

Abstract
Background
Chondrogenesis is the earliest stage of skeletal development and is a highly dynamic process, integrating the activities and functions of transcription factors, cell signaling molecules and extracellular matrix proteins. The molecular mechanisms underlying chondrogenesis have been extensively studied and multiple key regulators of this process have been identified. However, a genome-wide overview of the gene regulatory network in chondrogenesis has not been achieved.
Results
In this study, employing RNA sequencing, we identified 332 protein coding genes and 34 long non-coding RNA (lncRNA) genes that are highly selectively expressed in human fetal growth plate chondrocytes. Among the protein coding genes, 32 genes were associated with 62 distinct human skeletal disorders and 153 genes were associated with skeletal defects in knockout mice, confirming their essential roles in skeletal formation. These gene products formed a comprehensive physical interaction network and participated in multiple cellular processes regulating skeletal development. The data also revealed 34 transcription factors and 11,334 distal enhancers that were uniquely active in chondrocytes, functioning as transcriptional regulators for the cartilage-selective genes.
Conclusions
Our findings revealed a complex gene regulatory network controlling skeletal development whereby transcription factors, enhancers and lncRNAs participate in chondrogenesis by transcriptional regulation of key genes. Additionally, the cartilage-selective genes represent candidate genes for unsolved human skeletal disorders.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4378-y) contains supplementary material, which is available to authorized users.
Keywords: Cartilage, Chondrocyte, RNA-seq, lncRNA, Skeletal dysplasia, Gene expression, Human
Background
Chondrogenesis is the earliest stage of skeletal development and begins during embryogenesis with the proliferation and condensation of mesenchymal cells, followed by lineage commitment and differentiation, to yield the chondrocytes which populate and synthesize the scaffold for patterning the early skeletal elements. Subsequently, growth and mineralization of endochondral bones proceeds by establishment of the growth plates followed by the ordered differentiation, proliferation and hypertrophy of growth plate chondrocytes. Most of the terminally differentiated hypertrophic chondrocytes undergo apoptosis, while a subset can transdifferentiate to osteoblasts [1, 2]. Finally, the cartilage matrix is mineralized to form bone. This highly dynamic developmental process is regulated by transcription factors that include SOX9, RUNX2 and the GLI family of proteins, with synergistic regulation by multiple cell signaling pathways including the fibroblast growth factor (FGF), hedgehog, bone morphogenetic protein (BMP), transforming growth factor beta (TGF-beta) and WNT signaling pathways [3, 4]. Disruption of the genes that participate in chondrogenesis can lead to inherited skeletal disorders, as observed in humans and mice, which are characterized by abnormalities in the formation, linear growth and/or maintenance of the skeleton. The skeletal disorders comprise a broad range of severity from embryonic lethal phenotypes with extensive skeletal abnormalities to mild disorders in which short stature and early onset osteoarthritis are the main manifestations [5, 6].
Systematic identification of genes expressed in cartilage can uncover gene regulatory networks that are important for chondrogenesis and chondrocyte biology, providing fundamental insight into cartilage development and function. Indeed high-throughput gene expression profiling followed by data mining has been used to identify tissue-specific/selective genes in multiple human tissues, including cartilage [7–11]. Studies in cartilage have discovered genes that are highly and/or primarily expressed in chondrocytes and have been used as markers to investigate the differentiation potential of mesenchymal stem cells toward chondrocytes and other cell lineages [12]. Not surprisingly, a large number of the genes selectively expressed in chondrocytes are associated with human skeletal disorders [7], and the published gene expression data have been used as a resource to facilitate studies that have led to identification of additional genes associated with human skeletal phenotypes. For example, TRPV4, COL11A1 and ACAN gene mutations were identified as the causes of autosomal dominant brachyolmia, fibrochondrogenesis and an autosomal recessive form of spondyloepimetaphyseal dysplasia, respectively, based in part on their cartilage-selective (CS) expression patterns [13–15].
Microarray technology has been widely used for the high-throughput detection of gene expression. However, microarray gene expression applications are limited by low signal-to-noise ratios for genes expressed at low levels, signal saturation for highly expressed genes, and detection restricted to a predefined gene set. The development of high throughput sequencing methods has facilitated measuring most cellular transcript levels by RNA sequencing (RNA-Seq) [16]. In contrast to microarray technology, RNA-seq provides more sensitive and accurate measurement of gene expression, and is able to discover novel transcripts and genes. To provide a deeper measure of both quantitative and qualitative gene expression in human growth plate chondrocytes, we used RNA-seq to acquire a comprehensive gene expression profile of the tissue. These data were complemented by publically available RNA-Seq and ChIP-seq data sets, also generated on high throughput sequencing platforms, to reveal regulatory mechanisms that underlie chondrogenesis and linear growth. The data have provided a comprehensive understanding of the transcriptional network in chondrocytes and suggested novel regulatory networks and mechanisms underlying chondrogenesis and skeletal development.
Results
Identification of cartilage-selective (CS) genes in human fetal cartilage
We sought to identify CS genes using RNA-seq data from human tissues and cells. Cartilage gene expression profiles were generated from 14 to 18 week distal femur growth plate chondrocytes. RNA-seq data from other tissues/cells were generated by previously described studies [17–23] and accessed from public databases as detailed in Methods. There were 111 RNA-seq gene expression datasets representing 22 tissues or cell types in the comparison data set (Additional file 1: Dataset S1). With the exception of the data from CD4+ and CD8+ primary cells, which were obtained from adults, the remaining data were generated from human fetal tissues/cells. Therefore, the gene expression data mainly reflected human fetal transcription. Data quality were assessed by calculating the Pearson’s correlation coefficients between datasets, requiring that gene expression patterns be similar in the same or closely related tissues, which would be reflected by high Pearson’s correlation coefficients. As shown in Additional file 2: Figure S1, tissues/cells with same origins had high Pearson’s correlation coefficients and formed unique clusters, indicating that the data were of high quality and therefore represented the intrinsic gene expression properties of the tissues/cells studied.
Using the PaGeFinder algorithm, we identified 403 transcripts, representing 332 protein coding genes, which were selectively expressed in cartilage (Additional file 3: Dataset S2). A boxplot of the selective gene expression showed that there was a set of genes that were highly expressed in cartilage as compared with other tissues/cells (Additional file 2: Figure S2). Many well-studied cartilage marker genes were identified and listed as top CS genes with high specificity measure (SPM) values, such as the known CS genes encoding types II, IX and XI collagens, aggrecan and matrix metalloproteinase 13 (Additional file 3: Dataset S2). We found that 63 (42%) of the CS genes previously discovered in our similar microarray-based study [7] were also identified as CS in the current study (Additional file 4: Dataset S3). Another 25 genes from the previous study [7] had high SPM values (between 0.7 and 0.9) in the current study, indicating they were preferentially, but not selectively, expressed in cartilage. This comparison indicates that the RNA-seq approach was capable of recapitulating the prior findings [7] and able to reveal known and newly identified genes selectively expressed in human fetal cartilage. There were also 57 CS genes from the microarray study that showed expression in non-cartilage samples in the RNA-seq dataset, reflecting the differences between the microarray and RNA-seq techniques. RNA-seq has a broader dynamic range that is able to better detect low-abundance transcripts [24]. Thus genes expressed at low levels on microarrays have signals close to background and are set as non-expressed while by RNA-seq the low level expression can be measured, revealing that they may not be selectively expressed.
GO and KEGG pathway analysis indicated that the cartilage-selective gene set was enriched for genes known to participate in skeletal system development. These included selectively expressed genes involved in cellular processes necessary for chondrogenesis, such as regulation of cell proliferation and apoptosis, cellular biosynthesis, cell adhesion, blood vessel morphogenesis, transcriptional regulation and TGFβ signaling (Fig. 1a, b).
Fig. 1.
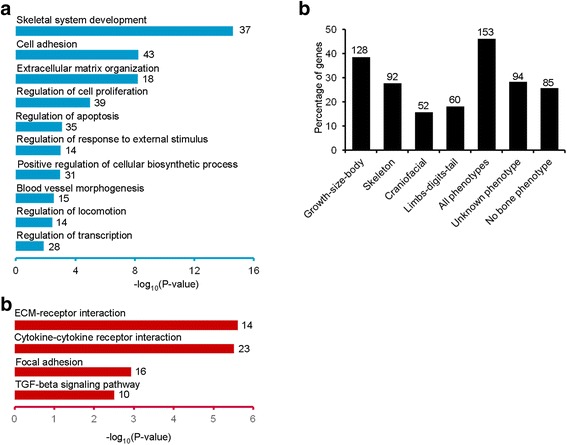
Cartilage-selective genes are required for proper bone formation. a Gene ontology (GO) analysis of cartilage-selective (CS) genes. b KEGG pathway analysis of cartilage-selective genes. The number of genes in each GO term is indicated. c CS genes associated with skeletal phenotypes in gene targeted mice
Cartilage-selective genes are required for skeletal development
As the GO analysis of the CS genes indicated that they have roles in skeletal development, it would be expected that disruption of their functions in mice and/or humans would lead to skeletal abnormalities. To assess this, we retrieved mouse phenotypes from the Mouse Genome Informatics Database (MGI, http://www.informatics.jax.org/) and found that mutations in 153/332 (46%) of the CS genes were associated with mouse skeletal defects (Fig. 1c; Additional file 3: Dataset S2). Using the MGI descriptors, the 153 genes were represented across a number of overlapping categories, with 128 genes associated with growth/size/body phenotypes, 92 genes associated with skeletal phenotypes, 52 genes associated with craniofacial phenotypes, and 60 genes associated with limb/digit/tail phenotypes. For the remaining 179 genes, 94 genes were not present in the MGI phenotype list and 85 genes did not show a skeletal phenotype when targeted in mice. Despite the latter observation, a review of the literature revealed that many of these genes do have roles in skeletogenesis, including human skeletal disorders due to mutations in SHOX, FBLN7 and VIT as examples [25–27]. In addition, although knockout mutations targeting Trpv4 or Comp do not show skeletal phenotypes in mice, dominant negative structural mutations in these genes result in skeletal disorders in humans [14, 28–30]. In this context, we compared the CS genes with the genes associated with human genetic skeletal disorders, expecting that many of the genes would be associated with skeletal dysplasias [5]. There were 32 of the CS genes (Additional file 3: Dataset S2) associated with a broad spectrum of 62 human skeletal disorders, 26 of which were also associated with mouse skeletal phenotypes. The human and mouse skeletal phenotypes highlight the essential roles of the CS genes in skeletal development but also reveal that loss-of-function mouse models do not identify the essential roles of all genes important for skeletogenesis.
Cartilage-selective gene products interact
To decipher relationships among the proteins encoded by the selective genes, a protein-protein interaction network was built with Cytoscape [31]. As shown in Fig. 2, proteins encoded by the genes showed extensive physical interactions and formed a large interaction network. Matrix metallopeptidases had complex interactions with extracellular matrix proteins, such as CS collagens and aggrecan, consistent with their central roles in processing these proteins and regulating bone mineralization. TGFβ family signaling molecules including BMP2/6, ACVR1 and GDF5 also showed physical interactions (Fig. 2), in agreement with their regulatory functions in chondrogenesis and bone formation [32]. Thus the proteins specified by the CS genes comprise a functional cartilage interactome.
Fig. 2.
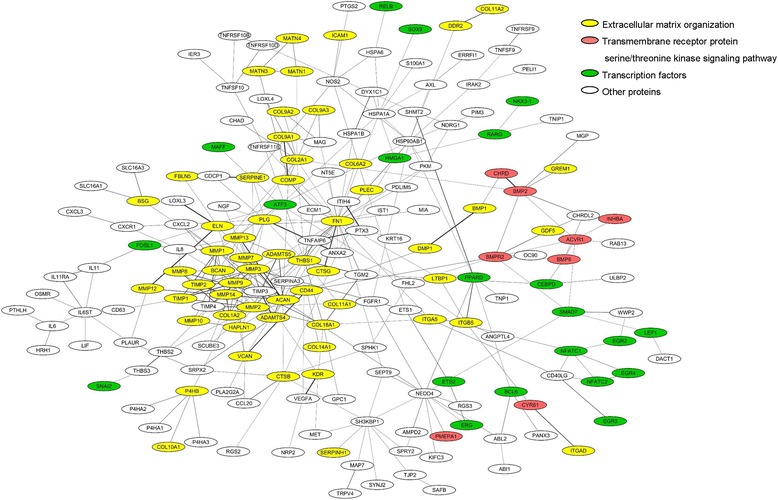
Protein-protein interaction network for the CS genes. CS proteins are represented as colored nodes. Node colors represent three GO groups as noted
Transcriptional regulation of cartilage-selective genes
We expected that CS transcription factors could function as master regulatory molecules to control downstream expression of additional selectively expressed genes. By comparing the CS genes with the transcription factor genes annotated in the Animal Transcription Factor Database (AnimalTFDB) [33], 34 selectively expressed cartilage transcription factors were identified [Figs. 1a, 2; Additional file 5: Dataset S4]. These transcription factors are known or now predicted to participate in multiple cellular activities that regulate chondrocyte development (Fig. 3a). To determine if these factors control CS gene transcription, we searched for transcription factor binding motifs in the promoter regions of the CS genes. As shown in Fig. 3b, the promoters were enriched in consensus-binding sites for transcription factors that were selectively expressed in cartilage, including SOX6, CREB3L2, EGR2/3/4, GLIS3, PLAGL1, RELB and SNAI1/2, all of which have been shown to play important roles in skeletal development [34–39]. Although RUNX2 was not identified as a CS gene, since it was also expressed in lymphocytes (Additional file 2: Figure S3), its binding motif was also enriched among the CS promoters (Fig. 3b), consistent with its known transcriptional regulatory role in chondrogenesis. These data suggest that expression of the selective genes might be directly regulated by a group of transcription factors that are uniquely expressed in cartilage.
Fig. 3.
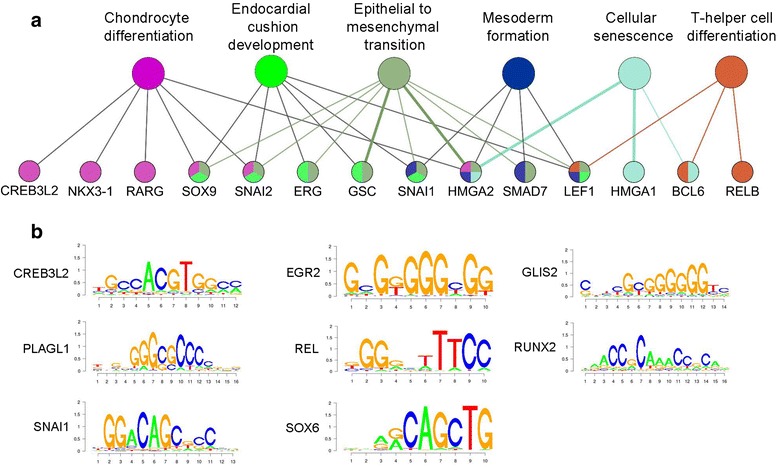
Cartilage-selective transcription factors and DNA binding motif analysis. a ClueGO network analysis for enrichment biological process modules within transcription factors. GO terms are represented as large nodes and genes are represented as small nodes. Node colors represent GO groups. b Transcription factor DNA binding motif analysis shows enrichment of motifs in cartilage-selective gene promoters. All P values for motif enrichment are less than 10−10
The binding motif of SOX6 was also enriched among the CS gene promoters, and SOX6 cooperates with SOX5 to secure SOX9 binding to CS enhancers in chondrocytes [40]. SOX9 is expressed in skeletogenic mesenchymal cells at early embryonic stages and regulates chondrogenesis through transcriptional activation of target genes [41, 42]. SOX9 mutations in humans produce campomelic dysplasia, a generally lethal osteochondrodysplasia with distinctive abnormalities in cartilage and bone [43, 44]. As has been previously observed in animal models, SOX9 is selectively expressed in cartilage and is required for expression of CS genes in chondrocytes [41, 42, 45]. In our analysis, 247 (74.4%) CS genes had SOX9 binding sites in their promoter regions (±2 kb of TSS). These CS genes included 25 transcription factor genes which by ChIP-seq had SOX9 bound to their TSS (Fig. 4a), with 13 of them showing strong SOX9 binding intensity (>25% quantile of all SOX9 binding sites). For instance, SOX9 strongly binds to the TSS of transcription factor genes Egr2 and Fosl1 (Fig. 4b, c), which are essential for skeletogenesis in mice [36, 46]. Because both SOX9 and EGR2 are highly expressed and regulate gene expression in mesenchymal stem cells (MSC) [41, 42, 47, 48], it is plausible to speculate that SOX9 controls chondrogenesis through functions as an upstream transcription factor that directly activates expression of downstream transcription factors.
Fig. 4.
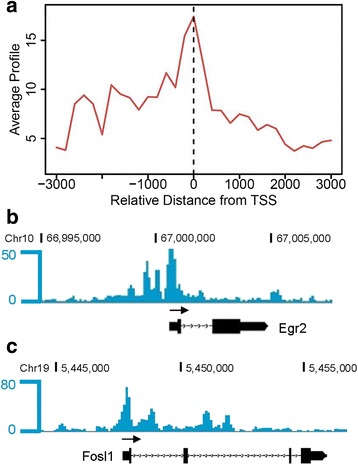
Sox9 binds to the cartilage-selective transcription factor gene promoters in mouse chondrocytes. a Average binding profile of Sox9 at the CS transcription factor gene TSS. The distance on the plot is measured in base pairs. b, c Genome browser views of Sox9 peaks along the Egr2 b and Fosl1 c loci
Cartilage-selective genes are regulated by active enhancers
Spatiotemporal gene expression is modulated by both transcription factors and epigenetic mechanisms such as enhancers [49]. Enhancers are distinct genomic regions bound by transcriptional activators and are marked by histone modifications such as histone H3 lysine 4 monomethylation (H3K4me1) and lysine 27 acetylation (H3K27ac) [50, 51]. Activation of tissue-specific enhancers provides a molecular mechanism that turns on gene expression in a cell type-specific manner [49]. Genome-wide studies in chondrocytes have identified a set of genes involved in chondrogenesis that are regulated by enhancers [52–54], so we asked whether CS gene expression might be activated by enhancers. Active enhancers in human chondrocytes were predicted by mapping the genome-wide distribution of H3K27ac [55], revealing 31,235 active chondrocyte enhancer regions marked by this chromatin modification (Additional file 6: Dataset S5). By comparing these data with enhancers across seven fetal tissues, we identified a subset of 11,334 enhancers that were uniquely present in chondrocytes. These enhancers were specifically enriched at CS gene loci including COL10A1, COL2A1 and COL11A1 (Fig. 5a and Additional file 2: Figure S4). Similar to other tissues, the chondrocyte enhancers were predominantly bound by H3K4me1 and H3K27ac (Fig. 5b, c). To predict enhancer target genes, we examined genome-wide enhancer-gene associations in chondrocytes with GREAT [56]. As shown in Fig. 6a, the chondrocyte unique enhancers were associated with genes involved in skeletal developmental processes including bone morphogenesis, chondrocyte differentiation, cellular carbohydrate biosynthesis, polysaccharide biosynthesis and endochondral ossification. 176 (53%) of the CS genes had active chondrocyte unique enhancers flanking their loci, suggesting that these genes are potentially enhancer regulated.
Fig. 5.
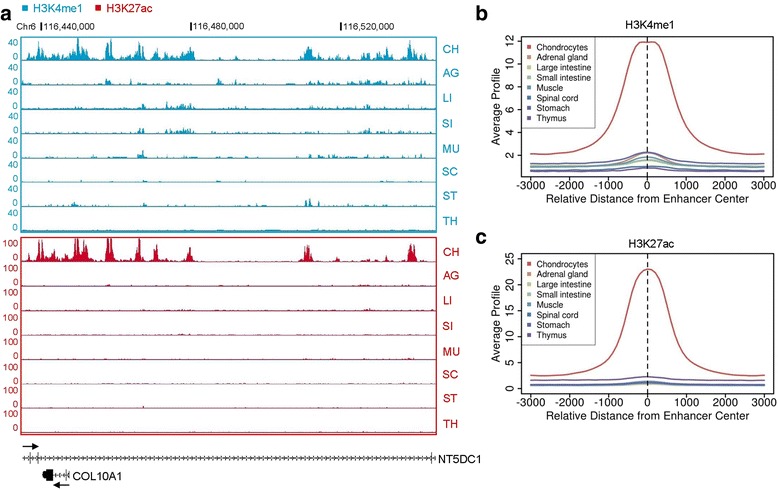
Identification of active enhancers in chondrocytes. a Genome browser views of H3K4me1 and H3K27ac peaks along the COL10A1 locus on chromosome 6. Chromosome coordinates are shown as black bars on top. Black arrows indicate the direction of transcription on a diagram of the gene below. CH, chondrocytes. AG, adrenal gland. LI, large intestine. SI, small intestine. MU, muscle. SC, spinal cord. ST, stomach. TH, thymus. b and c Genome-wide average of b H3K4me1 and c H3K27ac enrichment at chondrocyte enhancers in human fetal tissues and cells. The distance on the plot is measured in base pairs
Fig. 6.
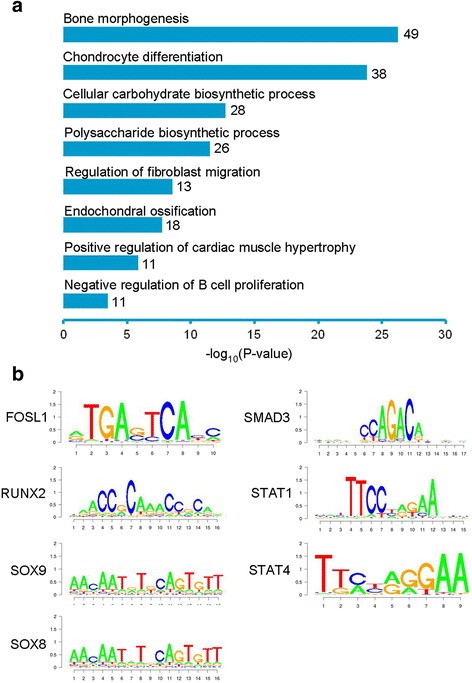
GO and DNA binding motif analysis of chondrocyte-unique enhancers. a GO analysis of chondrocyte enhancers. The number of genes in each GO term is indicated. b Enrichment of transcription factor DNA binding motifs on chondrocyte enhancers. All P values for motif enrichment are less than 10−10
With DNA motif analysis, we found that the chondrocyte unique enhancers were enriched for consensus-binding sites for transcription factors that have pivotal roles in chondrogenesis, including RUNX2, SOX8/9, SMAD3, STAT1/4 and FOSL1 (Fig. 6b), in agreement with the fact that they regulate transcription in part through binding to enhancers [52, 54, 57–60]. Furthermore, binding sites for additional transcription factors were also found to be enriched among the chondrocyte unique enhancers, suggesting roles in transcriptional regulation in chondrocytes that could be further explored experimentally (Additional file 6: Dataset S5).
Identification of cartilage-selective lncRNAs
LncRNAs are emerging as transcriptional regulators that participate in skeletogenesis through regulation of chondrocyte and osteoblast differentiation and homeostasis [61]. Since most lncRNAs are polyadenylated, and therefore can be measured by mRNA-seq, we analyzed the RNA-seq data and identified 34 lncRNAs genes selectively expressed in cartilage. LncRNAs have been shown to act as cis-regulators that modulate expression of neighboring genes [62, 63]. To identify potential regulatory targets, we used GREAT analysis to reveal protein coding genes neighboring the cartilage selective lncRNAs (Additional file 7: Dataset S6). Eight lncRNAs were adjacent to genes known to be involved in skeletal development including SOX9, GLI2, FOXA2, UMPS, SOD2, BHLHE41 and RAB11FIP4. Three lncRNAs (LINC00673, AC058791.1 and RP11–261P24.2) were embedded in active enhancer regions that were marked by H3K27ac and H3K4me1 in chondrocytes (Fig. 7a; Additional file 2: Figure S5), suggesting the lncRNA expression and enhancer activity might be functionally related. Among these, we were particularly interested in LINC00673 (annotated as a fusion gene with LINC00511 by ENSEMBL) because it is transcribed from a locus on human chromosome 17 that is ~280 kb downstream of SOX9. In human chondrocytes, LINC00673 was highly expressed and its promoter was enriched in H3K4me3, a marker for active transcription. The active enhancers at the LINC00673 gene locus span a ~0.5 Mb region downstream of SOX9. This region and the upstream region of SOX9 were marked with H3K27ac and shown to interact with the SOX9 promoter in melanoma cells that express SOX9 [64]. These data suggest that the LINC00673 locus might function as an enhancer to regulate SOX9 transcription (Fig. 7a).
Fig. 7.
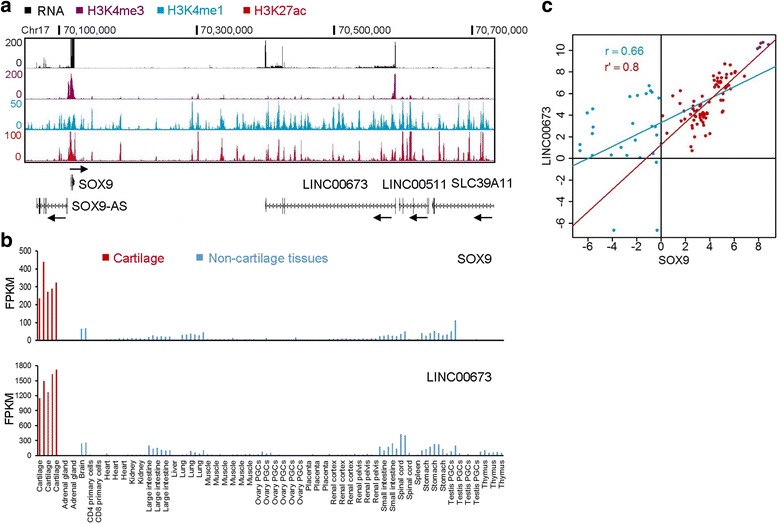
LINC00673 is co-expressed with SOX9. a Genome browser views of H3K4me1, H3K4me3, H3K27ac peaks and RNA expression track along SOX9 and LINC00673 loci on chromosome 17 in human chondrocytes. b Bar graphs showing SOX9 and LINC00673 expression in human tissues. Red bars indicate cartilage samples and blue bars indicate non-cartilage samples, respectively. Gene expression levels are shown as FPKM values. c Correlation between SOX9 and LINC00673 expression (Log2FPKM) in human tissues. Scatter plot of SOX9 against LINC00673 with regression lines showing a correlation between the two genes using the Pearson’s correlation coefficient. Red and blue dots indicates the tissues with SOX9 Log2FPKM value greater or less than 1, respectively. Cartilage samples are highlighted as purple dots. The blue regression line was generated with data from all tissues. The red regression line was generated with data from the tissues that have SOX9 FPKM value ≥1. The Pearson’s correlation coefficient for each regression line is shown
LINC00673 and SOX9 were highly expressed in chondrocytes, and also had low expression in brain, kidney, intestine, lung, spinal cord and stomach (Fig. 7b). Among all tissues, the expression levels of LINC00673 and SOX9 were positively correlated with each other, yielding a Pearson’s correlation coefficient 0.66. When we only considered the tissues/cells that express SOX9 (FPKM > 1), the Pearson’s correlation coefficient reached 0.8, indicating LINC00673 and SOX9 have orchestrated expression patterns across tissues (Fig. 7c). To validate the finding, we analyzed gene expression data using the UCLA Gene Expression Tool (UGET) [65], which reported the Pearson’s correlation coefficient for every pair of 54,675 probe sets on Affymetrix HG U133 Plus 2.0 arrays from the ∼12,000 human expression profiles. Among the 38,500 genes detected by the Affymetrix arrays, SOX9 was ranked as the gene with the second highest correlation with LINC00673 expression (the most highly correlated gene was LINC00673 itself), with a Pearson’s correlation coefficient 0.43. Reciprocally, LINC00673 was ranked as the gene that had the nineteenth highest correlation with SOX9 expression, with a Pearson’s correlation coefficient 0.39. The high Pearson’s correlation coefficients suggest a gene expression co-regulatory mechanism between SOX9 and LINC00673 (Additional file 7: Dataset S6).
Because SOX9 positively regulates transcription through binding to gene promoters and enhancers [54], we also considered the possibility that SOX9 could directly bind to LINC00673 and activate its transcription. To assess this, we examined SOX9 binding sites in mouse chondrocytes [52] and found that SOX9 bound to the promoter and structural gene for 2610035D17Rik, the orthologue of LINC00673 in mice, strongly suggesting that expression of the gene is regulated by SOX9 (Additional file 2: Figure S6). Because LINC00673 was selectively expressed in chondrocytes and was embedded in the enhancer region downstream of SOX9 gene, it is also possible that there is reciprocal regulation in which LINC00673 could work with enhancers to activate SOX9 and possibly other genes, and therefore participate in regulation of chondrogenesis.
Discussion
Organogenesis is a process of proliferation and differentiation of progenitor cells into diverse types of cells with distinct functions. Their unique functions are largely acquired through expression of tissue-specific/selective genes. Studies of gene expression patterns and regulatory mechanisms in particular tissues thus inform our understanding of tissue development and mechanisms of disease. Tissue-specificity refers to genes whose expression is restricted to one particular tissue or cell type, while tissue-selective expression includes genes that are expressed in one or a few generally biologically similar tissue types [66]. The data presented in this study described 332 cartilage-selective genes expressed in human fetal cartilage, providing unique insights into chondrocyte gene expression and regulation.
The significance of cartilage-selective gene identification
High expression of the CS genes in chondrocytes suggests they may have essential roles in skeletal development and skeletal disorders. In support of this hypothesis, 32 CS genes were linked to human skeletal disorders that result from mutations in these genes. Gene-targeting mice for 153 genes showed skeletal development defects. Among the list, there were well-known genes whose products have pivotal roles in chondrogenesis, such as cartilage collagens, cell signaling molecules and transcription factors. Base on this observation, we suggest that some of the newly identified CS genes may be good candidate genes for skeletal disorders in which the associated genes have yet to be identified. As new skeletal dysplasia loci are defined, the candidate genes can be compared with the CS genes to promote rapid identification of the mutated gene. Gene network analysis revealed complex interactions of CS gene products, data compatible with the hypothesis that CS genes are functionally related in the initiation and progression of complex diseases such as osteoarthritis. It may thus be useful to correlate gene mutations and dysregulation of gene expression among the chondrodysplasias with the CS gene interactome for further understanding of the pathogenesis of cartilage disorders [67]. In addition, we identified 94 genes that are less studied and for which knockout mice phenotypes have not been annotated by MGI (Additional file 3: Dataset S2). According to GO analysis, 39 of these genes are involved in cell signaling pathways and 27 genes encode secreted proteins. Their unique cartilage expression pattern suggests the genes might participate in chondrogenesis. For example, C2orf82 is highly expressed in human cartilage with an average FPKM value >1500 and encodes a type I single-pass transmembrane chondroitin sulfate proteoglycan. The gene showed a CS expression pattern with the highest expression in proliferating and prehypertrophic zones in embryonic and postnatal growth plate. Its expression was restricted to the uncalcified zone in adult articular cartilage, including chondrocyte clusters in human osteoarthritic cartilage. Its expression pattern was similar to the highly expressed CS genes Sox9, Acan and Col2a1, and could be induced by BMP-2, suggesting its functions may be associated with the development and maintenance of the chondrocyte phenotype [68]. As a second example, zinc-finger 385 family genes including ZNF385B/C/D were also selectively expressed in cartilage and another family member, ZNF385A, was also highly expressed in the tissue (Additional file 3: Dataset S2). Fluorescence-labeled Znf385C protein was highly expressed in maturing chondrocytes in the pharyngeal arches as well as in the cartilage associated with the ear and ventral fins in zebrafish [69]. Loss of Znf385C in zebrafish led to craniofacial defects and premature ossification of the Meckel’s cartilage, potentially through disturbance of p53-dependent cell cycle regulation [69]. Therefore, targeting some of the 94 understudied CS genes is likely to reveal new biology related to cartilage function.
Regulation of chondrogenesis by epigenetic mechanisms
Epigenetic mechanisms such as histone modifications and enhancers regulate transcription in skeletal development and disease [70, 71]. Genome-wide profiling of SOX9 indicated that it functions in part by binding to enhancers. Disruption of upstream distal intergenic regions especially the regions between -50 kb and -350 kb 5′ of SOX9 by deletion/translocation/inversion are associated with severe campomelic dysplasia [72, 73]. Systematically screening the 350 kb region upstream of SOX9 using histone modification markers and DNase I hypersensitivity assays identified ten potential enhancers, with three enhancers primarily active in chondrocytes and that activate SOX9 expression in the chondrocyte lineage [74]. SOX9, in complex with SOX5 and SOX6, reciprocally activate the enhancers, which provides a potential mechanism for autoregulation of its own expression in chondrocytes [74]. In the current study, genome-wide mapping of histone marks led to identification of 11,334 enhancers that were uniquely active in chondrocytes and associated with 176 of the CS genes. The identified active enhancers could be used as a resource to explore epigenetic regulation and higher-order chromatin organization in chondrocytes. The data also indicated that these enhancers were enriched in the consensus binding motifs for well-known transcription factors such as SOX9 and RUNX2, suggesting that transcription factor-bound enhancers contribute to the selective gene expression pattern in chondrocytes.
lncRNAs and chondrogenesis
lncRNAs are non-protein coding transcripts with lengths of more than 200 nucleotides, which are proposed to regulate transcription through both trans- and cis-regulatory mechanisms. Some lncRNAs tether to their target gene loci and affect transcriptional activity of neighboring genes through association with regulatory proteins. They could also bind to regions on other chromosomes and also control gene activity through recruiting regulatory proteins [75]. The roles for lncRNAs in skeletal development are emerging with a growing number of lncRNAs identified that participate in skeletal development and disease [61]. In our study, 34 lncRNAs were identified to be selectively expressed in human cartilage. Eight lncRNAs are adjacent to loci of protein coding genes that participate in skeletal development, suggesting the cartilage-selective lncRNAs may be involved in chondrogenesis through regulation of protein coding genes. We found that LINC00673 and SOX9 are co-regulated in human tissues, and that LINC00673 is embedded within a potential enhancer region for SOX9, suggesting LINC00673 may regulate SOX9 expression through a distal enhancer. In support of this hypothesis, by Circularized Chromosome Conformation Capture sequencing (4C–seq), the LINC00673 locus was found to be involved in long-range enhancer-promoter interactions at the SOX9 locus in SOX9-expressed melanoma cells but not in SOX9-inactive cells, consistent with the enhancer-promoter interactions driving SOX9 activation [64]. In addition, LINC00673 regulates target gene expression in gastric cancer cells by recruiting histone methyltransferase EZH2 and demethylase LSD1 to target gene promoters, suggesting that it can function as a transcriptional regulator through epigenetic mechanisms [76]. While the exact role of LINC00673 in SOX9 gene regulation is unknown, loss- and gain-of-function experiments could be used to directly address this question.
Cartilage-selective genes and mouse phenotypes
There were 85 CS genes that did not show a skeletal phenotype when targeted in mice (Figure 1C). Some of these genes may have overlapping functions with paralogous genes. For example, NKX3.1 is a transcription factor selectively expressed in cartilage. Inactivation of the Nkx3.1 gene produced no apparent skeletal phenotype, while knockout of its paralogue Nkx3.2 in mice led to a lethal skeletal dysplasia, with abnormalities of the vertebral column and craniofacial bones [77–80]. Nkx3.1 and Nkx3.2 double knockout mice showed enhanced vertebral defects compared with Nkx3.2 knockout mice, suggesting that Nkx3.1 and Nkx3.2 regulate skeletal development coordinately [81]. In addition, although knockout of some CS genes did not produce skeletal defects in mice, such mutations in humans produced skeletal disorders. As an example, knockout of Wisp3 in mice did not yield skeletal abnormalities, while human loss-of-function mutations in WISP3 produce the autosomal recessive skeletal disease progressive pseudorheumatoid dysplasia in humans (OMIM 208230). Depletion of the gene in zebrafish affected mandibular and pharyngeal cartilage development and over-expression Wisp3 in zebrafish indicated that it might function as a BMP and Wnt signaling modulator [82, 83]. As another example, COMP encodes a noncollagenous extracellular matrix protein that interacts with cartilage collagens and promotes formation of well-defined collagen fibrils [84]. In contrast to COMP knockout mice, which showed normal skeletal development, dominant missense mutations of human COMP result in autosomal dominant pseudoachondroplasia and multiple epiphyseal dysplasia type 1 [29, 85, 86]. Furthermore, pseudoachondroplasia knock-in mice carrying the COMP p.Thr583Met mutation that causes a skeletal phenotype in humans exhibited short-limb dwarfism and severe degeneration of articular cartilage [87]. COMP p.Asp469del knock-in mice developed a progressive short-limb dwarfism and hip dysplasia, with disorganized growth plate [88]. Finally, dominant structural mutations in TRPV4, which encodes a Ca2+ permeable ion channel that responds to chemical and physical stimuli, result in a broad spectrum of inherited skeletal disorders in humans [14, 28, 89–91]. Trpv4 knockout mice showed bladder dysfunction and hearing loss without obvious skeletal defects [92]. However, Trpv4 transgenic mice expressing the known pathogenic missense mutation p.Arg594His showed a severe skeletal dysplasia that recapitulated abnormalities of metatropic dysplasia in humans [93]. In the latter examples, dominant negative or gain-of-function mutations interrupt normal chondrocyte activities and lead to abnormal skeletal phenotypes that mouse knockouts did not reveal.
Limitations and future directions
The goal of this study was to identify key genes and transcriptional regulatory mechanisms in chondrogenesis using total RNA isolated from fetal distal femur cartilage, including all types of chondrocytes from the growth plate. As a consequence, we were not able to determine if the selective genes are uniformly expressed throughout the growth plate or are preferentially expressed in certain chondrocyte cell type(s). For example, it is known that SOX9 is expressed in reserve and proliferating chondrocytes and COL10A1 is expressed in hypertrophic chondrocytes [94, 95]. Laser capture microdissection followed by RNA-seq could further define the expression patterns of the cartilage-selective genes. In addition, due the limited number of tissue types and samples used in this study, and objective but arbitrary criteria for identifying cartilage selectivity, the CS gene list could change if the criteria for identifying CS selective genes are changed, or as additional fetal tissue expression profiles are added. Curation of additional RNA-seq data from human tissues and developmental stages will help to better define tissue selective genes and promote functional studies at spatial and temporal scales.
Conclusions
In summary, the current study identified 332 protein coding genes and 34 lncRNA genes that are selectively expressed in human fetal cartilage. Proteins transcribed from the cartilage-selective genes formed a complex interaction network and participate in multiple processes in skeletal development. The data further indicate that transcription factors, enhancers and lncRNAs work cooperatively to mediate transcriptional regulation in cartilage. In addition to known regulatory molecules, our study also discovered novel protein coding genes and lncRNAs that are likely to regulate chondrogenesis. Together, these findings help us to better understand the mechanisms underlying chondrogenesis and will be instrumental in further dissecting gene regulatory mechanisms in skeletal pattern formation, endochondral ossification and chondrocyte function.
Methods
Cartilage specimen collection and RNA isolation
Under a UCLA IRB-approved human subjects protocol, five independent 14–18 week distal human femur growth plate cartilage samples were obtained with informed consent. RNA was isolated and purified from the tissue as previously described [96]. Briefly, the fetal cartilage was digested in 0.3% bacterial collagenase, the chondrocytes were collected by centrifugation and total RNA was isolated with TRIZol (Thermo Fisher Scientific). Genomic DNA contamination was removed by DNase I (Thermo Fisher Scientific) digestion and the RNA was further purified with the RNeasy Mini Kit (Qiagen). All of the RNA samples had RIN (RNA Integrity Number) ≥8 (measured with the Agilent 2100 BioAnalyzer).
Gene expression analysis
PolyA-tailed mRNA was isolated from total RNA and used as the input for sequencing library construction with the TruSeq RNA Preparation Kit (Illumina). Sequencing was performed on the Illumina HiSeq 2000 instrument (Additional file 1: Dataset S1). The sequencing data analysis pipeline was described previously [97]. Briefly, RNA-seq reads were mapped to the human genome (hg19) using TopHat (v2.0.9), allowing up to 2-bp mismatches per read [98]. The mapped reads were analyzed using the Cufflinks (v2.2.1) package [99]. The transcript abundance in each sample was quantified with Cuffquant. The Cuffquant outputs for all samples were then joined and normalized with Cuffnorm. Gene expression level is represented as FPKM (fragments per kilobase of exon per million fragments mapped) values. Refseq gene annotation was obtained from the UCSC genome website (https://genome.ucsc.edu/). LncRNA gene annotation was downloaded from GENCODE [100]. Low abundance genes with FPKM values less than 1 were filtered before downstream analysis.
Identification of cartilage-selective (CS) genes
The PaGeFinder program has been used to define gene expression patterns from high-throughput transcriptomic data, which applies cosine similarity measurements to discover housekeeping and tissue-specific/selective genes [101]. The specificity measure (SPM) value produced by PaGeFinder was used as a criterion to estimate the relative expression specificity of a gene in a given sample. To identify CS genes, gene expression FPKM values from multiple samples within the same tissue/cell type were averaged, yielding a table with average FPKM values for all measured genes. The data were then processed with PaGeFinder and SPM values were generated. CS genes were defined as the subset of genes with average FPKM values ≥1 and SPM ≥0.9 in cartilage samples. For the cartilage-selective lncRNAs, to eliminate false-positives, lncRNAs that had sense strand exonic overlap with expressed protein-coding genes or that were embedded in gene introns for which there was intron-retention, were excluded from the cartilage-selective lncRNA list.
External data sources
The data used in this study are summarized in Additional file 1: Dataset S1. Some of the RNA-seq data from normal human tissues/cells have been reported by others [17–23]. Histone modification ChIP-seq data were downloaded from the NCBI epigenome roadmap project (http://www.ncbi.nlm.nih.gov/geo/roadmap/epigenomics/) [20]. Processed alignment files for histone ChIP-seq in human fetal tissues were also downloaded from the epigenome roadmap project website (http://egg2.wustl.edu/roadmap/data/byFileType/alignments/consolidated/). Genome-wide SOX9 binding data in mouse chondrocytes were described previously [52]. All downloaded data were processed using the UCSC hg19 build.
ChIP-seq data processing
Bowtie 0.12.7 was used to align sequenced reads to the human (hg19, for histone modifications) and murine genomes (mm9, for SOX9 ChIP-seq). Reads that aligned to more than one genomic location or that had more than 2 mismatches were discarded. The human genome was divided into 100 bp windows and peak calling was performed with MACS (v1.3.7.1) using its default P-value [102].
Prediction of active enhancers
Active enhancers were predicted based on H3K27ac ChIP-seq peaks that were >3 kb away from gene transcription start sites (TSS) and H3K4me3 peaks (a marker for active promoters) [55]. Enhancers within 1 kb of each other were merged. Chondrocyte-unique enhancers were defined as the enhancers only detected in chondrocytes in comparison with other tissues.
Gene ontology (GO) and network analysis
CS genes were submitted to DAVID for GO and KEGG pathway analysis [103]. GO analysis for chondrocyte-unique enhancers was performed with the Genomic Regions Enrichment of Annotations Tool (GREAT) using default settings [56]. Genome coordinates for CS lncRNAs were submitted to GREAT and protein coding genes flanking lncRNA loci were identified. Gene network analysis was performed with Cytoscape 3.2.1, using plug-ins ClueGO and GeneMANIA [31, 104, 105].
Motif analysis
Enrichment analysis of known transcription factor binding motifs was performed with SeqPos [106] using DNA sequences from gene promoter regions (±2 kb from TSS) or enhancers.
Data processing and visualization
Heatmaps, boxplots and scatter plots were generated with R. Sequencing data were visualized with the Integrated Genome Browser [107].
Additional files
Supplementary Dataset S1. Data used in the study. (XLSX 17 kb)
Dendrogram and heatmap of the Pearson’s correlation coefficient for human tissue gene expression (Log2FPKM). Only genes with FPKM > 1 in at least one sample were used for analysis. The cartilage samples are highlighted with a red frame. Figure S2. Box plots showing cartilage-selective gene expression in cartilage (red) and non-cartilage (blue) tissues. The thick horizontal line in each box represents the median gene expression value. Figure S3. Bar graph showing RUNX2 expression in human tissues. Red bars indicate cartilage samples and blue bars indicate non-cartilage samples, respectively. Gene expression levels are shown as FPKM values. Figure S4. Genome browser views of H3K4me1 and H3K27ac peaks along a COL2A1 and b COL11A1 loci. Chromosome coordinates are shown as black bars on top. Black arrows indicate the direction of transcription on a diagram of each gene below. CH, chondrocytes. AG, adrenal gland. LI, large intestine. SI, small intestine. MU, muscle. SC, spinal cord. ST, stomach. TH, thymus. Figure S5. Genome browser views of H3K4me1, H3K4me3, H3K27ac peaks and RNA expression track along lncRNA loci (A) AC058791.1 and (B) RP11–261P24.2 in human chondrocytes. Figure S6. Sox9 binds to the lncRNA 2610035D17Rik promoter and gene body in mouse rib chondrocytes. a Genome browser view of Sox9 binding along Sox9 and 2610035D17Rik loci on chromosome 11. b Shaded region in a was enlarged for a close view of Sox9 binding at the 2610035D17Rik promoter and first intron. (PDF 912 kb)
Supplementary Dataset S2. Cartilage-selective genes. (XLSX 45110 kb)
Supplementary Dataset S3. Data comparison between microarray and RNA-seq platforms. (XLSX 8 kb)
Supplementary Dataset S4. Cartilage-selective transcription factors and motif analysis. (XLSX 26 kb)
Supplementary Dataset S5. GO analysis of cartilage-unique enhancers. (XLSX 1028 kb)
Supplementary Dataset S6. Cartilage selective lncRNAs. (XLSX 17845 kb)
Acknowledgements
We thank Min Chen, Jer-Young Lin and Bob Goldberg (UCLA) for their assistance with the RNA-seq experiment.
Funding
This study was supported in part by NIH grants RO1AR062651 and RO1AR066124.
Availability of data and materials
The RNA-seq data sets generated in this study have been deposited in the Gene Expression Omnibus (GEO) under accession GSE107649.
Authors’ contributions
BL performed most of the experiments and analyses including data generation, computational analysis, methodological application and writing the original draft; KB participated in data generation, as well as review and editing of the manuscript; DK participated in conceptualizing the project, supervising the work, and reviewing and editing of the manuscript; DHC participated in conceptualizing the project, supervising the work, and reviewing and editing of the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Under a UCLA IRB-approved human subjects protocol, five independent 14-18 week distal human femur growth plate cartilage samples were obtained with informed consent. (Protocol ID: IRB#14-000177).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4378-y) contains supplementary material, which is available to authorized users.
Contributor Information
Bing Li, Email: thingen12@gmail.com.
Karthika Balasubramanian, Email: kbalasub@g.ucla.edu.
Deborah Krakow, Email: dkrakow@mednet.ucla.edu.
Daniel H. Cohn, Email: dcohn@mcdb.ucla.edu
References
- 1.Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014;10(12):e1004820. doi: 10.1371/journal.pgen.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. 2014;111(33):12097–12102. doi: 10.1073/pnas.1302703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem. 2006;97(1):33–44. doi: 10.1002/jcb.20652. [DOI] [PubMed] [Google Scholar]
- 4.Kozhemyakina E, Lassar AB, Zelzer E. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development. 2015;142(5):817–831. doi: 10.1242/dev.105536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015;167A(12):2869–2892. doi: 10.1002/ajmg.a.37365. [DOI] [PubMed] [Google Scholar]
- 6.Krakow D. Skeletal dysplasias. Clin Perinatol. 2015;42(2):301–319. doi: 10.1016/j.clp.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Funari VA, Day A, Krakow D, Cohn ZA, Chen Z, Nelson SF, Cohn DH. Cartilage-selective genes identified in genome-scale analysis of non-cartilage and cartilage gene expression. BMC Genomics. 2007;8:165. doi: 10.1186/1471-2164-8-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edqvist PH, Fagerberg L, Hallstrom BM, Danielsson A, Edlund K, Uhlen M, Ponten F. Expression of human skin-specific genes defined by transcriptomics and antibody-based profiling. J Histochem Cytochem. 2015;63(2):129–141. doi: 10.1369/0022155414562646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Megy K, Audic S, Claverie JM. Heart-specific genes revealed by expressed sequence tag (EST) sampling. Genome Biol. 2002;3(12):Research0074. doi: 10.1186/gb-2002-3-12-research0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pogue R, Sebald E, King L, Kronstadt E, Krakow D, Cohn DH. A transcriptional profile of human fetal cartilage. Matrix Biol. 2004;23(5):299–307. doi: 10.1016/j.matbio.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Freeman BT, Jung JP, Ogle BM. Single-cell RNA-Seq of bone marrow-derived Mesenchymal stem cells reveals unique profiles of lineage priming. PLoS One. 2015;10(9):e0136199. doi: 10.1371/journal.pone.0136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tompson SW, Bacino CA, Safina NP, Bober MB, Proud VK, Funari T, Wangler MF, Nevarez L, Ala-Kokko L, Wilcox WR, et al. Fibrochondrogenesis results from mutations in the COL11A1 type XI collagen gene. Am J Hum Genet. 2010;87(5):708–712. doi: 10.1016/j.ajhg.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, Lachman RS, Wilcox WR, Reyno S, Quadrelli R, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40(8):999–1003. doi: 10.1038/ng.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet. 2009;84(1):72–79. doi: 10.1016/j.ajhg.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gkountela S, Zhang KX, Shafiq TA, Liao WW, Hargan-Calvopina J, Chen PY, Clark AT. DNA Demethylation dynamics in the human prenatal Germline. Cell. 2015;161(6):1425–1436. doi: 10.1016/j.cell.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan L, Guo H, Hu B, Li R, Yong J, Zhao Y, Zhi X, Fan X, Guo F, Wang X, et al. Epigenomic landscape of human fetal brain, heart, and liver. J Biol Chem. 2016;291(9):4386–4398. doi: 10.1074/jbc.M115.672931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin S, Lee YK, Lim YC, Zheng Z, Lin XM, Ng DP, Holbrook JD, Law HY, Kwek KY, Yeo GS, et al. Global DNA hypermethylation in down syndrome placenta. PLoS Genet. 2013;9(6):e1003515. doi: 10.1371/journal.pgen.1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH roadmap Epigenomics mapping consortium. Nat Biotechnol. 2010;28(10):1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gkountela S, Li Z, Vincent JJ, Zhang KX, Chen A, Pellegrini M, Clark AT. The ontogeny of cKIT+ human primordial germ cells proves to be a resource for human germ line reprogramming, imprint erasure and in vitro differentiation. Nat Cell Biol. 2013;15(1):113–122. doi: 10.1038/ncb2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Consortium F. The RP, Clst, Forrest AR, Kawaji H, Rehli M, Baillie JK, de Hoon MJ, Haberle V, Lassmann T et al: a promoter-level mammalian expression atlas. Nature. 2014;507(7493):462–470. doi: 10.1038/nature13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One. 2014;9(1):e78644. doi: 10.1371/journal.pone.0078644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, Superti-Furga A, Scambler PJ, Winter RM. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet. 1998;19(1):70–73. doi: 10.1038/ng0198-70. [DOI] [PubMed] [Google Scholar]
- 26.Russell MW, Raeker MO, Geisler SB, Thomas PE, Simmons TA, Bernat JA, Thorsson T, Innis JW. Functional analysis of candidate genes in 2q13 deletion syndrome implicates FBLN7 and TMEM87B deficiency in congenital heart defects and FBLN7 in craniofacial malformations. Hum Mol Genet. 2014;23(16):4272–4284. doi: 10.1093/hmg/ddu144. [DOI] [PubMed] [Google Scholar]
- 27.Wilson R, Norris EL, Brachvogel B, Angelucci C, Zivkovic S, Gordon L, Bernardo BC, Stermann J, Sekiguchi K, Gorman JJ, et al. Changes in the chondrocyte and extracellular matrix proteome during post-natal mouse cartilage development. Mol Cell Proteomics. 2012;11(1):M111 014159. doi: 10.1074/mcp.M111.014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krakow D, Vriens J, Camacho N, Luong P, Deixler H, Funari TL, Bacino CA, Irons MB, Holm IA, Sadler L, et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am J Hum Genet. 2009;84(3):307–315. doi: 10.1016/j.ajhg.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, et al. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet. 1995;10(3):325–329. doi: 10.1038/ng0795-325. [DOI] [PubMed] [Google Scholar]
- 30.Briggs MD, Mortier GR, Cole WG, King LM, Golik SS, Bonaventure J, Nuytinck L, De Paepe A, Leroy JG, Biesecker L, et al. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am J Hum Genet. 1998;62(2):311–319. doi: 10.1086/301713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu M, Chen G, Li YP. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone research. 2016;4:16009. doi: 10.1038/boneres.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang HM, Chen H, Liu W, Liu H, Gong J, Wang H, Guo AY. AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res. 2012;40(Database issue):D144–D149. doi: 10.1093/nar/gkr965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saito A, Kanemoto S, Zhang Y, Asada R, Hino K, Imaizumi K. Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol Cell. 2014;53(1):127–139. doi: 10.1016/j.molcel.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Gridley T. Compensatory regulation of the Snai1 and Snai2 genes during chondrogenesis. J Bone Miner Res. 2013;28(6):1412–1421. doi: 10.1002/jbmr.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levi G, Topilko P, Schneider-Maunoury S, Lasagna M, Mantero S, Cancedda R, Charnay P. Defective bone formation in Krox-20 mutant mice. Development. 1996;122(1):113–120. doi: 10.1242/dev.122.1.113. [DOI] [PubMed] [Google Scholar]
- 37.Dimitri P, De Franco E, Habeb AM, Gurbuz F, Moussa K, Taha D, Wales JK, Hogue J, Slavotinek A, Shetty A, et al. An emerging, recognizable facial phenotype in association with mutations in GLI-similar 3 (GLIS3) Am J Med Genet A. 2016;170(7):1918–1923. doi: 10.1002/ajmg.a.37680. [DOI] [PubMed] [Google Scholar]
- 38.Tourtellotte WG, Milbrandt J. Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat Genet. 1998;20(1):87–91. doi: 10.1038/1757. [DOI] [PubMed] [Google Scholar]
- 39.Varrault A, Gueydan C, Delalbre A, Bellmann A, Houssami S, Aknin C, Severac D, Chotard L, Kahli M, Le Digarcher A, et al. Zac1 regulates an imprinted gene network critically involved in the control of embryonic growth. Dev Cell. 2006;11(5):711–722. doi: 10.1016/j.devcel.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Han Y, Lefebvre V. L-Sox5 and Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol Cell Biol. 2008;28(16):4999–5013. doi: 10.1128/MCB.00695-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee YH, Saint-Jeannet JP. Sox9 function in craniofacial development and disease. Genesis. 2011;49(4):200–208. doi: 10.1002/dvg.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16(21):2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, Pasantes J, Bricarelli FD, Keutel J, Hustert E, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994;79(6):1111–1120. doi: 10.1016/0092-8674(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 44.Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanovic M, Weissenbach J, Mansour S, Young ID, Goodfellow PN, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature. 1994;372(6506):525–530. doi: 10.1038/372525a0. [DOI] [PubMed] [Google Scholar]
- 45.Healy C, Uwanogho D, Sharpe PT. Regulation and role of Sox9 in cartilage formation. Dev Dyn. 1999;215(1):69–78. doi: 10.1002/(SICI)1097-0177(199905)215:1<69::AID-DVDY8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 46.Eferl R, Hoebertz A, Schilling AF, Rath M, Karreth F, Kenner L, Amling M, Wagner EF. The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO J. 2004;23(14):2789–2799. doi: 10.1038/sj.emboj.7600282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tamama K, Barbeau DJ. Early growth response genes signaling supports strong paracrine capability of mesenchymal stem cells. Stem Cells Int. 2012;2012:428403. doi: 10.1155/2012/428403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22(1):85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 49.Smallwood A, Ren B. Genome organization and long-range regulation of gene expression by enhancers. Curr Opin Cell Biol. 2013;25(3):387–394. doi: 10.1016/j.ceb.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohba S, He X, Hojo H, McMahon AP. Distinct transcriptional programs underlie Sox9 regulation of the mammalian Chondrocyte. Cell Rep. 2015;12(2):229–243. doi: 10.1016/j.celrep.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herlofsen SR, Bryne JC, Hoiby T, Wang L, Issner R, Zhang X, Coyne MJ, Boyle P, Gu H, Meza-Zepeda LA, et al. Genome-wide map of quantified epigenetic changes during in vitro chondrogenic differentiation of primary human mesenchymal stem cells. BMC Genomics. 2013;14:105. doi: 10.1186/1471-2164-14-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu CF, Lefebvre V. The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 2015;43(17):8183–8203. doi: 10.1093/nar/gkv688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang A, Yue F, Li Y, Xie R, Harper T, Patel NA, Muth K, Palmer J, Qiu Y, Wang J, et al. Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell. 2015;16(4):386–399. doi: 10.1016/j.stem.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyer MB, Benkusky NA, Pike JW. The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J Biol Chem. 2014;289(23):16016–16031. doi: 10.1074/jbc.M114.552216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mullen AC, Orlando DA, Newman JJ, Loven J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell. 2011;147(3):565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152(1–2):157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 60.Letimier FA, Passini N, Gasparian S, Bianchi E, Rogge L. Chromatin remodeling by the SWI/SNF-like BAF complex and STAT4 activation synergistically induce IL-12Rbeta2 expression during human Th1 cell differentiation. EMBO J. 2007;26(5):1292–1302. doi: 10.1038/sj.emboj.7601586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huynh NP, Anderson BA, Guilak F, McAlinden A. Emerging roles for long noncoding RNAs in skeletal biology and disease. Connect Tissue Res. 2017;58(1):116–141. [DOI] [PMC free article] [PubMed]
- 62.Guil S, Esteller M. Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol. 2012;19(11):1068–1075. doi: 10.1038/nsmb.2428. [DOI] [PubMed] [Google Scholar]
- 63.Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539(7629):452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat Commun. 2015;6:6683. doi: 10.1038/ncomms7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Day A, Dong J, Funari VA, Harry B, Strom SP, Cohn DH, Nelson SF. Disease gene characterization through large-scale co-expression analysis. PLoS One. 2009;4(12):e8491. doi: 10.1371/journal.pone.0008491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang S, Li Y, Be X, Howes S, Liu W. Detecting and profiling tissue-selective genes. Physiol Genomics. 2006;26(2):158–162. doi: 10.1152/physiolgenomics.00313.2005. [DOI] [PubMed] [Google Scholar]
- 67.Reginato AM, Olsen BR. The role of structural genes in the pathogenesis of osteoarthritic disorders. Arthritis Res. 2002;4(6):337–345. doi: 10.1186/ar595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinonen J, Taipaleenmaki H, Roering P, Takatalo M, Harkness L, Sandholm J, Uusitalo-Jarvinen H, Kassem M, Kiviranta I, Laitala-Leinonen T, et al. Snorc is a novel cartilage specific small membrane proteoglycan expressed in differentiating and articular chondrocytes. Osteoarthr Cartil. 2011;19(8):1026–1035. doi: 10.1016/j.joca.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 69.Hochgreb-Hagele T, Koo DE, Bronner ME. Znf385C mediates a novel p53-dependent transcriptional switch to control timing of facial bone formation. Dev Biol. 2015;400(1):23–32. doi: 10.1016/j.ydbio.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 70.Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, et al. Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012;22(6):1059–1068. doi: 10.1101/gr.133546.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang L, Lawson KA, Teteak CJ, Zou J, Hacquebord J, Patterson D, Ghatan AC, Mei Q, Zielinska-Kwiatkowska A, Bain SD, et al. ESET histone methyltransferase is essential to hypertrophic differentiation of growth plate chondrocytes and formation of epiphyseal plates. Dev Biol. 2013;380(1):99–110. doi: 10.1016/j.ydbio.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Velagaleti GV, Bien-Willner GA, Northup JK, Lockhart LH, Hawkins JC, Jalal SM, Withers M, Lupski JR, Stankiewicz P. Position effects due to chromosome breakpoints that map approximately 900 kb upstream and approximately 1.3 Mb downstream of SOX9 in two patients with campomelic dysplasia. Am J Hum Genet. 2005;76(4):652–662. doi: 10.1086/429252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leipoldt M, Erdel M, Bien-Willner GA, Smyk M, Theurl M, Yatsenko SA, Lupski JR, Lane AH, Shanske AL, Stankiewicz P, et al. Two novel translocation breakpoints upstream of SOX9 define borders of the proximal and distal breakpoint cluster region in campomelic dysplasia. Clin Genet. 2007;71(1):67–75. doi: 10.1111/j.1399-0004.2007.00736.x. [DOI] [PubMed] [Google Scholar]
- 74.Yao B, Wang Q, Liu CF, Bhattaram P, Li W, Mead TJ, Crish JF, Lefebvre V. The SOX9 upstream region prone to chromosomal aberrations causing campomelic dysplasia contains multiple cartilage enhancers. Nucleic Acids Res. 2015;43(11):5394–5408. doi: 10.1093/nar/gkv426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang M, Hou J, Wang Y, Xie M, Wei C, Nie F, Wang Z, Sun M. Long noncoding RNA LINC00673 is activated by SP1 and exerts Oncogenic properties by interacting with LSD1 and EZH2 in gastric cancer. Mol Ther. 2017; [DOI] [PMC free article] [PubMed] [Retracted]
- 77.Schneider A, Brand T, Zweigerdt R, Arnold H. Targeted disruption of the Nkx3.1 gene in mice results in morphogenetic defects of minor salivary glands: parallels to glandular duct morphogenesis in prostate. Mech Dev. 2000;95(1–2):163–174. doi: 10.1016/S0925-4773(00)00355-5. [DOI] [PubMed] [Google Scholar]
- 78.Lettice LA, Purdie LA, Carlson GJ, Kilanowski F, Dorin J, Hill RE. The mouse bagpipe gene controls development of axial skeleton, skull, and spleen. Proc Natl Acad Sci U S A. 1999;96(17):9695–9700. doi: 10.1073/pnas.96.17.9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tribioli C, Lufkin T. The murine Bapx1 homeobox gene plays a critical role in embryonic development of the axial skeleton and spleen. Development. 1999;126(24):5699–5711. doi: 10.1242/dev.126.24.5699. [DOI] [PubMed] [Google Scholar]
- 80.Akazawa H, Komuro I, Sugitani Y, Yazaki Y, Nagai R, Noda T. Targeted disruption of the homeobox transcription factor Bapx1 results in lethal skeletal dysplasia with asplenia and gastroduodenal malformation. Genes to cells: devoted to molecular & cellular mechanisms. 2000;5(6):499–513. doi: 10.1046/j.1365-2443.2000.00339.x. [DOI] [PubMed] [Google Scholar]
- 81.Herbrand H, Pabst O, Hill R, Arnold HH. Transcription factors Nkx3.1 and Nkx3.2 (Bapx1) play an overlapping role in sclerotomal development of the mouse. Mech Dev. 2002;117(1–2):217–224. doi: 10.1016/S0925-4773(02)00207-1. [DOI] [PubMed] [Google Scholar]
- 82.Nakamura Y, Weidinger G, Liang JO, Aquilina-Beck A, Tamai K, Moon RT, Warman ML. The CCN family member Wisp3, mutant in progressive pseudorheumatoid dysplasia, modulates BMP and Wnt signaling. J Clin Invest. 2007;117(10):3075–3086. doi: 10.1172/JCI32001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kutz WE, Gong Y, Warman ML. WISP3, the gene responsible for the human skeletal disease progressive pseudorheumatoid dysplasia, is not essential for skeletal function in mice. Mol Cell Biol. 2005;25(1):414–421. doi: 10.1128/MCB.25.1.414-421.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Halasz K, Kassner A, Morgelin M, Heinegard D. COMP acts as a catalyst in collagen fibrillogenesis. J Biol Chem. 2007;282(43):31166–31173. doi: 10.1074/jbc.M705735200. [DOI] [PubMed] [Google Scholar]
- 85.Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, Mortier GR, Rimoin DL, Lachman RS, Gaines ES, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995;10(3):330–336. doi: 10.1038/ng0795-330. [DOI] [PubMed] [Google Scholar]
- 86.Svensson L, Aszodi A, Heinegard D, Hunziker EB, Reinholt FP, Fassler R, Oldberg A. Cartilage oligomeric matrix protein-deficient mice have normal skeletal development. Mol Cell Biol. 2002;22(12):4366–4371. doi: 10.1128/MCB.22.12.4366-4371.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pirog-Garcia KA, Meadows RS, Knowles L, Heinegard D, Thornton DJ, Kadler KE, Boot-Handford RP, Briggs MD. Reduced cell proliferation and increased apoptosis are significant pathological mechanisms in a murine model of mild pseudoachondroplasia resulting from a mutation in the C-terminal domain of COMP. Hum Mol Genet. 2007;16(17):2072–2088. doi: 10.1093/hmg/ddm155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suleman F, Gualeni B, Gregson HJ, Leighton MP, Pirog KA, Edwards S, Holden P, Boot-Handford RP, Briggs MD. A novel form of chondrocyte stress is triggered by a COMP mutation causing pseudoachondroplasia. Hum Mutat. 2012;33(1):218–231. doi: 10.1002/humu.21631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Camacho N, Krakow D, Johnykutty S, Katzman PJ, Pepkowitz S, Vriens J, Nilius B, Boyce BF, Cohn DH. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genet A. 2010;152A(5):1169–1177. doi: 10.1002/ajmg.a.33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nishimura G, Dai J, Lausch E, Unger S, Megarbane A, Kitoh H, Kim OH, Cho TJ, Bedeschi F, Benedicenti F, et al. Spondylo-epiphyseal dysplasia, Maroteaux type (pseudo-Morquio syndrome type 2), and parastremmatic dysplasia are caused by TRPV4 mutations. Am J Med Genet A. 2010;152a(6):1443–1449. doi: 10.1002/ajmg.a.33414. [DOI] [PubMed] [Google Scholar]
- 91.Lamande SR, Yuan Y, Gresshoff IL, Rowley L, Belluoccio D, Kaluarachchi K, Little CB, Botzenhart E, Zerres K, Amor DJ, et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat Genet. 2011;43(11):1142–1146. doi: 10.1038/ng.945. [DOI] [PubMed] [Google Scholar]
- 92.Tabuchi K, Suzuki M, Mizuno A, Hara A. Hearing impairment in TRPV4 knockout mice. Neurosci Lett. 2005;382(3):304–308. doi: 10.1016/j.neulet.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 93.Weinstein MM, Tompson SW, Chen Y, Lee B, Cohn DH. Mice expressing mutant Trpv4 recapitulate the human TRPV4 disorders. J Bone Miner Res. 2014;29(8):1815–1822. doi: 10.1002/jbmr.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lefebvre V, Li P, de Crombrugghe B. A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 1998;17(19):5718–5733. doi: 10.1093/emboj/17.19.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Frutos CA, Vega S, Manzanares M, Flores JM, Huertas H, Martinez-Frias ML, Nieto MA. Snail1 is a transcriptional effector of FGFR3 signaling during chondrogenesis and achondroplasias. Dev Cell. 2007;13(6):872–883. doi: 10.1016/j.devcel.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 96.Krakow D, Sebald ET, Pogue R, Rimoin LP, King L, Cohn DH. Analysis of clones from a human cartilage cDNA library provides insight into chondrocyte gene expression and identifies novel candidate genes for the osteochondrodysplasias. Mol Genet Metab. 2003;79(1):34–42. doi: 10.1016/S1096-7192(03)00064-7. [DOI] [PubMed] [Google Scholar]
- 97.Marques F, Tenney J, Duran I, Martin J, Nevarez L, Pogue R, Krakow D, Cohn DH, Li B. Altered mRNA splicing, Chondrocyte gene expression and abnormal skeletal development due to SF3B4 mutations in Rodriguez Acrofacial Dysostosis. PLoS Genet. 2016;12(9):e1006307. doi: 10.1371/journal.pgen.1006307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pan JB, Hu SC, Wang H, Zou Q, Ji ZL. PaGeFinder: quantitative identification of spatiotemporal pattern genes. Bioinformatics. 2012;28(11):1544–1545. doi: 10.1093/bioinformatics/bts169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 104.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mostafavi S, Ray D, Warde-Farley D, Grouios C, Morris Q. GeneMANIA: a real-time multiple association network integration algorithm for predicting gene function. Genome Biol. 2008;9(Suppl 1):S4. doi: 10.1186/gb-2008-9-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12(8):R83. doi: 10.1186/gb-2011-12-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nicol JW, Helt GA, Blanchard SG, Jr, Raja A, Loraine AE. The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25(20):2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Dataset S1. Data used in the study. (XLSX 17 kb)
Dendrogram and heatmap of the Pearson’s correlation coefficient for human tissue gene expression (Log2FPKM). Only genes with FPKM > 1 in at least one sample were used for analysis. The cartilage samples are highlighted with a red frame. Figure S2. Box plots showing cartilage-selective gene expression in cartilage (red) and non-cartilage (blue) tissues. The thick horizontal line in each box represents the median gene expression value. Figure S3. Bar graph showing RUNX2 expression in human tissues. Red bars indicate cartilage samples and blue bars indicate non-cartilage samples, respectively. Gene expression levels are shown as FPKM values. Figure S4. Genome browser views of H3K4me1 and H3K27ac peaks along a COL2A1 and b COL11A1 loci. Chromosome coordinates are shown as black bars on top. Black arrows indicate the direction of transcription on a diagram of each gene below. CH, chondrocytes. AG, adrenal gland. LI, large intestine. SI, small intestine. MU, muscle. SC, spinal cord. ST, stomach. TH, thymus. Figure S5. Genome browser views of H3K4me1, H3K4me3, H3K27ac peaks and RNA expression track along lncRNA loci (A) AC058791.1 and (B) RP11–261P24.2 in human chondrocytes. Figure S6. Sox9 binds to the lncRNA 2610035D17Rik promoter and gene body in mouse rib chondrocytes. a Genome browser view of Sox9 binding along Sox9 and 2610035D17Rik loci on chromosome 11. b Shaded region in a was enlarged for a close view of Sox9 binding at the 2610035D17Rik promoter and first intron. (PDF 912 kb)
Supplementary Dataset S2. Cartilage-selective genes. (XLSX 45110 kb)
Supplementary Dataset S3. Data comparison between microarray and RNA-seq platforms. (XLSX 8 kb)
Supplementary Dataset S4. Cartilage-selective transcription factors and motif analysis. (XLSX 26 kb)
Supplementary Dataset S5. GO analysis of cartilage-unique enhancers. (XLSX 1028 kb)
Supplementary Dataset S6. Cartilage selective lncRNAs. (XLSX 17845 kb)
Data Availability Statement
The RNA-seq data sets generated in this study have been deposited in the Gene Expression Omnibus (GEO) under accession GSE107649.