER-E. 2015. [Google Scholar]
- 32.Clarke K. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol. 1993;18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 33.Kruskal J, Wish M. Quantitative applications in the social science: multidimensional scaling. Thousand Oaks: SAGE Publications Ltd; 1978. [Google Scholar]
- 34.Chen H. VennDiagram: Generate high-resolution venn and euler plots. R package version 1.0.1432016.
- 35.Boudova L, Kazakov D, Šíma R, Vanecek T, Torlakovic E, Lamovec J, et al. Cutaneous lymphoid hyperplasia and other lymphoid infiltrates in the breast nipple. Am J Dermatopathol. 2005;27:375–386. doi: 10.1097/01.dad.0000179463.55129.8a. [DOI] [PubMed] [Google Scholar]
- 36.Hii SF, Lawrence AL, Cuttell L, Tynas R, Abd Rani PA, Slapeta J, Traub RJ. Evidence for a specific host-endosymbiont relationship between ‘Rickettsia sp. genotype RF2125’ and Ctenocephalides felis orientis infesting dogs in India. Parasit Vectors. 2015;8:169. doi: 10.1186/s13071-015-0781-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangoulidis D, Meyer H, Kahlhofer C, Splettstoesser WD. ‘Real-time’ PCR-based detection of Coxiella burnetii using conventional techniques. FEMS Immunol Med Microbiol. 2012;64(1):134–136. doi: 10.1111/j.1574-695X.2011.00900.x. [DOI] [PubMed] [Google Scholar]
- 38.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148:257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 39.Chakravorty S, Helb D, Burday M, Connell N, Alland D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods. 2007;69:330–339. doi: 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS One. 2011;6(12):e27310. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van De Peer Y, Chapelle S, De Wachter R. A quantitative map of nucleotide substitution rates in bacterial rRNA. Nucleic Acids Res. 1996;24(17):3381–3391. doi: 10.1093/nar/24.17.3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Claesson MJ, Wang Q, O'Sullivan O, Greene-Diniz R, Cole JR, Ross RP, O'Toole PW. Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Res. 2010;38(22):e200. doi: 10.1093/nar/gkq873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siegwald L, Touzet H, Lemoine Y, Hot D, Audebert C, Caboche S. Assessment of common and emerging bioinformatics pipelines for targeted metagenomics. PLoS One. 2017;12(1):e0169563. doi: 10.1371/journal.pone.0169563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar PS, Brooker MR, Dowd SE, Camerlengo T. Target region selection is a critical determinant of community fingerprints generated by 16S pyrosequencing. PLoS One. 2011;6(6):1–8. doi: 10.1371/journal.pone.0020956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vishnivetskaya TA, Layton AC, Lau MC, Chauhan A, Cheng KR, Meyers AJ, et al. Commercial DNA extraction kits impact observed microbial community composition in permafrost samples. FEMS Microbiol Ecol. 2014;87(1):217–230. doi: 10.1111/1574-6941.12219. [DOI] [PubMed] [Google Scholar]
- 46.Halos L, Bord S, Cotte V, Gasqui P, Abrial D, Barnouin J, et al. Ecological factors characterizing the prevalence of bacterial tick-borne pathogens in Ixodes ricinus ticks in pastures and woodlands. Appl Environ Microbiol. 2010;76(13):4413–4420. doi: 10.1128/AEM.00610-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruaud P, Vigneron A, Lucchetti-Miganeh C, Ciron PE, Godfroy S, Cambon-Bonavita MA. Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl Environ Microbiol. 2014;80(15):4626–4639. doi: 10.1128/AEM.00592-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murrell A, Dobson SJ, Yang X, Lacey E, Barker SC. A survey of bacterial diversity in ticks, lice and fleas from Australia. Parasitol Res. 2003;89(4):326–334. doi: 10.1007/s00436-002-0722-4. [DOI] [PubMed] [Google Scholar]
- 49.Takano A, Fujita H, Kadosaka T, Konnai S, Tajima T, Watanabe H, et al. Characterization of reptile-associated Borrelia sp. in the vector tick, Amblyomma geoemydae, and its association with Lyme disease and relapsing fever Borrelia spp. Environ Microbiol Rep. 2011;3(5):632–637. doi: 10.1111/j.1758-2229.2011.00280.x. [DOI] [PubMed] [Google Scholar]
- 50.Sentausa E, El Karkouri K, Michelle C, Caputo A, Raoult D, Fournier PE. Genome sequence of Rickettsia tamurae, a recently detected human pathogen in Japan. Genome Announc. 2014;2(5):e00838–e00814. doi: 10.1128/genomeA.00838-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imaoka K, Kaneko S, Tabara K, Kusatake K, Morita E. The first human case of Rickettsia tamurae infection in Japan. Case Rep Dermatol. 2011;3(1):68–73. doi: 10.1159/000326941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kho KL, Koh FX, Tay ST. Molecular evidence of potential novel spotted fever group rickettsiae, Anaplasma and Ehrlichia species in Amblyomma ticks parasitizing wild snakes. Parasit Vectors. 2015;8:112. doi: 10.1186/s13071-015-0719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of tick samples used. Table S2. Summary of Qubit DNA concentrations and qPCR bacterial numbers estimates. Table S3. Summary of DNA sequencing of bacterial 16S rRNA gene. (XLSX 18 kb)
Data Availability Statement
The tick exoskeletons were donated to the Australian National Insect Collection (ANIC) in Canberra, Australia under database numbers ANIC 48 006553 to ANIC 48 006558. Nucleotide sequence data from this study are available in the GenBank (National Center for Biotechnology Information, NCBI) database under accession numbers MG004673–MG004678 (Rickettsia gltA), MG004679–MG004680 and MG452684 (Borrelia 16S rRNA gene), and MG004681–MG004686 (Bothriocroton cox1) and the SRA database under BioProject accession number: PRJNA411926 (https://www.ncbi.nlm.nih.gov/bioproject/411926). BIOM, PRIMER and associated files are available at LabArchives (10.6070/H4W094FD).