

Summary
During environmental adaptation bacteria use small regulatory RNAs (sRNAs) to repress or activate expression of a large fraction of their proteome. We extended the use of the in vivo RNA proximity ligation method towards probing global sRNA interactions with their targets in Pseudomonas aeruginosa and verified the method with a known regulon controlled by the PrrF1 sRNA. We also identified two sRNAs (Sr0161 and ErsA) that interact with the mRNA encoding the major porin OprD responsible for the uptake of carbapenem antibiotics. These two sRNAs base pair with the 5′ UTR of oprD leading to increase in resistance of the bacteria to meropenem. Additional proximity ligation experiments and enrichment for Sr0161 targets identified the mRNA for the regulator of type III secretion system. Interaction between the exsA mRNA and Sr0161 leads to a block in the synthesis of a component of the T3SS apparatus and an effector. Another sRNA, Sr006, positively regulates, without Hfq, the expression of PagL, an enzyme responsible for deacylation of lipid A, reducing its pro-inflammatory property and resulting in polymyxin resistance. Therefore, an analysis of global sRNA-mRNA interactions can lead to discoveries of novel pathways controlling gene expression that are likely integrated into larger regulatory networks.
Keywords: sRNA, post-transcriptional regulation, OprD, PagL, regulation of T3SS
Abbreviated Summary
We used the in vivo RNA proximity ligation method and probed global sRNA interactions with their targets in Pseudomonas aeruginosa. New post-transcriptional regulators of determinants of pathogenicity and antibiotic susceptibility were discovered.
Introduction
A group of bacterial non-coding small RNAs (sRNAs) represent an important arm of global regulatory networks coordinating the expression of multiple genes to optimize the fitness of a microorganism in a particular niche or in response to environmental stressors. These RNA regulators are grouped based on the location of their targets. The cis-acting group includes riboswitches each physically linked to the 5′ region of a single target through base paired structures controlling translation or the stability of the mRNA portion upon an interaction with a ligand (Winkler et al., 2005). Another class of cis-acting regulatory RNAs is transcribed from a strand opposite to the RNA, allowing base pairing though complementarity of the two partially overlapping RNAs. The best studied examples of this group are found among molecular systems controlling plasmid segregation during bacterial cell division, where the expression of a toxin protein is translated only in the absence of the antitoxin sRNA; the antitoxin transcribed from the strand opposite to and partially overlapping the toxin gene disrupts translation and stability of the toxin mRNA by base pairing near the sites of the initiation of translation (Gerdes et al., 1997). The largest group of sRNA regulators are the so-called trans-acting sRNA; these are transcribed usually from intergenic regions and can have multiple targets. The base pairing region of the trans-acting sRNAs share limited complementarity with their target and may involve different regions of the molecules. The base-pairing segments of the majority of mRNAs targeted by sRNA are located in the 5′ untranslated region (5′ UTR), and the sRNA-mRNA interaction frequently occludes the ribosome binding sites (Storz et al., 2011, Gottesman et al., 2011). The base pairing, facilitated by the interaction with the RNA chaperone Hfq, blocks translation and recruits ribonuclease, leading to repression of gene expression. Several exceptions of positive regulation by the sRNAs have been also described, where sRNA base pairing leads to release of the sites of initiation of translation by formation of alternative base paired structures with the 5′ end of the mRNA (Fröhlich et al., 2009, Papenfort et al., 2015).
Recently, several investigators have addressed the difficulty of identifying targets of sRNAs using computational tools by developing several experimental approaches towards this goal (Waters et al., 2017, Melamed et al., 2016, Lalaouna et al., 2015, Han et al., 2016). Our laboratory has developed and used the method referred to as Global sRNA target Identification by Ligation and Sequencing (GRIL-seq) to identify sRNA interactions with a variety of transcripts, including mRNAs or sequestering RNAs (“sponges”) (Han et al., 2016). The critical steps used by GRIL-seq include induced expression of a single sRNA in bacteria which is also expressing T4 RNA ligase and subsequent enrichment for transcripts containing the sRNA or, chimeras created by ligation of these sRNAs to base-paired RNAs. The isolated RNA is subjected to RNA sequencing and mapping of the ligated transcripts to the bacterial genome. This allows identification of targets of a specific sRNA. We used GRIL-seq to identify targets of an iron regulated sRNA (PrrF1) (Wilderman et al., 2004) and discovered several unusual regulatory mechanisms, including interaction with a “sponge” derived from the 3′end of the katA transcript, thus antagonizing the regulatory interaction of the 5′ end of the same mRNA.
To obtain a more global picture of the interactions between the various sRNAs and their targets, we modified the original GRIL-seq method omitting the enrichment step and to facilitate sequencing coverage, depleting the majority of rRNAs from the samples. A computational pipeline was created which allowed us to map the chimeras created by ligation of the 3′ hydroxyl ends of mRNAs to 5′ monophosphate ends of sRNAs and vice versa, thus providing a more comprehensive view of interactions between the individual RNAs in the cell. We refer to the experimental method and the linked data analysis High-throughput GRIL-seq (Hi-GRIL-seq). We validated this approach by first demonstrating that it is capable of detecting the same interactions as GRIL-seq where the samples are enriched for specific sRNA-containing chimeras. Moreover, examination of individual target mRNAs can be traced back to sRNA regulation and we provide two examples of how Hi-GRIL-seq can yield new insights into the interplay between sRNAs and mRNAs within post-transcriptional regulatory networks involving control of expression of important virulence factors.
Results
An assessment of the global sRNA regulatory landscape
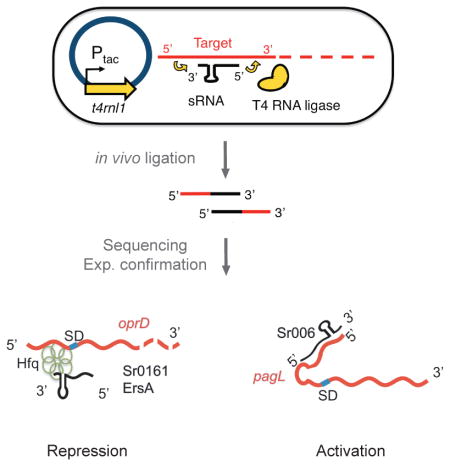
To investigate the regulatory range of individual sRNAs, we developed GRIL-seq, an in vivo approach to capture interactions between a single sRNA and its multiple targets, based on preferential ligation of the base-paired transcripts following ectopic expression of T4 RNA ligase (Han et al., 2016). The regulated expression of an sRNA gene allowed us to probe interactions with different transcripts that would vary in abundance when the bacteria are propagated under different growth conditions. We wished to extend this method to a more global approach testing whether endogenously expressed sRNAs can be ligated to their targets without sRNA overexpression or subsequent enrichment for a single sRNA species. Given the performance of current deep sequencing platforms, it should be possible to identify chimeras created by ligation of any sRNA bound to its target by simple sequencing of the entire RNA pool, depleted of the abundant rRNAs. We tested this extension of the GRIL-seq method, referred to as Hi-GRIL-seq, and here we demonstrate its utility as a useful tool for the discovery of global RNA interactions under the steady-state growth conditions. The principles of Hi-GRIL-seq including the data analysis pipeline are outlined in Figure 1. The major steps include culturing of the bacteria expressing the T4 RNA ligase in the test conditions, RNA isolation, rRNA removal, construction of a library for RNA-seq, paired-end sequencing, and BLAST (Altschul et al., 1990) -based analysis to map the location and abundance of chimeric reads.
Fig. 1.

Schematic overview of Hi-GRIL-seq.
Induction of T4 RNA ligase expression from the Ptac promoter with IPTG leads to the expression of the enzyme and the formation of chimeras between base paired endogenous sRNAs and their targets. Following isolation of total RNA and rRNA depletion, a cDNA library for Illumina sequencing is constructed and sequenced. RNA interactions between sRNAs and their targets are identified by a BLAST-based analysis pipeline. Global chimeras are visualized in a two-dimensional dot plot, in which the location of the dot represents the genomic coordinate of the participating RNAs. To examine the targets of a particular RNA, the coverage of its targets can be visualized. To further zoom in on a particular interaction between the two RNAs, the exact location of ligation junctions in the chimeras are mapped and visualized.
To compare Hi-GRIL-seq and GRIL-seq we tracked the interactions of the iron-regulated sRNA PrrF1, a well-characterized regulator of iron homeostasis in P. aeruginosa. This sRNA was used in the development and validation of the original GRIL-seq method (Han et al., 2016). In the present study, instead of expressing PrrF1 under the control of an artificially (L-arabinose) inducible promoter in cells co-expressing the T4 RNA ligase, we relied on induction of expression of this sRNA by shifting cells from iron replete to iron limiting conditions and comparing the identity of the chimeras formed between PrrF1 and its targets.
To confirm that under conditions of iron depletion, PrrF1 is induced and ligated to its regulatory targets, we first identified ligation products formed by PrrF1 and its two known target transcripts (sodB and rubA1) following PCR with reverse transcription (RT-PCR), using sRNA and target gene-specific primer pairs (Fig. 2A). We confirmed the ligation junctions by sequencing of individual amplicons. The presence of unique amplicons was dependent on induction of the T4 RNA ligase gene t4rnl1 by isopropyl β-D-1-thiogalactopyranoside (IPTG). The strength of this chimera signal was dose-dependent on iron depletion and thereby PrrF1 induction by different concentrations of the iron chelator 2,2′-dipyridyl (2,2′-DIP), as shown in Supporting Information Fig. S1. Therefore, T4 RNA ligase is able to ligate endogenously expressed sRNAs with their targets.
Fig. 2.

Detection of chimeras containing PrrF1 and its targets under iron limiting conditions.
A. In vivo expressed T4 RNA ligase links endogenous PrrF1 to its known targets sodB (left panel) and rubA1 (right panel). The chimeras were detected by gel electrophoresis of PCR products following reverse transcription (RT-PCR) as described in Experimental procedures. Iron chelator 2,2′-dipyridyl (2,2′-DIP) treatment was used to deplete the iron from the media. Amplicons were cloned and sequenced to determine the ligation junctions between PrrF1 and its targets, and their locations in sodB and rubA1 are shown in red numbers. The figure shows results of two biological replicates. The relative position of each RNA in the tested chimeras is shown schematically and the numbers indicate the locations of junctions based on the sequence of the amplicons indicated by stars. Also shown are the expression of PrrF1 and the control housekeeping gene rpoD.
B. Transcripts in sequenced PrrF1-containing chimeric reads identified by Hi-GRIL-seq. The lines and their height represent the location of the junction and the number of chimeric reads at that location. Two biological replicate experiments are shown for iron depletion group (with 2,2′-DIP) and control group (without 2,2′-DIP). Bold numbers on the left indicate the scales of the tracks.
We performed Hi-GRIL-seq of ligated RNA, recovered from biological duplicate samples of P. aeruginosa grown in media where iron was limiting (created by the addition of 2,2′-DIP) compared to RNA isolated from cells grown in iron replete media that has not been treated with 2,2′-DIP (control condition). Following removal of the majority of rRNAs, cDNA libraries were constructed. Sequencing of these samples using the Illumina platform yielded 64,539,855 and 65,434,990 reads for the iron depletion samples and 79,896,070 and 75,931,749 reads for the iron replete samples. 85–87% of the reads could be mapped to the annotated P. aeruginosa PAO1 genome. For accurate identification, a read was called chimeric only when the precise ligation junction between the sRNA and its target could be identified. In total, 0.26–0.29% of the reads were identified to be chimeric reads. We detected no motifs flanking the ligation junctions and the potential bias of ligase’s preference for certain nucleotides was minimal (Supporting Information Fig. S2). The frequency of chimeras was calculated relative to annotated genes and intergenic regions. The list of interactions, where at least 10 chimeras under one of the conditions were identified, can be found in Supporting Information Table S1. Fig. 2B shows the distribution of transcripts found in the PrrF1-containing chimeras mapped onto the genomic location of corresponding genes on the PAO1 chromosome. Out of the top 10 targets of PrrF1/2 identified by the GRIL-seq method (Han et al., 2016), 8 of them were also identified as the top 15 ranked targets by Hi-GRIL-seq experiments in the iron-depletion condition (Table S2). The unique targets identified by each method may reflect the abundance of mRNAs at the time of induction of RNA ligase. For example, iron limitation in the media by the chelator, used in Hi-GRIL-seq to induce PrrF1/2 expression may, lead to the expression of additional targets that were poorly or not expressed under conditions of ectopic expression of PrrF1 in iron rich media, employed in the GRIL-seq experiments. The identity of the top PrrF1/2 targets was similar between the iron depletion condition and the control condition, indicating that low-level expression of this sRNA occurs even when iron is not limiting (Fig. 2B). However, PrrF1/2-containing chimeras were markedly enriched when the expression of PrrFs was induced with 2,2′-DIP (note differences in Fig. 2B scales). For example, the chimeric read count for the target sdhC is 50 and 56 for the duplicate iron depletion groups while the number of chimeras dropped to 2 and 5 for the control groups. Therefore, Hi-GRIL-seq is able to capture sRNA-target interactions in sRNA inducing stress conditions or under conditions when the interactions are abundant. However, its target detection power for sRNA of basal expression level is limited by the sequencing depth. Increasing read depth should uncover new interactions, particularly when the sRNA or its targets are not in high abundance. For interactions involving sRNAs that are highly expressed in the tested condition, increasing read depth has less impact (Supporting Information Fig. S3A).
Identification of new interactions by Hi-GRIL-seq
The global sRNA regulon uncovered by Hi-GRIL-seq can be used to identify specific interactions between sRNAs and any transcript of interest. To illustrate this point, we examined two P. aeruginosa mRNAs encoding an antibiotic resistance and virulence determinant, oprD and pagL, respectively (Fig. 3). OprD is an outer membrane porin required for the entry of basic amino acids, peptides and carbapenem antibiotics into the periplasm. Loss of OprD leads to resistance to carbapenem in clinical isolates and enhanced fitness in animal infection models (Pirnay et al., 2002, Skurnik et al., 2013b, Wolter et al., 2004). It has been previously suggested that oprD is regulated at the post-transcriptional level (Ducret et al., 2016, Li et al., 2012). PagL encodes a lipid A 3-O-deacylase. Deacylation of lipid A modifies its ability to induce Toll-like receptor 4 signaling (Miller et al., 2005).
Fig. 3.

Two-dimensional display of RNAs found in chimeras detected by Hi-GRIL-seq.
A. Genomic location of the transcripts in chimeras found in RNAs isolated from P. aeruginosa grown in iron-depleted medium
B. Chimeras in RNAs from bacteria grown in iron replete medium.
The position of the points on the x and y axes correspond to the genome coordinates of interacting RNAs; with the x-axis representing the 5′ sequence within each chimeric ligation product while the y-axis indicates the coordinates of the 3′ sequence. Shown are chimeras with a frequency of at least 10 reads. The size of the points is proportional to the abundance of chimeras detected by sequencing, with a scale shown on the right. Chimeras involving PrrF1/2 are labeled blue. Chimeras containing the mRNA target oprD are labeled red. Chimera containing the mRNA target pagL is labeled green.
Hi-GRIL-seq and the analysis of the oprD-containing chimeras identified transcripts mapping to two genomic regions on the P. aeruginosa PAO1 chromosome, detected in both iron depletion and control groups. The first sRNA is transcribed from a region located between PA0160 and opdC (Fig. 4A). It is annotated as a gene, possibly encoding a short peptide. However, functional studies described below using transcripts with mutations suggest that it is a regulatory sRNA and we will refer to it as Sr0161. The sites of initiation and termination of Sr0161 transcription (184,211–184,458 bp on the plus strand) were confirmed by 3′ and 5′ RACE. The average Sr0161-oprD chimeric read count identified by Hi-GRIL-seq of the four samples (from RNA isolated from duplicate cultures grown under iron replete and iron limiting conditions) was 28.8 ± 3.9. The analysis of the sequences with the oprD-Sr0161 junctions showed that the ligation occurred between the cleavage products of both the sRNA and mRNA. The majority (93%) of the chimeras consisted of oprD at the 5′ end and Sr0161 at the 3′ end, while the rest were ligation products where the sRNA was at the 5′end (Fig. 4A). The majority of the oprD ligation junctions resided within 300 bp of the 5′ CDS (Fig. 4A). The second sRNA identified by ligation to oprD mRNA is a previously described sRNA ErsA (Ferrara et al., 2015) (Fig. 4B) with an average chimeric read count of 18.8 ± 3.0. Here, 70% of the products were generated by ligase catalyzed joining of the 3′hydroxyl group of the oprD fragments and the 5′ monophosphate of various ends of ErsA degradation products, while the remaining had the inverse configuration. The majority of the junctions were either in the 5′ UTR or 5′ CDS of the oprD transcript.
Fig. 4.

Location of ligation junctions in selected chimeras identified by Hi-GRIL-seq.
A. The junctions between Sr0161 and oprD, mapped to the genomic location of the corresponding genes.
B. Location of the junctions in ErsA-oprD chimeras.
C. Location of the junctions between Sr006-pagL chimeras.
Junctions of the sRNA fragments in the chimeric reads are shows as black peaks, while mRNA junctions are shown as red peaks. The percentages of the sequenced chimeric reads with different configurations (sRNA on either the 5′ end or 3′ end of the chimera) are shown in the insets. Two biological replicate experiments are shown for the iron depletion group and control group. Bold numbers on the left are the scales of the tracks.
ErsA was reported to negatively regulate algC at the post-transcriptional level (Ferrara et al., 2015), however in all Hi-GRIL-seq samples, the count of algC-ErsA chimeric reads were below 5. This may reflect the low abundance of ErsA and algC in P. aeruginosa PAO1 grown under the experimental conditions used in this study. We therefore examined the interaction between these two RNAs under conditions where ErsA and algC are more abundant, for example, when P. aeruginosa are exposed to D-cycloserine, which induces envelope stress leading to increased alginate production (Ferrara et al., 2015, Lizewski et al., 2002, Wood et al., 2009). D-cycloserine treatment of P. aeruginosa with expression of the T4 RNA ligase, resulted in readily identifiable formation of EsrA-algC chimeras (Supporting Information Fig. S4). Similarly, when examining the interaction of ErsA with algC mRNA in the highly mucoid strains PA2192 and PDO300, carrying mutations in the anti-sigma factor mucA, chimeras between the two transcripts can also be detected but were not recovered from their non-mucoid isogenic parental strains (Supporting Information Fig. S4). Therefore, ErsA controls the synthesis of polysaccharide via AlgC, and the extent of its regulatory effect depends on the levels of the target mRNA.
A previously annotated sRNA Sr006 (Wurtzel et al., 2012) transcribed from the intergenic region between 182,570–182,693 bp on the minus strand was found in the chimeras with the pagL mRNA (average chimeric read count of the four experiments is 201.3 ± 36.2). The 5′ and 3′ ends of the Sr006 were confirmed by RACE mapping. Unlike the chimeras involving oprD, those between the Sr006 and pagL mRNA were formed exclusively by ligation of 5′ phosphates of pagL fragments to 3′ hydroxyls derived from fragments of sRNA Sr006 (Fig. 4C). The ligation junctions mapped to the 5′ UTR of pagL (Fig. 4C).
In order to confirm the ligations, RT-PCR with primers from each of the two interacting RNAs was performed. First, we determined the kinetics of expression of each sRNA in two P. aeruginosa strains (PAO1 and PA14). The expression of ErsA was increased in stationary phase of growth compared to exponential phase while no phase dependent expression was observed for Sr0161 and Sr006 (Supporting Information Fig. S5). We then sampled RNA from late exponential phase of growth and identified PCR products using sRNA and mRNA specific primers only in cells where the T4 RNA ligase was induced. Sequencing of these amplicons confirmed that they indeed consisted of sRNA/mRNA chimeras (Supporting Information Fig. S6).
Negative post-transcriptional regulation of oprD by sRNA
Base-pairing of sRNAs with mRNAs usually facilitates either degradation or stabilization of the transcripts and GRIL-seq does not differentiate between the potential positive or negative consequences of their interaction.
First we tested whether deletion or overexpression of the sRNA genes affects the translation of oprD mRNA. We created a recombinant construct, containing (5′ to 3′) Ptac promoter followed by the predicted 5′ UTR and the coding sequence for OprD, ending with additional codons for the FLAG-tag epitope. This IPTG-inducible tagged protein construct was integrated into the chromosome of a P. aeruginosa lacking the sRNA genes and carrying either the sRNA overexpression plasmid or control vector. This construct allowed us to test whether the OprD protein levels were affected by posttranscriptional regulation exerted by the sRNAs. Western blot analysis showed that induction of Sr0161 and ErsA caused reductions in OprD protein expression (Fig. 5A).
Fig. 5.

sRNA Sr0161 and ErsA are negative regulators of oprD. The effects of overexpression of the sRNAs on the OprD protein, oprD mRNA and meropenem susceptibility was analyzed in P. aeruginosa strains with chromosomal deletions of sr0161 or ersA genes.
A. Western immunoblot analysis of FLAG-tagged OprD following overexpression of Sr0161 (left panel, Sr0161++) or ErsA (right panel, ErsA++). A Ptac directed construct, consisting of a fusion between the 5′ UTR and the entire coding sequence of oprD with the codons of the FLAG tag peptide was inserted into the chromosome of wild type P. aeruginosa PAO1 or the sRNA deletion mutants; these strains were the recipients of the empty vector (Vec.) or sRNA overexpression plasmid (Sr0161++ or ErsA++). The sRNA was first expressed for 1 hr following the addition of the L-arabinose inducer, then expression of the FLAG-tagged OprD was induced with IPTG for 1 hr in Sr0161 expressing strains and 3 hrs in the ErsA strains. The loading control is shown in a blot probed with antibody against RpoA.
B. Effect of sRNA overexpression on levels of oprD mRNA. Left panel: RT-qPCR analysis of oprD mRNA following overexpression of full length and truncated (171 to 248 bp) Sr016, labeled as Sr0161++ and Sr0161-BP++, respectively. Right panel: the analysis of oprD transcript levels following overexpression of ErsA (ErsA++). OprD mRNA was also analyzed in wildtype strains and sRNA deletion strains carrying the empty vector (Vec). The housekeeping gene proC was used to calculate relative expression and rpoD was used as a control. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, and **, *** denote P < 0.01 and P < 0.001.
C. Survival of P. aeruginosa overexpressing Sr0161 and ErsA. P. aeruginosa (wild type and sRNA mutants) were grown in the presence of meropenem at 0.25 mg L−1 (left panel) and 0.5 mg L−1 (right panel). The bacterial density in cultures was calculated following a spectrophotometric measurement of optical density at 600 nm (OD600) and using a conversion formula, where a culture of OD600 1.0 equals approximately 109 CFU ml−1. Overnight cell cultures were diluted to equivalent of 105 CFU ml−1 in LB and the sRNA expression was induced with L-arabinose. At the same time, meropenem was added to the indicated concentration. After 6 hours, the bacterial viability (as colony forming units, CFUs) was determined following serial dilution and plating on LB agar plates and overnight incubation. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, and *, **, *** denote P<0.05, P<0.01 and P < 0.001.
We further tested the effects of the sRNAs on steady state levels of oprD mRNA levels. Total RNA was extracted from P. aeruginosa Δsr0161 or ΔersA and levels of oprD mRNAs were determined by RT-qPCR (Fig. 5B). The levels of oprD mRNA were quantified relative to those of the housekeeping gene proC (Savli et al., 2003). There was a 2-fold increase of oprD mRNA comparing Δsr0161 to the parent strain while the deletion of ersA did not cause a significant change in oprD level. However, overexpression of either Sr0161 or ErsA significantly decreased the level of oprD mRNA. Collectively these results show that Sr0161 and ErsA are sRNA negative regulators of oprD expression, affecting its translation and stability.
The OprD porin of P. aeruginosa is the major channel for entry of the carbapenem antibiotics into the periplasm. Therefore, sRNA-dependent oprD repression should lead to increased resistance against this class of antibiotics. We therefore tested the meropenem susceptibility of P. aeruginosa Δsr0161 and ΔersA and the same strains overproducing the sRNA at two concentrations of the antibiotic and compared the effect to a strain with deleted oprD gene (Fig. 5C). Loss of Sr0161 resulted in a modest decrease in meropenem resistance while strains lacking ErsA were significantly more susceptible to the antibiotic than the wild type PAO1. Overexpression of Sr0161 led to increased resistance, while overexpression of ErsA resulted in only a modest enhancement in survival at the lower antibiotic concentration (0.25 mg L−1) (Fig. 5C, left). Unexpectedly, the CFU of ΔersA strain overexpressing ErsA could not be detected at the higher antibiotic concentrations (0.5 mg L−1) (Fig. 5C, right). It is conceivable that pleotropic effects of ErsA on other targets, affecting meropenem susceptibility, also contributed to this modest effect besides negative regulation of OprD protein expression.
Base pairing interactions between the oprD mRNA and its sRNA regulators
We have generated a model for the most stable favorable pairing between the oprD and Sr0161/ErsA transcripts (Fig. 6A,C). Both sRNAs appear to interact with a partially overlapping sequence of oprD leading to, in each case, the formation of a base paired region which includes the ribosome binding site (the Shine Dalgarno sequence, S/D) but not the AUG initiation codon.
Fig. 6.

Interaction of Sr0161 and ErsA with oprD mRNA.
A. The predicted base-pairing region of Sr0161 and oprD using the IntaRNA algorithm. The start codon is underlined and the Shine-Dalgarno sequence (S/D) is marked and underlined. The substitution mutations are indicated by arrows and nucleotides.
B. Expression of the oprD::lacZ fusion in response to wild type (WT), mutant (M) Sr0161 or the compensatory mutation in the 5′ UTR of oprD (M′). Effect of overexpression of the sRNA was assessed in P. aeruginosa Δsr0161 expressing, under the control of the Ptac promoter, a fusion between the 5′ UTR and the first 20 amino acids codons of oprD and the entire coding sequence of lacZ. Bacteria were grown to a mid-logarithmic phase (OD600 0.5) and sRNA expression was induced by the addition of L-arabinose for 1 hr. Afterwards, the expression of the lacZ reporter was induced by addition of IPTG. Following incubation for an additional 2 hrs, the level of β-galactosidase activity was determined in each culture. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, where *** denotes P < 0.001.
C. The predicted base-pairing region of ErsA and oprD. The start codon is indicated as underlined and the Shine-Dalgarno sequence (S/D) is marked and underlined. A two-nucleotide mutation in ErsA (M) was generated and is indicated by an arrow and nucleotides.
D. The effect of ErsA and ErsA-M on oprD::lacZ expression. The growth conditions were the same as described in B, except the expression of the lacZ reporter was extended to 4 hrs. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, where ** denotes P < 0.01.
To validate the predicted base-pairing region between Sr0161, ErsA and their target mRNA, we used the β-galactosidase (lacZ) translational fusion to oprD to assess the effects of mutations in the predicted base pairing regions. A single nucleotide substitution (G for C) in the middle of the predicted base-pairing segment of Sr0161 abolished the repression of the translational oprD-lacZ reporter. A compensatory mutation (C for G) in the 5′ UTR restored the repression of lacZ expression (Fig. 6A,B). Similarly, disruption of the base paired region between ErsA and oprD by two substitutions (CC to GG) abolished repression (Fig. 6C,D). A compensatory mutation in the 5′ UTR of oprD could not be constructed because of its effect on translation initiation due to the disruption of the Shine-Dalgarno sequence.
Identification of additional targets of Sr0161 by GRIL-seq
Since we obtained low counts of Sr0161 chimeric reads by Hi-GRIL-seq, other targets could be missed due to the limitation of sequencing depth. Therefore, we used the original GRIL-seq protocol to further extend the repertoire of the Sr0161 mRNA targets, since this approach involves ligation of an expressed sRNA to its targets and an additional step to enrich the transcripts for the sRNAs containing chimeras (Han et al., 2016). Total RNA was isolated from the Δsr0161 strain co-expressing T4 RNA ligase and Sr0161. Following enrichment using complementary oligonucleotides (Supporting Information Fig. S7), the samples were sequenced using the Illumina platform. We obtained 143,383 and 130,811 chimeric reads for the two samples respectively, accounting for 0.51% and 0.54% of the total reads. Among them, 22,270 and 20,504 were reads of chimeric fragments containing sequences from oprD mRNA and Sr0161. The genomic coverage of the top 10 candidate targets are shown in Fig. 7A and Supporting Information Fig. S8. For all of these targets, most of the obtained chimeric reads showed Sr0161 at the 3′ end of the fragment. We designed primers against the 7 mRNA targets, and used RT-PCR to confirm that they are present as chimeras in bacteria expressing T4 RNA ligase (Supporting Information Fig. S8). The RT-qPCR data shows that besides oprD, the RNA levels of 4 other targets (opdP, braC, lecB, exsA) are also decreased following Sr0161 overexpression and modestly increased by Sr0161 deletion (Fig. 7B). The most dramatic effect of Sr0161 overexpression has been seen on the opdP transcript. It has been previously reported that OpdP (PA4501 or OccD3) also contributes to carbapenem uptake and, loss of OpdP causes an increase in carbapenem resistance in strains also lacking OprD (Isabella et al., 2015). However, we were unable to detect a difference in meropenem resistance caused by Sr0161-dependent inhibition of opdP (Supporting Information Fig. S9), suggesting that although it is a target of Sr0161, reduced expression of OpdP by induction of Sr0161 may affect other porin related functions but not show a noticeable change of carbapenem resistance.
Fig. 7.

Identification of additional Sr0161 targets by GRIL-seq.
A. Genome coverage of candidate targets in sequenced chimeric reads containing Sr0161. The lines and their height represent the location of the junction and the number of chimeric reads at that location. Two biological replicates (S1, S2) are shown. Bold numbers on the left are the scales of the tracks.
B. Effect of overexpression of Sr0161 on transcript levels of selected putative Sr0161 targets measured by RT-qPCR. Control vector or a plasmid carrying the L-arabinose inducible Sr0161 (Sr0161++) was conjugated into wild type P. aeruginosa PAO1 or the Δsr0161 mutant. Following 1 hr induction of sRNA, at the mid-logarithmic, cells were collected for RNA extraction and RT-qPCR. The housekeeping gene proC was used to calculate relative expression and rpoD was used as control. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, and **, *** denotes P<0.01 and P < 0.001.
Another target was exsA, the key transcriptional regulator of the type III secretion system (T3SS). ExsA is an activator protein and controls the expression of the T3SS by specific interactions with regulatory sequences located upstream of the genes coding for the secretion apparatus and the secreted effectors (Yahr et al., 2006). We therefore examined whether the Sr0161-dependent repression of ExsA affected the expression of its regulon. Western blot analysis of P. aeruginosa overexpressing Sr0161 in low calcium T3SS inducing medium demonstrated that this sRNA reduced the levels of the PscC outer membrane component of the T3SS machinery as well as that of the secreted exoenzyme S (ExoS), as shown in Figure 8.
Fig. 8.
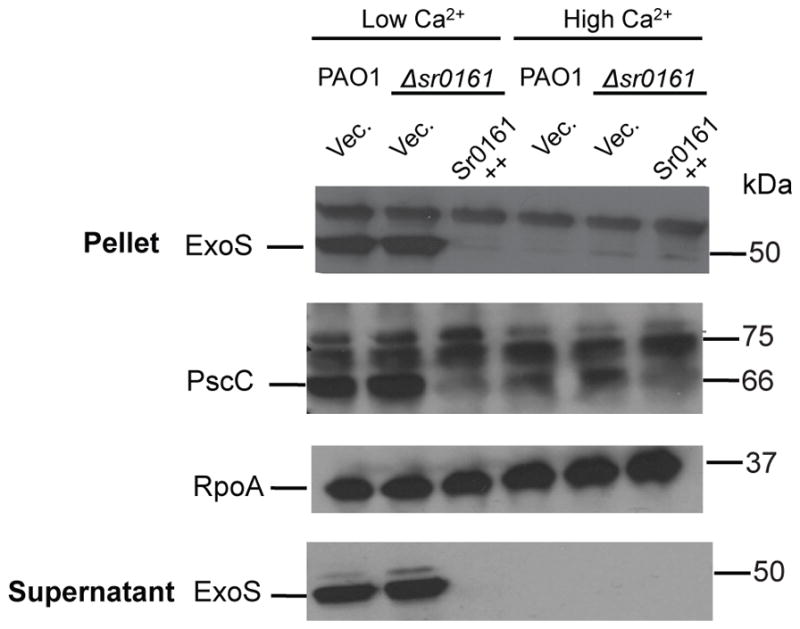
Sr0161 is a regulator of the T3SS. Cultures of P. aeruginosa were grown in T3SS inducing (low Ca2+) and non-inducing (high Ca 2+) media containing the L-arabinose inducer of sRNA expression (Sr0161++). Normalized cell pellets and their supernatant were used in western immunoblot and they were probed with antibodies against PscC and ExoS. A blot using antibodies against RpoA served as a loading control.
Positive post-transcriptional regulation of pagL by sRNA
Another target of an sRNA involved in P. aeruginosa virulence and host adaptation identified by Hi-GRIL-seq is pagL (Fig. 3 and Fig. 4C), which was ligated to the sRNA Sr006 (average count 201.3 ± 36.2). To test whether the interaction between Sr006 and the mRNA of pagL had any regulatory effect, we performed western immunoblots on cells expressing PagL with a C-terminal FLAG tag in the wild type PAO1 and Δsr006 strains overexpressing Sr006 or carrying the empty plasmid (Fig. 9A). Deletion of Sr006 caused no noticeable changes in PagL, while its overexpression leads to a significant increase in FLAG-tagged PagL protein expression (Fig. 9A). Analogous findings were made when examining pagL mRNA levels by RT-qPCR (Fig. 9B), where the overexpression of Sr006 leads to a significant accumulation of pagL mRNA. Therefore, Sr006 is a positive post-transcriptional regulator of pagL.
Fig. 9.
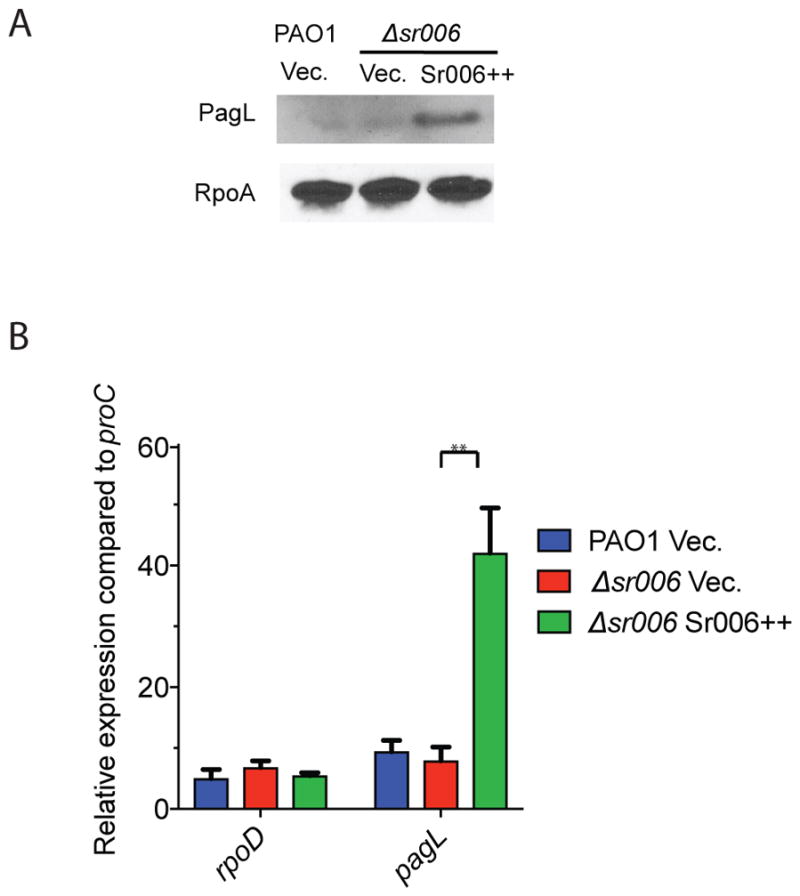
sRNA Sr006 is a positive regulator of pagL.
A. Western immunoblot analysis of Sr006’s effect on the expression of FLAG-tagged PagL fusion protein. A Ptac directed construct, consisting of a fusion between the 5′ UTR and the entire coding sequence of pagL with the codons of the FLAG tag peptide was inserted into the chromosome of wild type P. aeruginosa PAO1 or the Δsr006 mutant; these strains were then the recipients of the empty vector (Vec.) or Sr006 overexpression plasmid (Sr006++). The sRNA was expressed for 1 hr following the addition of the L-arabinose inducer and expression of the FLAG-tagged PagL was induced with IPTG for 1 hr. Samples were collected for western immunoblot analysis. RpoA was used as loading control.
B. Impact of Sr006 on pagL transcript levels. RT-qPCR was used to assess the pagL mRNA levels in wild type PAO1 or Δsr006. Cells were grown to mid-logarithmic phase and Sr006 expression was induced for 1 hr before samples were collected for RNA isolation and qRT-PCR. Housekeeping gene proC was used to calculate relative expression and rpoD was used as control. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, and ** denotes P < 0.01.
The predicted base-pairing model for Sr006-pagL interaction suggests the involvement of a region starting at 32 nucleotides upstream of the AUG codon. To confirm that this base pairing is responsible for enhancement of pagL expression, we mutated two nucleotides (CC to GG) in the sRNA and created compensatory substitutions (GG to CC) in the base-paired region of the pagL 5′UTR (Fig. 10A). We used a pagL-lacZ translational reporter construct to assess the consequences of these mutations on the expression of the reporter gene. β-galactosidase assays showed that the two-nucleotide substitution mutation in the predicted base-pairing area of Sr006 disrupted its positive regulatory effect on pagL-lacZ translational fusion expression (Fig. 10B). Expression of the mutant Sr006 increased the β-galactosidase activity of translational fusion carrying a compensatory mutation in the 5′ UTR of pagL. We did not identify additional mRNA targets of Sr006 in the tested condition even after carrying out a specific GRIL-seq experiment with Sr006 overexpression and an enrichment for Sr006 chimeras (data not shown). Therefore, Sr006 appears to control only pagL, although we cannot exclude the possibility that other growth conditions may uncover additional regulatory targets.
Fig. 10.

Base pairing between Sr006 and pagL mRNA.
A. The predicted base-pairing regions of Sr006 and the 5′ UTR of the pagL mRNA. using the IntaRNA algorithm. The start codon at +1 is underlined. The locations of the substitution mutations are indicated by arrows.
B. A Ptac directed pagL::lacZ construct consisting of the 5′ UTR and the first 18 amino acids codons of pagL fused to lacZ was introduced into P. aeruginosa Δsr006. The effect of the WT and a two-nucleotide mutation in Sr006 (M) was determined in this reporter strain. To assess the ability of the compensatory mutation in the base-pairing region to restore sRNA regulation, the 5′ UTR of pagL was mutated in the reporter construct (M′) and tested for the effect of Sr006 overexpression. Bacterial growth and induction conditions were identical to those described in Fig. 6B. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, and ** denotes P < 0.01.
To investigate whether regulation of PagL expression by Sr006 can lead to structural changes in the lipid component of LPS, we isolated lipid A from wild type P. aeruginosa PAO1 carrying an empty vector, an sr006 deletion strain carrying an empty vector, and the deletion strain over-expressing Sr006. All strains were grown in M9 succinate minimal media supplemented with 8μM Mg2+, a growth condition that activates the two-component regulatory system, PhoP/PhoQ. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) in the negative ion mode was used to analyze the structural characteristics of the isolated lipid A. Deacylation of the 3-hydroxy decanoic acid (3-OH C10) acyl chain from lipid A by the PagL enzyme can be observed by MALDI-MS (correlates to a to Δm/z of 170). Lipid A structures present in each sample were estimated based on their known structures and molecular weights. The canonical PAO1 lipid A species is penta-acylated with fatty acid chains ranging from 10–12 carbons in length (m/z 1446 and 1462). Hexa-acylated lipid A synthesis intermediates differ by the addition of a 10- or 16-carbon acyl chain. These hexa-acylated species can be distinguished using mass spectrometry, allowing identification of the specific PagL substrate (hexa-acylated lipid A with a hydroxy-C10 acyl chain, represented by m/z 1616 and 1632). Compared to PAO1 wildtype (Fig. 11A), the absence of sr006 did not appear to alter the levels of hexa-acylated lipid A species containing C10 (Fig. 11B). However, overexpression of Sr006 reduced the intensity of hexa-acylated lipid A species (Fig. 11C), suggestive of an increase in PagL deacylation. The decrease in 3-OH C10-modified hexa-acylated lipid A species observed with Sr006 over-expression was validated using gas chromatography with flame ion detection (GC-FID) of triplicate samples (data not shown). These results strongly suggest that post-transcriptional regulation of PagL expression by Sr006 has physiological consequences, leading to conversion of lipid A to a less pro-inflammatory form.
Fig. 11.

MS analysis of PAO1 strains grown in M9 succinate minimal media supplemented with 8μM Mg2+.
PagL-mediated deacylation is represented in red. m/z 1616 and 1632 represent hexa-acylated lipid A species with hydroxy-C10 addition, the canonical substrate of PagL. m/z 1446 and 1462 represent penta-acylated lipid A species after deacylation of the C10 chain by PagL. Shown samples from P. aeruginosa PAO1 carrying the empty vector (A), an sr006 deletion mutant with an empty vector (B) and the same deletion strain carrying an overexpressing sr006 plasmid (C).
It has been previously reported that in Salmonella enterica, PagL increases the polymyxin B resistance of lipopolysaccharide (LPS) aminoarabinose modification-defective strains (Kawasaki et al., 2007). We wished to test whether Sr006-dependent regulation of PagL affects the polymyxin B resistance in P. aeruginosa as well. We found that in a P. aeruginosa strain lacking the LPS aminoarabinose modification (arn) operon, loss of PagL decreased its resistance to polymyxin B, while overexpression of Sr006 lead to increased resistance (Fig. 12). This result suggests that Sr006 regulates LPS modification through affecting PagL expression.
Fig. 12.
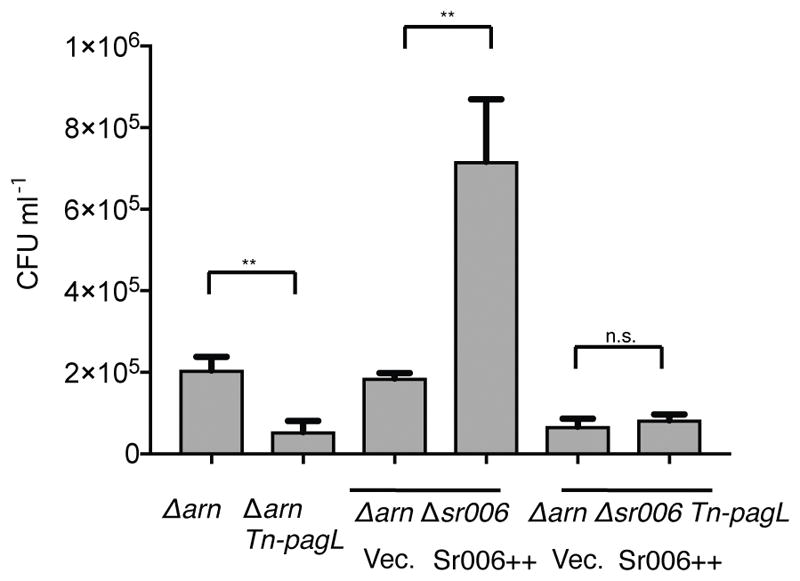
Overexpression of Sr006 increases polymyxin B resistance of strains lacking the LPS arabinose modification operon (Δarn).
Following L-arabinose induction of Sr006 (Sr006++) in ΔarnΔsr006 or ΔarnΔsr006 Tn-pagL strain, polymyxin B was added to the cultures for 3 hours. Cells were diluted and plated on LB plates to enumerate the survivors. Data are shown as mean ± SD for three biological replicates. Statistical comparisons were performed using Student’s t-test. Differences were considered statistically significant at P-values < 0.05, where n.s. and ** denote not significant and P < 0.01, respectively. Δarn is a deletion mutant of LPS arabinose modification operon. Tn-pagL is a transposon insertion mutant of pagL.
We evaluated whether the negative and positive regulation by Sr0161 and Sr006 respectively, was dependent on the RNA chaperone Hfq. First, we used RT-PCR to detect chimeras between the sRNAs and their mRNA targets in wild type P. aeruginosa or Δhfq each expressing T4 RNA ligase. We observed no specific amplicons of chimeras formed by Sr0161 and the oprD mRNA in Δhfq (Supporting Information Fig. S10A) while the absence of Hfq had no effect on the formation of Sr006-pagL chimeras (Supporting Information Fig. S10B). We further tested mRNA levels of target genes by RT-qPCR in the Δhfq strain overexpressing the sRNA or carrying the empty vector. The negative regulation of oprD by Sr0161 was abolished in the hfq deletion strain (Supporting Information Fig. S10C), consistent with the role of this RNA chaperone in sRNA-mediated gene regulation. In contrast, deletion of hfq had no effect on up-regulation of pagL by Sr006 (Supporting Information Fig. S10D). The regulation of pagL by Sr006 appears to be independent of Hfq and it may require a different RNA chaperone (Attaiech et al., 2017) or no other factor.
Sequence conservation of sRNAs and their targets
Previous studies have suggested that two features characteristic of trans-acting sRNAs play an important role in regulation of gene expression: the conservation of the base-pairing regions and their accessibility (Peer et al., 2011). We determined the conservation of the sRNAs and the 5′ UTR of the targets by calculating the positional entropy as previously described (Peer et al., 2011) (Supporting Information Fig. S11). The entropy score was calculated based on alignment of similar sequences from available complete and draft genomes (from 2226 strains) of P. aeruginosa (Winsor et al., 2016) that were found using BLAST (Altschul et al., 1990). No single nucleotide polymorphisms (SNPs) were found in the base-pairing region of either the sRNAs or the 5′ UTR of the target mRNAs. A limited number of SNPs was observed outside the base-pairing region of Sr006 and 5′ UTR of oprD while the 5′ UTR of pagL was almost completely conserved. These results suggest that, in addition to sequences involved in base pairing, other regions of the transcripts of sRNA and their targets are under selective pressure to maintain sequence conservation, possibly to allow optimal base pairing or serving as recognition sites for accessory factors such as Hfq or ProQ. Sr0161 shows considerable variability, particularly in the 5′ two thirds of the transcript (Supporting Information Fig. S11A). In the Hi-GRIL-seq data, the majority of the Sr0161 chimeras are formed by the ligation of the 3′ hydroxyl group of the target fragments and the 5′ monophosphate of the Sr0161 degradation products at the +171–175 bp downstream of transcription start site (Fig. 4A). We tested whether this 3′ degradation product of Sr0161 alone was sufficient to repress oprD. The 3′ end of Sr0161 starting from the +171 bp containing the base-pairing region (Sr0161-BP) was cloned and this construct, when over-expressed in P. aeruginosa, was able to decrease the RNA level of oprD (Fig. 5B) with a concomitant increase of the resistance to meropenem (Supporting Information Fig. S9B) similar to the effect of the full length Sr0161. Therefore, the 5′ end of Sr0161 is not required for inhibition of oprD and it is conceivable that it is not under the same selection pressure for conservation as the 3′ end.
The predicted secondary structure of Sr0161 and Sr006 and the 5′ UTR of their respective main targets oprD and pagL by the RNA structure software (Supporting Information Fig. S12A–D) showed that the base-pairing regions are all in less structured and more accessible areas. While a putative element associated with Hfq binding could be identified in Sr0161 and oprD, none were apparent in Sr006 or the 5′ UTR of pagL, confirming that the positive regulatory effect resulting from the interaction between these two RNAs is Hfq independent.
Discussion
The recognition of an important regulatory mechanism in bacteria based on base pairing of non-coding sRNAs with complementary regions of mRNAs made it possible to describe the activities of regulatory networks more completely, consisting of both transcriptional and post-transcriptional components. Here we extended GRIL-seq, the proximity RNA ligation method for sRNA target identification (Han et al., 2016), towards a more comprehensive analysis of sRNA-target RNA interactions. The Hi-GRIL-seq approach differs from GRIL-seq by being able to identify all chimeras created by the expressed T4 RNA ligase in live bacterial cells, followed by RNA-seq using rRNA depleted samples. Hi-GRIL-seq does not require previous knowledge of the sRNA sequence. By comparing RNA ligation products isolated from cultures grown in iron replete and iron-limiting medium, we confirmed that most of the previously identified targets of the iron-regulated sRNA PrrF1 could be detected by Hi-GRIL-seq. The main limitation of the Hi-GRIL-seq approach is sequencing depth, since in most cases, chimeras between the sRNAs and their targets represent a very small fraction of total sequencing reads generated by RNA-seq. It is reasonable to expect that there will be further improvements in the performance of sequencing platforms to increase the depth of sequencing and consequently, enhance the ability of Hi-GRIL-seq to identify a wider range of sRNA-mRNA interactions. However, as we illustrate here, the coverage available by the use of a current sequencing tool (Illumina NextSeq) is sufficient to identify biologically important novel sRNA-mRNA regulatory interactions. In addition, a more important consideration for using Hi-GRIL-seq as a discovery tool for sRNA mediated regulatory network is to sample RNAs from bacteria with a particular genetic background or grown under conditions when a specific sRNA and its targets are expressed at sufficient levels to provide substrates for the formation of chimeras by T4 RNA ligase.
In this work, we showed how Hi-GRIL-seq can be used to uncover novel interactions of sRNAs with specific mRNAs. We found that the levels of a transcript encoding the OprD porin responsible for the uptake of basic amino acids, peptides and carbapenem antibiotics, are controlled by two trans-acting sRNAs (Sr0161 and ErsA). We confirmed their post-transcriptional regulatory functions by demonstrating that the concentration of the oprD mRNA, the OprD protein level and the antibiotic (meropenem) susceptibility phenotypes are all reduced when the expression of each sRNA is increased. The previously described regulatory target of ErsA, the transcript for the alginate biosynthetic gene algC, was not identified by Hi-GRIL-seq in the non-mucoid strain PAO1, however, interaction between ErsA and algC can be readily detected in several mucoid, alginate producing strains.
Several previous studies have examined the regulatory mechanisms controlling the expression of OprD and suggesting a role for Hfq and an sRNA-based regulatory mechanism. The transcription of the oprD gene is negatively regulated by two metal responsive two-component systems (CzcRS and CopRS) and this regulatory effect requires the Hfq protein (Ducret et al., 2016). Hfq appears to directly facilitate binding of the response regulator CzcR to the promoter of the oprD gene. The molecular mechanism for the involvement of an RNA chaperone in transcriptional repression has not been elucidated to date. The PhrS sRNA, identified by Hfq co-immunoprecipitation was shown upon over-expression to increase the intracellular concentration of OprD (Sonnleitner et al., 2008). Whether this effect is indirect or the consequence of positive regulation via base-pairing interaction between PhrS and oprD has not been determined. It is therefore conceivable that Hfq and multiple sRNAs, including the two identified in this study, regulate OprD. The transcription of the sRNAs may be under regulatory control of environmental signals that reflect the diverse functions of this porin ranging from the uptake of nutrients, antibiotics and a role in recognition by host defenses (Skurnik et al., 2013a, Skurnik et al., 2013b).
Following enrichment for Sr0161 containing chimeras and sequencing (GRIL-seq), we identified additional targets of this sRNA and for braC, opdP, lecP and exsA, and confirmed its regulatory effects using P. aeruginosa lacking or over-expressing the sRNA (Fig. 7). Of particular interest is the interaction of Sr0161with the transcript encoding the T3SS regulator ExsA, adding another element to the complex regulatory network controlling this virulence mechanism by various transcriptional and post-transcriptional mechanisms (Yahr et al., 2006, Diaz et al., 2011). The role of Hfq in post-transcriptional regulation of the T3SS has been established by demonstrating its role in stabilizing the RsmY sRNA (Sorger-Domenigg et al., 2007). RsmY is an antagonist of the RsmA protein, a positive regulator of the T3SS (Burrowes et al., 2006). Therefore, Hfq appears to indirectly and adversely affect T3SS expression by causing an increase in levels of RsmY. Our finding that Sr0161 directly interacts with the 5′UTR of the exsA mRNA leading to its degradation and consequently a block in the synthesis of at least one component of the T3SS machinery and a T3SS effector, implicates another sRNA-mediated mechanism for regulating this virulence mechanism during infection.
Sr006 is another sRNA uncovered by the application of Hi-GRIL-seq, regulating the expression of a determinant of pathogenesis. Over-expression of this sRNA causes an increase in the levels of the pagL transcript, encoding a lipid A 3-O-deacylase, which leads to a reduction in the pro-inflammatory signaling activity of LPS (Moskowitz et al., 2010). As expected, over-expression of Sr006 in P. aeruginosa results in increased lipid A deacylation (Fig. 11) and a corresponding enhancement of polymyxin B resistance (Fig. 12). PagL levels were shown to be elevated in the isolates from young CF patients compared to strains from other sources and mutations in pagL are not uncommon among isolates from patients with a severe respiratory disease (Greipel et al., 2016, Ernst et al., 2006). A decrease of expression of this sRNA during the chronic phase of the infection could lead to an increase in the highly inflammatory hexa-acylated lipid A in the bacterial LPS, contributing to the severe lung damage seen in CF patients with more advanced disease.
Unlike the negative control of oprD translation or stability by two sRNAs, pagL is positively regulated by Sr006 without a need for Hfq. Regulation of expression of the PagL protein, likely at the level of transcription of Sr006 in response to its interaction with the infected host may represent a mechanism allowing P. aeruginosa to recognize the host environment. An increase in the levels of pagL, in response to contact with epithelial cells has been previously demonstrated (Gellatly et al., 2012). Whether this effect on transcript levels is due to increased transcription of the pagL gene or a stabilizing effect by Sr006 on the mRNA, has not been determined.
We identified several interactions between individual sRNA which may result in regulatory functions. Among these, the CrcZ sRNA, involved in post-transcriptional regulation of catabolic genes (Sonnleitner et al., 2014) interacted with PrrF1 as well as Sr0161 (Figure 2B and 7A). It is conceivable that CrcZ can recognize both of these sRNAs by a base-pairing mechanism, and sequesters them from their mRNA targets by a “sponging” mechanism. A more likely explanation is that the formation of the chimeras between CrcZ and PrrF1 or Sr061 by RNA ligase is through its interaction with Hfq (Sonnleitner et al., 2017), where simultaneous binding of the sRNA to this protein may lead to the formation of a covalently linked product due to the proximity of their 3′ and 5′ ends.
In conclusion, the work described here demonstrates that Hi-GRIL-seq, a modification of the T4 RNA ligase catalyzed proximity ligation method, is well suited for studies of sRNA-mediated regulation of gene expression at a genome-wide level. By focusing on a handful of examples, we demonstrated that Hi-GRIL-seq is a promising tool for the discovery of novel sRNA-mediated regulatory pathways. Refining this method, particularly when improved platforms with an increased sequencing depth to detect interactions between less abundant sRNA and their targets become available, should greatly facilitate progress in our attempts to fully define complex regulatory networks involved in bacterial adaptation to different environments including an infected host.
Experimental Procedures
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in Supporting Information Table S3. Unless otherwise noted, P. aeruginosa PAO1 was used for plasmid conjugation and mutant construction. All deletion and substitution mutants of P. aeruginosa were created with the pEXG2 plasmid as described previously (Rietsch et al., 2005). A list of the oligonucleotides used for the constructions is provided in Supporting Information Table S4. Cloning was performed with either splicing by overlap extension (SOEing) PCR (Horton et al., 1989) and T4 DNA ligation or a Gibson Assembly cloning kit (New England Biolabs). The sRNA overexpression vectors were created by coning PCR-generated amplicons starting from the sRNA transcription start sites to their native transcription terminators, inserted into the pKH6 (Han et al., 2016) between the XbaI and HindIII sites.
Media and growth conditions
P. aeruginosa cultures were grown with shaking at 37 °C, in Luria–Bertani (LB) broth. Antibiotics were added to the culture as required at the following concentrations unless otherwise noted: for P. aeruginosa: 150 μg ml−1 carbenicillin, 75 μg ml−1 gentamicin, 75 μg ml−1 tetracycline and 25 μg ml−1 irgasan; for E. coli: 50 μg ml−1 carbenicillin, 15 μg ml−1 gentamicin and 10 μg ml−1 tetracycline. For lipid A analysis, P. aeruginosa were grown in M9 minimal media supplemented with 20 mM succinate and 8μM MgSO4.
For RT-PCR detection of PrrF1 chimeras and for Hi-GRIL-seq, P. aeruginosa PAO1 carrying pKH13-t4rnl1 was grown overnight in LB broth with carbenicillin at 37 °C. These cultures were then used to inoculate the same medium to an OD600 0.01. When the cultures reached OD600 0.5. T4 RNA ligase was induced by addition of IPTG to 1mM. After 1 hr, the iron chelator 2,2′-dipyridyl (2,2′-DIP) was added to 300μM for 20 min for the iron depletion group while the controls were not treated with the chelator. The cells were then collected for RNA extraction. For GRIL-seq, PAO1Δsr0161 carrying two expression plasmids pKH13-t4rnl1 and pKH6-Sr0161, was grown as described previously (Han et al., 2016). Briefly expression of T4 RNA ligase was induced for 1 hr with 1mM IPTG at OD600 0.5, followed by induction of sRNA Sr0161 for 20 min with 0.2% L-arabinose for 20 min.
RNA isolation and reverse transcription
Bacteria were harvested by centrifugation for 1 min at 16,000 x g when they reached a density equivalent to OD600 1.0 and the pellets were rapidly frozen in liquid nitrogen. Total RNA was isolated using the phenol-free total RNA purification kit (Amresco), according to the manufacturer’s instructions, with the modification that 50μl of nuclease-free water (Ambion) was applied to the column for RNA elution. A NanoDrop spectrophotometer (Thermo Fisher) was used to measure RNA concentration. To remove DNA from total RNA samples, 10μg of RNA sample was treated in a 50 μl reaction with a Turbo DNA-free kit (Ambion). To prepare template for RT-PCR and RT-qPCR, cDNA was synthesized with a SuperScript III First-Strand Synthesis system (Invitrogen) with random hexamers.
Detection of sRNA-target chimeras by RT-PCR
cDNA reverse transcribed from 100ng of total RNA was used as template for RT-PCR with a GoTaq Green Master Mix (Promega). Primers specific for sRNA and mRNA respectively were used to amplify chimeras and are listed in Supporting Information Table S4. Cycling conditions were: 95°C for 3 min; 30 cycles of 94 °C for 25 s, 55–66 °C for 25 s and 72 °C for 60 s; and a final extension cycle of 72 °C for 5 min. For detection of chimeras formed by Sr0161 and the mRNA of exsA, PA3801 and wbpH, PCR was performed with 32 cycles instead of 30. The PCR products were separated by 2% agarose gel electrophoresis, and cloned into pJET1.2 vector (Thermo Fisher) and sequenced with the pJET1.2 forward and reverse primer.
Preparation of the cDNA library for Illumina sequencing
After total RNA isolation, the ribosome integrity number (RIN) was determined with an RNA picochip on a 2100 Agilent Bioanalyzer. The RINs ranged from 9 to 10. For Hi-GRIL-seq, the Ribo Zero rRNA removal kit for Gram-negative bacteria (Illumina) was used for rRNA depletion. For GRIL-seq, the enrichment of chimeric sRNA was carried out as previously described (Han et al., 2016), with the exception that poly(A)-tailed Sr0161 complementary oligonucleotides were used to capture chimeric sRNA, their sequence is listed in Table 4. After rRNA depletion or sRNA enrichment, cDNA libraries were prepared with the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs). RNA-seq was performed on an Illumina NextSeq platform by the Biopolymers Facility at Harvard Medical School.
GRIL-seq data analysis
A custom BLAST-based chimera pipeline was used to analyze the GRIL-seq data. The first and last 25 nucleotides at both ends of the Illumina reads were mapped to a reference genome using a modified version of the Rockhopper software system (McClure et al., 2013). Reads were considered to be potentially chimeric if the two ends mapped to different regions of the genome at least 1,000 nucleotides apart and the mappings contained no more than three mismatches. This allowed rapid identification of a subset of the reads that were likely to be chimeric. These potential chimeric reads were further aligned to the genome using BLAST version 2.6.0. BLASTN was run with the expectation value (e-value) set to 0.01, reward value set to 1, penalty value set to −2 and other parameters set to default (States et al., 1991). The precise nucleotide locations of the chimeric junctions were determined from the BLAST results by the following criteria: (1) the read did not have a long top hit (read length-20, e-value ≤ 1.0e-50) suggesting that the read was likely to correspond to a single fragment rather than a chimeric read; (2) the read had at least two BLAST hits with e-value ≤ 0.01, and the two hits were either adjacent, or with up to 5 nucleotides of gap or overlap; (3) for reads that aligned to multiple chimeric locations in the genome, the most significant alignment as determined by e-value was used; and (4) reads that mapped to different regions of at least 2,000 nucleotides apart were used for downstream analysis. We evaluated different distance thresholds (Supporting Information Fig. S14), and found that 2,000 is efficient in removing chimeras that are likely to be preferential ligations within the same transcript. After determining the chimeric junctions, we calculated the frequency of junctions corresponding to annotated protein coding genes, RNAs, and intergenic regions, as reported in Supporting Information Table S1. When reporting top candidate targets for an sRNA (Supporting Information Table S2 for PrrF1/2), we considered these as chimeric reads if the sRNA corresponded to either the 5′ end or the 3′ end of the chimera. Chimeric junctions located in the untranslated region and the coding region of a target gene were combined when calculating the abundance of chimeric reads of a target with a given sRNA. Visualization of chimeric junctions throughout the genome was performed using R and IGV (Thorvaldsdóttir et al., 2013).
Western immunoblots
The plasmid pPtac-miniCTX::FLAG was first constructed for making IPTG inducible c-terminal FLAG tagged fusion protein. Briefly, plasmid miniCTX1 was linearized with KpnI and SacI and a synthesized 1.6 kb fragment (gBlocks gene fragment, IDT), containing the sequence for lacIq, Ptac, a multiple cloning site followed by a C-terminal FLAG tag and stop codon, was inserted (Supporting Information Table S4). The lacIq and Ptac sequence is the same as in pPtac-miniCTX::lacZTL (Han et al., 2016). The 5′UTR and ORF without a stop codon of oprD and pagL were inserted into the pPtac-miniCTX::FLAG at EcoRI and BamHI restriction sites. The construct was introduced into the chromosome of various P. aeruginosa strains at the CTX attB site. For western blot analysis, overnight cultures of P. aeruginosa containing the FLAG tagged fusion protein and the pKH6 empty vector, or the sRNA overexpression plasmid, were grown in LB with gentamicin and tetracycline. The overnight cultures were diluted to OD600 0.01 in the same medium and incubated at 37°C to OD600 0.5. sRNA was induced for 1 hr with 0.2% L-arabinose and then FLAG fusion protein was induced with 0.1 mM IPTG for 1 hr or 3 hrs, followed by sample collection. Total proteins were resolved by SDS-PAGE and transferred to a nitrocellulose membrane. FLAG tagged fusion proteins were detected with rabbit anti-FLAG antibody (Sigma). The loading control RpoA was detected with mouse anti-E. coli RNA Polymerase α subunit antibody (Yang et al., 2015).
Western immunoblot was performed to detect the protein level of ExoS and PscC in strains expressing high or low levels of Sr0161. P. aeruginosa overnight cultures were diluted to an equivalent of OD600 0.02 in low-calcium or high-calcium LB media. Low-calcium medium was achieved by treating LB media with nitrilotriacetic acid (NTA) to 10 mM. For high-calcium medium, CaCl2 was added to the NTA-treated LB to a final concentration of 20mM (Wolfgang et al., 2003). The media was supplemented with 0.2% L-arabinose to induce Sr0161 expression. Cultures were incubated at 37°C for 6 hours and reached OD600 2.2–2.4. All samples were normalized based on OD600. For each sample, half of the culture was centrifuged and the cell pellets were resuspended in Laemmli sample buffer (Bio-Rad). The supernatant of the other half of the sample was precipitated with 10% trichloroacetic acid, washed twice with acetone, and resuspended in sample buffer. Western immunoblot was performed similarly as described above except that ExoS and PscC were detected with respective primary polyclonal antibody (Hoang et al., 2011, Lee et al., 2005).
RT-qPCR
For determining target RNA levels upon induction of sRNA, overnight cultures of PAO1 or PAO1 sRNA deletion mutants, carrying either the empty plasmid pKH6 or pKH6-sRNA, were grown in LB with gentamicin. The following morning, the cultures were diluted to an OD600 0.02 and the bacteria were grown at 37°C with shaking. When OD600 reached 0.5, sRNA was overexpressed for 1 hr by adding L-arabinose to 0.2%. RNA extraction, DNA removal and cDNA synthesis were performed as described above. Quantitative PCR was carried out with the PerfeCTa SYBR Green FastMix (Quanta Biosciences) on a Mastercycler Realplex2 system (Eppendorf) according to the manufacture’s instructions. The primers used for RT-qPCR are listed in Supporting Information Table S4. RNA level of genes relative to the housekeeping gene proC (Savli et al., 2003) were calculated.
β-galactosidase assay
The lacZ translational fusion was constructed as described previously (Han et al., 2016). Briefly, the sequence consisting of the 5′ UTR and the codons for the first 20 amino acids of oprD or the sequence of the 5′ UTR and the codons for the first 18 amino acids of pagL were amplified by PCR and inserted between the EcoRI and HindIII sites of pPtac-miniCTX::lacZTL. The base-pairing region mutation constructs were created by SOEing PCR with primers containing the changes. The translational lacZ reporter constructs were integrated into the chromosome at the attB site. Overnight cultures of cells containing the lacZ reporter construct and the pKH6 empty vector or sRNA overexpression plasmid were grown in LB with gentamicin and tetracycline. The overnight cultures were diluted to OD600 0.02 in the same medium and incubated for additional 4 hrs and reached OD600 0.5–1. The sRNA expression was induced for 1 hr with 0.2% L-arabinose and then lacZ fusion protein was induced with 0.1 mM IPTG for 2 or 4 hrs, followed by β-galactosidase assay, as performed in an earlier publication (Griffith et al., 2002).
5′ and 3′ rapid amplification of cDNA ends (RACE)
5′ and 3′ RACE was carried out using the FirstChoice RLM-RACE Kit (ThermoFisher) according to the manufacture’s instructions with modifications as described in a previous paper (Sharma et al., 2014). For 5′ RACE, the 5′ monophosphate end of Tobacco Acid Pyrophosphatase (TAP) treated RNA was ligated to the 5′ RACE adapter by T4 RNA ligase. Samples without TAP treatment was used as control. Random-primed reverse transcription reaction and nested PCR amplified the 5′ end of the sRNA. For 3′ RACE, the total RNA was polyadenylated using Poly (A) Polymerase enzyme (New England Biolabs). Polyadenylated RNA was then reversed transcribed and PCR amplified for the 3′ end of the sRNA. The PCR products were cloned into pJET1.2 vector (Thermo Fisher) and sequenced to confirm the initiation and termination site of sRNA transcription. Primers used for 5′ and 3′ RACE are listed in Supporting Information Table S4.
Lipid A structural analysis
For a mass spectrometric analysis, cell pellets were processed with an ammonium hydroxide-isobutyric acid-based procedure (El Hamidi et al., 2005) to extract the lipid A. Briefly, approximately 50 ml of cell culture was pelleted, resuspended in 400 μL of 70% isobutyric acid and 1M ammonium hydroxide (5:3, vol/vol), and heated for 1 hour at 100°C. After incubation, samples were centrifuged at 2000 x g for 15 minutes and supernatants were transferred to a fresh tube with endotoxin-free water (1:1 vol/vol). Samples were frozen on dry ice and lyophilized overnight. Dried material was washed twice with methanol and lipid A was extracted with 100 μL of a mixture of chloroform, methanol, and water (3:1:0.25, vol/vol/vol). After extraction, 1 μL of the lipid A concentrate was spotted on a stainless steel matrix-assisted laser desorption-time of flight (MALDI-TOF) plate followed by 1 μL of 10 mg/mL norharmane matrix in chloroform:methanol (2:1, vol:vol) (Sigma-Aldrich, St. Louis, MO). Samples were air dried and then analyzed on a Bruker Microflex mass spectrometer (Bruker Daltonics, Billerica, MA) in the negative-ion mode with reflectron mode. Electrospray tuning mix (Agilent, Palo Alto, CA) was used for mass calibration. Spectral data were processed and analyzed with Bruker Daltonics FlexAnalysis software. For gas chromatography analysis, LPS was extracted from biological triplicate replicates of each strain as described previously (Somerville Jr et al., 1996). Briefly, 80 mg of cell pellet was resuspended in 90% phenol and water (1:1, vol/vol) and incubated for 1 hour at 70°C with constant shaking. Samples were cooled and centrifuged at 9,000 x g for 10 minutes. The supernatant was transferred to a fresh glass tube and washed twice with diethyl ether. The lower phase containing LPS was collected and lyophilized. Fatty acids of the resultant product were converted to methyl esters by methanolysis (2M HCl in methanol; Alltech, Lexington, KY) with 18 hours incubation at 90°C. Samples were extracted twice with hexane. GC analysis was conducted using an HP 5890 series II with a 7673 autoinjector. Fatty acid methyl esters (Matreya BAME/bacterial acid methyl esters, CP mix no.1114) and pentadecanoic acid (C15; Sigma, St. Louis, MO) were used as standard of a known fatty acid mixture and internal standard for concentration control, respectively.
Prediction of interactions and secondary structures of sRNAs and target mRNAs
We used the IntaRNA (Busch et al., 2008) algorithm to predict base pairing between sRNA and target RNAs. RNAstructure (Reuter et al., 2010) software was used to predict secondary structure of the sRNAs and target RNAs.
Computing positional conservation of sRNA and target sequence
Similar sequences were identified by BLAST from 2,226 complete and draft genomes of P. aeruginosa (Winsor et al., 2016). A multiple sequence alignment of the similar sequences was performed using Clustal Omega. A conservation entropy score for each position was calculated as previously described (Peer et al., 2011).
Data availability
The sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive under accession codes SAMN07284097-SAMN7284100.
Supplementary Material
Acknowledgments
We thank members of the Lory lab for their comments on the work. We thank Thomas Dougherty for comments on the manuscript and Christian Lorenz for the gift of the P. aeruginosa arn mutant. This work was supported by an NIH grant R21 AI125972 and Grant LORY16GO from the Cystic Fibrosis Foundation to SL, NIH R01AI123820 to RKE and R15 GM102755 to BT. The authors do not have any conflicts of interest to report.
Footnotes
Author contributions
YFZ, SL, KH, CEC, RKE designed the study, YFZ, CEC performed the experiments, YFZ, KH, SL, RKE and BT analyzed the data and YFZ, SL wrote the manuscript.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Attaiech L, Glover JNM, Charpentier X. RNA chaperones step out of Hfq’s shadow. Trends Microbiol. 2017;25:247–249. doi: 10.1016/j.tim.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Burrowes E, Baysse C, Adams C, O’Gara F. Influence of the regulatory protein RsmA on cellular functions in Pseudomonas aeruginosa PAO1, as revealed by transcriptome analysis. Microbiology. 2006;152:405–418. doi: 10.1099/mic.0.28324-0. [DOI] [PubMed] [Google Scholar]
- Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008;24:2849–2856. doi: 10.1093/bioinformatics/btn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz MR, King JM, Yahr TL. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front Microbiol. 2011;2:89–89. doi: 10.3389/fmicb.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducret V, Gonzalez M, Scrignari T, Perron K. OprD repression upon metal treatment requires the RNA chaperone Hfq in Pseudomonas aeruginosa. Genes. 2016;7:82–82. doi: 10.3390/genes7100082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hamidi A, Tirsoaga A, Novikov A, Hussein A, Caroff M. Microextraction of bacterial lipid A: easy and rapid method for mass spectrometric characterization. J Lipid Res. 2005;46:1773–1778. doi: 10.1194/jlr.D500014-JLR200. [DOI] [PubMed] [Google Scholar]
- Ernst RK, Adams KN, Moskowitz SM, Kraig GM, Kawasaki K, Stead CM, et al. The Pseudomonas aeruginosa lipid A deacylase: selection for expression and loss within the cystic fibrosis airway. J Bacteriol. 2006;188:191–201. doi: 10.1128/JB.188.1.191-201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara S, Carloni S, Fulco R, Falcone M. Post-transcriptional regulation of the virulence-associated enzyme AlgC by the σ22-dependent small RNA ErsA of Pseudomonas aeruginosa. Environ Microbiol. 2015 doi: 10.1111/1462-2920.12590. [DOI] [PubMed] [Google Scholar]
- Fröhlich KS, Vogel J. Activation of gene expression by small RNA. Curr Opin Microbiol. 2009;12:674–682. doi: 10.1016/j.mib.2009.09.009. [DOI] [PubMed] [Google Scholar]
- Gellatly SL, Needham B, Madera L, Trent MS, Hancock RE. The Pseudomonas aeruginosa PhoP-PhoQ two-component regulatory system is induced upon interaction with epithelial cells and controls cytotoxicity and inflammation. Infect Immun. 2012;80:3122–3131. doi: 10.1128/IAI.00382-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes K, Gultyaev AP, Franch T, Pedersen K, Mikkelsen ND. Antisense RNA-regulated programmed cell death. Annu Rev Genet. 1997;31:1–31. doi: 10.1146/annurev.genet.31.1.1. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Storz G. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol. 2011;3:a003798–a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greipel L, Fischer S, Klockgether J, Dorda M, Mielke S, Wiehlmann L, et al. Molecular epidemiology of mutations in antimicrobial resistance loci of Pseudomonas aeruginosa isolates from airways of cystic fibrosis patients. Antimicrob Agents Chemother. 2016;60:6726–6734. doi: 10.1128/AAC.00724-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith KL, Wolf RE. Measuring β-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem Biophys Res Commun. 2002;290:397–402. doi: 10.1006/bbrc.2001.6152. [DOI] [PubMed] [Google Scholar]
- Han K, Tjaden B, Lory S. GRIL-seq provides a method for identifying direct targets of bacterial small regulatory RNA by in vivo proximity ligation. Nat Microbiol. 2016;2:16239–16239. doi: 10.1038/nmicrobiol.2016.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang HH, Nickerson NN, Lee VT, Kazimirova A, Chami M, Pugsley AP, Lory S. Outer membrane targeting of Pseudomonas aeruginosa proteins shows variable dependence on the components of Bam and Lol machineries. MBio. 2011;2:e00246–00211. doi: 10.1128/mBio.00246-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Isabella Vincent M, Campbell Arthur J, Manchester J, Sylvester M, Nayar Asha S, Ferguson Keith E, et al. Toward the rational design of carbapenem uptake in Pseudomonas aeruginosa. Chem Biol. 2015;22:535–547. doi: 10.1016/j.chembiol.2015.03.018. [DOI] [PubMed] [Google Scholar]
- Kawasaki K, China K, Nishijima M. Release of the lipopolysaccharide deacylase PagL from latency compensates for a lack of lipopolysaccharide aminoarabinose modification-dependent resistance to the antimicrobial peptide polymyxin B in Salmonella enterica. J Bacteriol. 2007;189:4911–4919. doi: 10.1128/JB.00451-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalaouna D, Massé E. Identification of sRNA interacting with a transcript of interest using MS2-affinity purification coupled with RNA sequencing (MAPS) technology. Genomics data. 2015;5:136–138. doi: 10.1016/j.gdata.2015.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VT, Smith RS, Tümmler B, Lory S. Activities of Pseudomonas aeruginosa effectors secreted by the Type III secretion system in vitro and during infection. Infect Immun. 2005;73:1695–1705. doi: 10.1128/IAI.73.3.1695-1705.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Luo YF, Williams BJ, Blackwell TS, Xie CM. Structure and function of OprD protein in Pseudomonas aeruginosa : From antibiotic resistance to novel therapies. Int J Med Microbiol. 2012;302:63–68. doi: 10.1016/j.ijmm.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizewski SE, Lundberg DS, Schurr MJ. The transcriptional regulator AlgR is essential for Pseudomonas aeruginosa pathogenesis. Infect Immun. 2002;70:6083–6093. doi: 10.1128/IAI.70.11.6083-6093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, et al. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013;41:e140–e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed S, Peer A, Faigenbaum-Romm R, Gatt YE, Reiss N, Bar A, et al. Global mapping of small RNA-target interactions in bacteria. Mol Cell. 2016;63:884–897. doi: 10.1016/j.molcel.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- Moskowitz SM, Ernst RK. Endotoxins: Structure, Function and Recognition. Springer; 2010. The role of Pseudomonas lipopolysaccharide in cystic fibrosis airway infection; pp. 241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K, Vanderpool CK. Target activation by regulatory RNAs in bacteria. FEMS Microbiol Rev. 2015;39:362–378. doi: 10.1093/femsre/fuv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peer A, Margalit H. Accessibility and evolutionary conservation mark bacterial small-RNA target-binding regions. J Bacteriol. 2011;193:1690–1690. doi: 10.1128/JB.01419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirnay JP, De Vos D, Mossialos D, Vanderkelen A, Cornelis P, Zizi M. Analysis of the Pseudomonas aeruginosa oprD gene from clinical and environmental isolates. Environ Microbiol. 2002;4:872–882. doi: 10.1046/j.1462-2920.2002.00281.x. [DOI] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11:129–129. doi: 10.1186/1471-2105-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences. 2005;102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savli H, Karadenizli A, Kolayli F, Gundes S, Ozbek U, Vahaboglu H. Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J Med Microbiol. 2003;52:403–408. doi: 10.1099/jmm.0.05132-0. [DOI] [PubMed] [Google Scholar]
- Sharma R, Arya S, Patil SD, Sharma A, Jain PK, Navani NK, Pathania R. Identification of Novel Regulatory Small RNAs in Acinetobacter baumannii. PLoS One. 2014;9:e93833–e93833. doi: 10.1371/journal.pone.0093833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurnik D, Roux D, Aschard H, Cattoir V, Yoder-Himes D, Lory S, Pier GB. A comprehensive analysis of in vitro and in vivo genetic fitness of Pseudomonas aeruginosa using high-throughput sequencing of transposon libraries. PLoS Pathog. 2013a;9:e1003582. doi: 10.1371/journal.ppat.1003582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurnik D, Roux D, Cattoir V, Danilchanka O, Lu X, Yoder-Himes DR, et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc Natl Acad Sci U S A. 2013b;110:20747–20752. doi: 10.1073/pnas.1221552110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville JE, Jr, Cassiano L, Bainbridge B, Cunningham MD, Darveau RP. A novel Escherichia coli lipid A mutant that produces an antiinflammatory lipopolysaccharide. J Clin Invest. 1996;97:359. doi: 10.1172/JCI118423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Bläsi U, Tielen P, Rosenau F, Jaeger KE. Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa Carbon Catabolite Repression. PLoS Genet. 2014;10:e1004440–e1004440. doi: 10.1371/journal.pgen.1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Prindl K, Bläsi U. The Pseudomonas aeruginosa CrcZ RNA interferes with Hfq-mediated riboregulation. PLoS One. 2017;12:e0180887. doi: 10.1371/journal.pone.0180887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnleitner E, Sorger-Domenigg T, Madej MJ, Findeiss S, Hackermuller J, Huttenhofer A, et al. Detection of small RNAs in Pseudomonas aeruginosa by RNomics and structure-based bioinformatic tools. Microbiology. 2008;154:3175–3187. doi: 10.1099/mic.0.2008/019703-0. [DOI] [PubMed] [Google Scholar]
- Sorger-Domenigg T, Sonnleitner E, Kaberdin VR, Bläsi U. Distinct and overlapping binding sites of Pseudomonas aeruginosa Hfq and RsmA proteins on the non-coding RNA RsmY. Biochem Biophys Res Commun. 2007;352:769–773. doi: 10.1016/j.bbrc.2006.11.084. [DOI] [PubMed] [Google Scholar]
- States DJ, Gish W, Altschul SF. Improved sensitivity of nucleic acid database searches using application-specific scoring matrices. Methods. 1991;3:66–70. [Google Scholar]
- Storz G, Vogel J, Wassarman Karen M. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters SA, McAteer SP, Kudla G, Pang I, Deshpande NP, Amos TG, et al. Small RNA interactome of pathogenic E. coli revealed through crosslinking of RNase E. The EMBO journal. 2017;36:374–387. doi: 10.15252/embj.201694639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilderman PJ, Sowa NA, FitzGerald DJ, FitzGerald PC, Gottesman S, Ochsner UA, Vasil ML. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc Natl Acad Sci U S A. 2004;101:9792–9797. doi: 10.1073/pnas.0403423101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FSL. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016;44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang MC, Lee VT, Gilmore ME, Lory S. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell. 2003;4:253–263. doi: 10.1016/s1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- Wolter DJ, Hanson ND, Lister PD, YL, PZ, NH, SA Insertional inactivation of oprD in clinical isolates of Pseudomonas aeruginosa leading to carbapenem resistance. FEMS Microbiol Lett. 2004;236:137–143. doi: 10.1016/j.femsle.2004.05.039. [DOI] [PubMed] [Google Scholar]
- Wood LF, Ohman DE. Use of cell wall stress to characterize σ22 (AlgT/U) activation by regulated proteolysis and its regulon in Pseudomonas aeruginosa. Mol Microbiol. 2009;72:183–201. [Google Scholar]
- Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, et al. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Path. 2012;8:e1002945–e1002945. doi: 10.1371/journal.ppat.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahr TL, Wolfgang MC. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol. 2006;62:631–640. doi: 10.1111/j.1365-2958.2006.05412.x. [DOI] [PubMed] [Google Scholar]
- Yang N, Ding S, Chen F, Zhang X, Xia Y, Di H, et al. The Crc protein participates in down-regulation of the Lon gene to promote rhamnolipid production and rhl quorum sensing in Pseudomonas aeruginosa. Mol Microbiol. 2015;96:526–547. doi: 10.1111/mmi.12954. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive under accession codes SAMN07284097-SAMN7284100.