

Abstract
The burgeoning field of gene-by-environment (G × E) interactions has revealed fascinating biological insights, particularly in the realm of stress-, anxiety-, and depression-related disorders. In this review we present an integrated view of the study of G × E interactions in stress and anxiety disorders, including the evolution of genetic association studies from genetic epidemiology to contemporary large-scale genome-wide association studies and G × E studies. We convey the importance of consortia efforts and collaboration to gain the large sample sizes needed to move the field forward. Finally, we discuss several robust and well-reproduced G × E interactions and demonstrate how epidemiological identification of G × E interactions has naturally led to a plethora of basic research elucidating the mechanisms of high-impact genetic variants.
Keywords: anxiety, depression, posttraumatic stress disorder, stress, trauma, epigenetics, gene-by-environment interaction, genome-wide association study
INTRODUCTION
In the study of mental health, the complex interplay of experience, environment, and genetics in both health and disease makes it challenging to interpret the contribution of any particular genetic variant to disease. The disciplines of psychology and psychiatry, perhaps more than any other fields of medicine, are faced with understanding diseases of incredible complexity, from the genetics underpinning neural circuits and hormonal signaling, to the influence of environment-dependent experience on shaping these pathways, to the way biological processes create the mind. These challenges have led to the application of genetic association studies to mental health diseases. By necessity, the complexity of cognitive and emotional disorders, and the crucial role of the environment in these diseases, has quickly led to the study of gene-by-environment (G × E) interactions in mental health. In this review we focus on G × E interactions in stress- and anxiety-related disorders, but we also present a broad overview of genetic association studies.
The fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5; Am. Psychiatr. Assoc. 2013) has drawn a distinction between stress and anxiety, separating trauma- and stressor-related disorders [e.g., posttraumatic stress disorder (PTSD), acute stress disorder] from anxiety disorders (e.g., generalized anxiety disorder, phobias, panic attack). However, representing fear and anxiety along a continuum with shared symptomology may paint a more accurate picture (Craske et al. 2009). Furthermore, for PTSD, comorbidity with other psychiatric disorders may be the rule rather than the exception (Brady et al. 2000). Depressive disorders may also be better defined in relation to stress and anxiety, as evidenced by the diagnostic comorbidity of both PTSD and anxiety disorders with depression, and the well-established relationship between early life stress and the development of depression. At present it is unclear whether individuals with comorbid disorders exhibit a unique disease etiology as compared to individuals with just one disorder, or whether the comorbidity of stress, anxiety, and depression is simply a product of the diagnostic criteria we use to identify each of these disorders.
In addition to discussing genetic associations and robust G × E findings, below we also consider the disease selection criteria and environmental measures used in genetic association studies. Because sample size is the primary hindrance to achieving significant genetic associations, studies may tend to group together subjects that have distinct diseases in an effort to maximize statistical power. We focus primarily on PTSD as the disease outcome, because it is the best-studied stress and anxiety disorder; however, we also consider depression because of the association between stress and depression, and the insightful G × E studies that have confirmed this outcome.
In this review of G × E associations in stress and anxiety disorders, we outline the progress from the origins of genetic epidemiology to the genome-wide association studies (GWAS) and G × E studies of today. Though we do not cover every facet of genetic association studies, we aim to convey the continuity of this field and lay out a logical path moving forward. We also present the theory underpinning genetic association studies and the challenges that have arisen from these fundamentals. Finally, we discuss specific G × E examples to illustrate how findings are validated and how basic neurobiology research has dovetailed with genetic association studies to understand the biology underlying genetic variants identified in G × E studies.
AN OVERVIEW OF GENETIC ASSOCIATION STUDIES
Genetic Epidemiology
Understanding G × E studies requires an overview of genetic association approaches in general and of the progress in the field. The first step in studying disease genetics is determining the heritability of a particular disorder, which has historically been carried out through epidemiological studies. One of the earliest studies of psychiatric heritability was conducted in 1911, when Canon and Rosanoff used family pedigrees to search for patterns of Mendelian inheritance in psychiatric patients (Zhang 2011). This was a precursor to large-scale genetic epidemiology studies (e.g., twin-, family-, adoption-, and other population-based studies) that have provided a necessary first step in establishing heritability and exploring genetic interactions in stress and anxiety disorders. A meta-analysis from Hettema et al. (2001) examined family and twin studies for panic disorder, generalized anxiety disorder, phobias, and obsessive-compulsive disorder. Each disorder was found to be heritable, with odds ratios from familial studies of 4–6, and heritability estimates from twin studies of 0.43 for panic disorder and 0.32 for generalized anxiety disorder (Hettema et al. 2001). For PTSD, twin studies estimate heritability at 0.3–0.4 (Cornelis et al. 2010). These genetic epidemiology studies, together with others, have established the influence of genetic inheritance on the development of PTSD, and other stress and anxiety disorders. Once heritability is established, the next step is to identify specific genetic regions associated with disease, which is what linkage approaches aim to accomplish. But before we delve into specific approaches, we need to explore what genetic variants might contribute to disease and what kinds of associations are theoretically possible.
Hypotheses of Genetic Association Studies
A genetic variant is any portion of an individual’s DNA sequence that differs from the reference human genome sequence. The majority of genetic association studies focus on single nucleotide polymorphisms (SNPs) as the source of genetic variation, so we concentrate on these. However, chromosomal rearrangements (duplications, deletions, inversions, and translocations) can also be quite common, and SNP-based GWAS can be extended to query copy number variation (McCarroll 2008, Mills et al. 2011). Evolutionary models of complex diseases posit that both common variation and rare variation in the genome contribute to disease (Cichon et al. 2009). A common SNP is defined to have a minor allele frequency (MAF) of at least 5%, whereas a rare variant is defined by a MAF of 1% or less; at the extreme, a rare variant may only be present in a single individual. The MAF is defined as the frequency of the least common allele in a population.
The common disease–common variant hypothesis posits that some portion of disease heritability must lie in common variants, and it assumes that testing SNPs in enough cases and controls can collectively identify common SNPs with small individual effects on disease status. It is more challenging to draw statistically significant conclusions about rare variants, as their prevalence is very low; however, the 1000 Genomes Project and other large-scale efforts have allowed us to query SNPs with a MAF in the population as low as 0.01% (Schizophr. Work. Group Psychiatr. Genom. Consort. 2014). The rich catalog of human variation that has been produced by the HapMap Project and the 1000 Genomes Project has greatly contributed to the advancement of genetic association studies (Abecasis et al. 2012, Int. HapMap Consort. 2003). To understand how the efforts of large consortia are essential to progress in the field of genetic association, we first briefly discuss the mechanism of genetic association studies.
Genome-Wide Association Studies: Basic Tenets
The purpose of GWAS is to identify loci in the genome where genetic variation is associated with the presence of disease. These disease-associated variants are thought to increase the risk of developing the related disorder (Hirschhorn et al. 2002), but mechanistic studies are required to confirm the influence of a genetic variant on disease pathophysiology. In contrast to G × E studies, GWAS query the main effect of a genetic variant. The statistical definition of a main effect is the effect of an independent variable on a dependent variable, averaging across all other independent variables involved. In GWAS terms, that is equivalent to determining the association of a particular genetic variant with a disease or an endophenotype measure, averaging across all other variables. G × E studies are an extension of GWAS, wherein G × E studies also consider the environment as a variable. In a G × E framework, the environment can be considered the pathogenic or etiologic factor, and the genetic variant is contributing to the susceptibility to that environmental pathogen (Kim-Cohen et al. 2006). However, G × E studies and GWAS are similar in that the same limitations of genetic association studies are present in both—namely, the limitation posed by our ability to measure variation in the genome.
At present, measuring SNPs is by far the most cost-effective manner to genotype individuals. Efforts by industry and large consortia have made genotyping an individual much cheaper. In particular, Illumina and the Psychiatric Genomics Consortium (PGC) have collaborated to produce the PsychArray, a SNP genotyping array that can be purchased for ~$100 and contains ~600,000 probes to test for common variants and SNPs specific for psychiatric disorders.
It is assumed that most genetic susceptibility to a disease is acquired through a de novo mutation in an ancestor. As a consequence of meiotic crossover, the disease-causing mutation is inherited along with the surrounding DNA sequence as this mutation is passed down to successive generations (Figure 1) (Ardlie et al. 2002). Identifying a particular genetic variant (such as a SNP) that is inherited along with one such inherited block of DNA (referred to as a haplotype) allows us to determine the presence of that haplotype by testing only that SNP (tag SNP; Figure 1). We can use the association of tag SNPs with disease to infer that the haplotype linked with the tag SNP is associated with disease, and that within that haplotype there is a genetic variant driving disease etiology or susceptibility. This nonrandom association of genetic variants within haplotypes is called linkage disequilibrium (LD), and it is the basic principle that underlies genetic association studies (Cichon et al. 2009).
Figure 1.

A schematic demonstration of linkage disequilibrium. The top chromosome represents an ancestor, where a de novo mutation (red triangle) first appears. This mutation is passed down to descendent chromosomes, but it is inherited along with the nearby ancestral DNA sequence (haplotype). The linkage of alleles that lie within a haplotype is referred to as linkage disequilibrium. One such allele that is highly linked to the haplotype is chosen to be a tag single nucleotide polymorphism (tag SNP) ( green triangle).
A critical point in understanding GWAS is the determination of which SNPs are used for analysis, given that there are tens of millions of SNPs within the genome. Tag SNPs are not necessarily disease-causal, or disease-related mutations; they simply act as proxies for the haplotypes with which they segregate and are co-inherited. A process called imputation allows us to infer SNPs in a genotyped individual by comparing the individual’s haplotype structure with that of reference genomes. In other words, the distribution of haplotypes in the patient genome is compared to fully sequenced reference genomes with the same (or similar) haplotype structure, and SNPs are inferred based on the sequence of the reference genome. The imputation process is also crucial for comparison of multiple data sets and meta-analyses, as it allows SNPs to be “called” (or determined) independent of the specific tag SNPs utilized in the array (Halperin & Stephan 2009).
For these reasons, the richness of the reference data sets is integral to progress in all types of genetic association studies. The HapMap Project and 1000 Genomes Project have sequenced multiple individuals from major global populations to catalog haplotypes and high-fidelity tag SNPs. SNP calling depends on imputation, and imputation is only as effective as the reference genomes are complete. The latest schizophrenia GWAS mega-analysis used the 1000 Genomes Project reference panel to call variants with MAFs as low as 0.01. Richer reference data sets may allow us to probe for less-frequent alleles in the population. New sequencing technologies will greatly enhance studies of genetic association (see sidebar Future of Genome Sequencing). Sequencing cases and controls directly will allow us to identify rare variation and its contribution to disease. Moving forward, large-scale consortia-based efforts will continue to be crucial to progress in psychiatry-specific genetic association studies.
FUTURE OF GENOME SEQUENCING.
Emerging whole-genome sequencing technologies are likely to reduce the cost of whole-genome sequencing to a point where large-scale sequencing approaches can be used to directly genotype patients in studies of genetic association. Complete sequences will allow the full evaluation of rare variants, common variants, and chromosomal rearrangements as well as their association with disease. To fully characterize repetitive DNA sequences and chromosomal rearrangements, sequencing technologies need to be able to sequence long stretches of DNA. Second-generation sequencing platforms can produce a range of read lengths, with an upper limit of approximately 1 kb. However, it is difficult to map shorter reads to very repetitive regions of the genome because there are often not enough unique base pairs for a confident alignment. To achieve longer read lengths, a variety of unique approaches and technologies are being developed. Emerging methodologies such as third-generation sequencing technologies utilizing single-molecule visualization methods and engineered proteins, and computational programs that generate long reads from shorter reads, in silico, are paving the way toward this goal.
Genome-Wide Association Studies: Consortia Efforts and Statistical Power
The need for greater statistical power in psychiatric genetics led to the formation of the PGC. As in other disciplines of biomedical research, it quickly became apparent that many variants identified through GWAS are of very small effect size—i.e., their contribution to disease is small. Thus, discovering these variants of small effect requires very large samples. Logue et al. (2015) calculated that tens of thousands of subjects will be required to discover disease-associated SNPs with MAFs of 5–20% in the population (Figure 2). The PGC has facilitated collaborative efforts in studies of genetic association for schizophrenia, bipolar disorder, and major depressive disorder (MDD), among others. Schizophrenia represents the major GWAS success in psychiatry so far. To date, the latest schizophrenia GWAS meta-analysis has revealed over 108 loci as being genome-wide significant (Schizophr. Work. Group Psychiatr. Genom. Consort. 2014). An association at a genome-wide significance level means that a genetic variant is associated with cases over controls, with p < 5 × 10−8, based on a conservative multiple test correction of p = 0.05 divided by 1 million SNP tests. This p-value is based on statistical estimates assuming that all common SNPs have been tested. Although other nonfrequentist statistical measures (Bayesian approaches) have been used that also have merit (Sham & Purcell 2014), the majority of studies to date utilize significance testing with p-values as the measure of statistical significance. In this review we refer to genome-wide significance in GWAS for those variants that reach the p-value threshold of 5 × 10−8.
Figure 2.
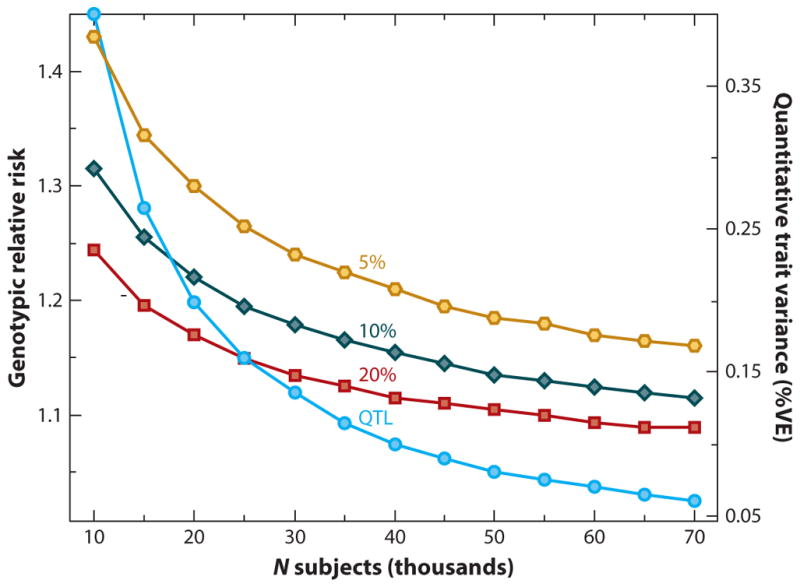
Sample size calculation as a function of power: an example calculation demonstrating the sample sizes necessary to achieve 80% power for case-control and quantitative-trait association analyses, for SNPs with MAFs of 5%, 10%, and 20% that have relative risks between 1.1 and 1.4. Calculation assumes PTSD prevalence of 15%, additive model, a type I error rate of 5 × 10−8, and perfect LD between marker and trait allele for MAF > 5%. Calculations were based on a 1:3 PTSD case-control ratio for quantitative traits such as PTSD symptoms. Abbreviations: LD, linkage disequilibrium; MAF, minor allele frequency; PTSD, posttraumatic stress disorder; QTL, quantitative trait locus; SNP, single nucleotide polymorphism; VE, environmental variance.
Success in GWAS for stress- and anxiety-related disorders has been limited. To date, five independent GWAS for PTSD have been carried out and replicated. These studies identified both genes [RORA, COBL, TLL1 (a long noncoding RNA), and PRTFDC1] and intergenic regions as significant hits, and other studies have begun to show the association of these loci with functional intermediate phenotypes (Almli et al. 2015, Guffanti et al. 2013, Logue et al. 2013, Nievergelt et al. 2015, Xie et al. 2013). Each study also replicated the association of the identified SNP in these genes with PTSD in an independent cohort. However, only a subset of these hits achieved genome-wide significance. This points to a need for greater sample sizes. The PTSD working group of the PGC is moving toward a huge GWAS effort of over 10,000 cases and 40,000 trauma-exposed controls (Logue et al. 2015), with the possibility of reaching even 100,000 total samples within the next few years. This effort will be the first large-scale GWAS for PTSD. It may replicate the findings from previous studies with smaller cohorts, and it may identify more genome-wide significant SNPs associated with PTSD. Taken together, such works offer the best hope for determining the overall genetic architecture underlying mental disorders including PTSD. However, genetic association studies should first answer the important question of how much effort the field should invest into searching for main effects with GWAS versus focusing on G × E studies. G × E studies offer both the promise of better understanding and discovering of environmental pathogens, and the hope of determining genetic risks that are not observable when examining only main effects.
Gene-by-Environment Interactions: Rationale
In 2009, the Psychiatric GWAS Consortium Steering Committee (a subset of the PGC) laid out a proposed workflow for genetic association studies (Figure 3) (Psychiatr. GWAS Consort. Steer. Comm. 2009). The rationale behind genetic association studies has been clear for decades and G × E efforts follow naturally in the progression of GWAS analyses. The next step in the study of genetic association is to identify disease-causal genetic variants by determining how these variants influence the susceptibility or resilience of an individual to particular environmental pathogens. In stress- and anxiety-related disorders, environmental measures generally include instruments that query levels of overall trauma, childhood trauma, and other stressful experiences. Disease status, as determined by physician diagnosis, can also be used as a variable in G × E studies.
Figure 3.
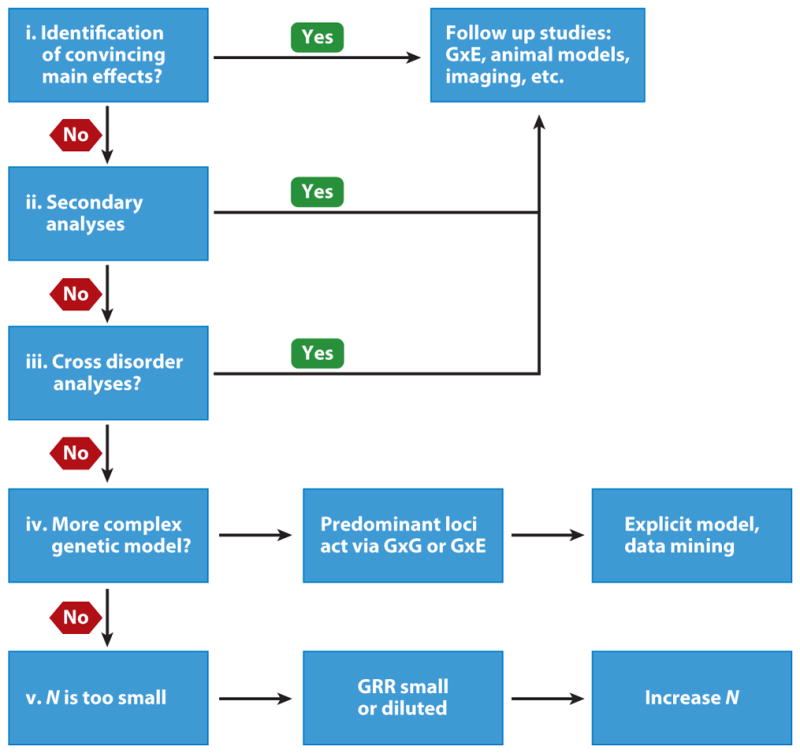
A proposed workflow for genetic association efforts put forward by the PGC. Abbreviations: G × E, gene-by-environment; G × G, gene-by-gene; GRR, genotypic relative risk; GWAS, genome-wide association study; PGC, Psychiatric Genomics Consortium.
The decision tree in Figure 3 is the path for a hypothetical within-disorder GWAS mega-analysis. For PTSD, mega-analyses have not been carried out as of yet, but smaller GWAS analyses have already discovered genome-wide significant SNPs. There is debate over whether G × E studies should be pursued for variants that have not been found to have a significant main effect in GWAS analyses. There are two points to consider here: G × E analyses may uncover hidden interactions that are not discovered in GWAS, and the sample size limitation for PTSD GWAS, at present, may prevent us from investigating targets because of a perceived lack of significance. However, even with the caveat of small sample sizes, main effect loci have been discovered, and the path forward should certainly involve studies of G × E. Note, however, that of the remaining possibilities in Figure 3, cross-disorder analyses and more complex genetic models may still be true for genes involved in stress- and anxiety-related disorders. As we discuss below, cross-disorder analyses are still an important consideration, and a more complex model based on G × E interactions is yielding new insights.
Another strong argument for pursuing G × E studies comes from the latest GWAS for MDD. Although this GWAS had approximately 9,000 cases and controls, no SNPs achieved genome-wide significance (Ripke et al. 2013). Increased sample sizes for MDD have not resulted in greater significance for SNPs, whereas in schizophrenia and other diseases adding more subjects clearly resulted in more genome-wide significant SNPs associated with disease (Figure 4). One interpretation is that depression is etiologically heterogeneous, and cases with a stronger genetic component are diluted by cases with more environmental contribution. Another interpretation is that MDD is not strongly influenced by genetic variation. Larger sample sizes to sufficiently power experiments and find genome-wide significant SNPs may be a costly endeavor, and they may not yield any associations we expect; on the other hand, G × E offers the possibility to leverage more sophisticated measures of environmental influence to identify the contributions of both genetics and environment to the development of psychiatric disease. PTSD, as an example of a stress disorder, has yet to be explored at a deep level in GWAS; however, smaller studies have already identified and replicated genome-wide significant associations in GWAS and significant G × E associations, and larger pursuits are under way.
Figure 4.
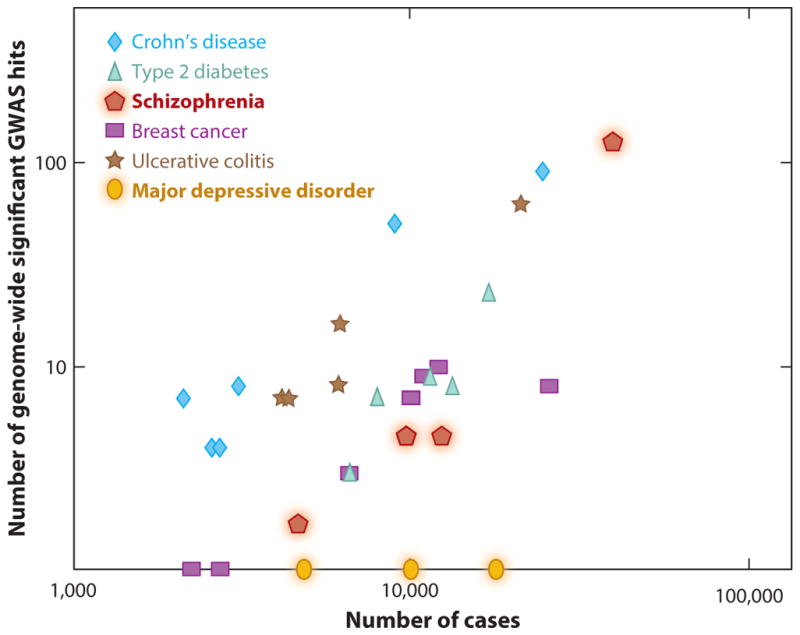
Genome-wide association study (GWAS) power has increased with greater sample sizes across many diseases, except for major depressive disorder. The graph demonstrates progress in GWAS for a variety of diseases, including schizophrenia and major depressive disorder. For all disorders except major depression, increased sample sizes have resulted in more genome-wide significant GWAS hits ( y axis, GWAS single nucleotide polymorphisms discovered with a p-value < 5 × 10−8; x axis, number of cases used in each study).
GWAS provide an unbiased approach by evaluating common variations across the genome to identify risk loci. For polygenic disorders, multiple variants likely work together to form the genetic contribution to a disorder. However, studying genetic variants alone does not address the environmental component we know to be critically important in psychiatric disease. Moreover, as evidenced by GWAS for MDD, increasing sample sizes to search for main effects may not lead to improved variant detection as it did for schizophrenia, making the search for G × E interactions more relevant. In reality, increasing the sample sizes of GWAS studies will translate to increased sample sizes for G × E approaches, as long as care is taken to obtain additional information of environmental measures in a clinical setting.
Gene-by-Environment Interactions: Basic Tenets and Limitations
The study of G × E interactions speaks to a question at the core of mental health and the study of human disease in general: To what extent do individual genetic variation and environmental context influence the etiology of disease? The conception of G × E represents the realization that for many disorders, the effect of an external stimulus—be it an infectious agent, cancer, or physical or psychological trauma—depends on the unique genetic makeup of each individual. In the realm of stress and anxiety disorders, genetic variation may predispose individuals to resilience or susceptibility to environmental stressors, which may then result in the development of psychiatric disorders. This also means that without exposure to those environmental stressors, the negative outcome may not occur; thus, it is the interaction between genes and environment that is critical for the expression of the phenotype of interest.
G × E interactions represent our understanding of the shared influence that genes and the environment play in the development of mental disorders. Statistically, an interaction between two variables means that the outcome (disease) depends on both variables. For example, without knowing the genetic variants present in an individual, it is impossible to know the relative risk for development of PTSD in the aftermath of a traumatic event; vice versa, without knowing what traumatic experiences the individual has encountered, it is impossible to know whether he or she will develop PTSD based on genetics alone. Thus, both components must be known to evaluate the etiology of disease.
The potential bias of G × E correlation is also an important consideration. Genetic variation and environmental influences may not be independent entities; that is, an individual’s genetics may predispose him or her to seek out particular environments. For example, an individual predisposed to high levels of anxiety may find himself or herself engaging in substance abuse in order to alleviate anxiety symptoms temporarily. Thus, the environment may be correlated with the genotype and therefore not be an independent variable. Such correlations are deeply embedded biases that have to be tolerated in human studies of G × E interaction. However, the identification of such correlations may lead to improved models for discovery of G × E interactions (Dick 2011).
One can conceive of two general forms for SNP-based G × E interactions (Figure 5) (Dick 2011). In the first case (Figure 5a), a fan-shaped interaction, the presence of a risk allele increases the probability of disease negligibly at baseline; however, as the individual experiences increased environmental risk, these risk alleles translate to a much higher probability of disease. In a second model (Figure 5b,c), a crossover interaction indicates that a particular SNP may not only be deleterious, but its influence depends on the environmental risk exposure. In Figure 5b, the so-called risk allele actually is protective in a low-risk environment (as compared to the wild type); disease probability is increased as environmental risk is increased. Meanwhile, individuals with none of these risk alleles have an increased probability of disease when the environmental risk is low but a decreased probability of disease in a high-risk environment. These complex interactions demonstrate that certain genetic variants can play very different roles depending on the environmental context in which they evolved, and the definition of risk allele and normal allele may not be so clear-cut.
Figure 5.

Three simple types of gene-by-environment (G × E) interactions: (a) a fan-shaped interaction, (b) a crossover interaction with the wild-type alleles demonstrating an increased probability of disease in low-risk environments and a decreased probability of disease in high risk-environments, and (c) a crossover interaction with the wild-type allele posing no increased or decreased risk.
The level of environmental loading at which crossing over occurs is likely variable; however, the importance of these models is in the biology they convey. To truly understand the effect of a genetic variant, one must take into account the full gamut of environments from positive to negative. A bias in our data sets today is the focus on negative environmental measures. This bias arises from our interest in asking disease-relevant biological questions. However, as Figure 5b and 5c show, it is conceivable that alleles that predispose someone to disease in a high-risk environment may also be beneficial by reducing susceptibility to disease in a low-risk environment. In the absence of more explicit measures of positive environment, in addition to negative risk factors, a full understanding of how a genetic variant interacts with the environment will be difficult, particularly with respect to resilience. However, some studies are starting to address this question more directly. For example, Dunn et al. (2014) used a measure of post-traumatic growth to find a SNP in the RGS2 gene that conferred resilience among Hurricane Katrina victims.
Major criticisms of G × E studies center on their lack of reproducibility, positive-results bias, insufficient sample sizes, and improper design of the statistical models used. These concerns suggest that the false positive rate in G × E studies may be much higher than we suspect, which harkens back to the days of candidate gene findings, many of which failed replication when subject to unbiased GWAS analyses (Zannas & Binder 2014). Keller (2014) put forth a fundamental criticism of the linear models used to eliminate confounding variables. In the vast majority of G × E studies to date, confounders are entered as covariates in a general linear model; however, to properly control for the impact of confounders, a covariate × environment term and a covariate × gene term must be included in the same model. This demonstrates that fundamental approaches to G × E are maturing, and the development of genome-wide approaches to G × E will help us evaluate the significance of current findings in a more rigorous, unbiased manner. Recent nonlinear statistical approaches to whole-genome G × E and GWAS analytic methods are making progress (Almli et al. 2014). Issues of power and sample size require consortia efforts, and we should pay attention to the standardization of environmental measures; however, advances in fundamental statistical methodologies should be applied to all future studies and reanalyses of past studies. This points to the need for closer communication between neuroscientists and statistical geneticists. In light of the increased attention to G × E approaches, some journals have laid out strict criteria for publication, which is an excellent step to improve the false positive rate of published findings (Hewitt 2012).
DEFINING ENVIRONMENTS AND PHENOTYPES IN STUDIES OF GENETIC ASSOCIATION
Environment
A countless number of possible factors can influence mental health outcomes. In particular, certain clinically measured environmental factors have been shown to be important risk factors for many psychiatric conditions. For example, exposure to trauma and significant stressful life events are well-established risk factors for the development of many major mental disorders, including depression, anxiety disorders, PTSD, and substance use disorders (Brewin et al. 2000, Hettema et al. 2005, Kilpatrick et al. 2003). Important considerations in studies of G × E include the measurement of these environmental factors and the scaling of these measures in calculations.
Very often, the primary method for measuring environmental variables is self-report questionnaires. Questionnaires provide a quick, inexpensive approach for assessing a wide range of constructs for researchers. However, there are a number of limitations to the use of self-report instruments. The most important issue is whether the questionnaire is valid and fully captures the construct of interest in the study. Self-report questionnaires are also limited to the information remembered and willingly provided by the individual. Retrospective biases, as well as social desirability and response bias, are all factors that affect the information obtained through questionnaires. An alternative to self-report questionnaires is the use of structured or semistructured clinical interviews conducted by trained research interviewers (or clinicians), which provide a more thorough assessment of psychiatric symptoms. Again, these instruments are limited to the information provided by the individual, but additional questioning and expertise by the interviewer often allow clarification of the construct under study and reduce the chance to code symptoms incorrectly due to ambiguous wording of questions. Standardized clinical interviews, although often more valid than self-report questionnaires, are much more expensive and more lengthy to administer, and therefore they limit the feasibility of large-scale data collection.
The developmental stage in which environmental factors occur also affects risk. With regard to trauma, for example, research has shown that early exposure to trauma or abuse in childhood is particularly detrimental and leads to a wide range of negative mental health outcomes, putting individuals at risk for psychopathology and related outcomes (e.g., suicide, psychiatric hospitalization) (Gillespie et al. 2009, MacMillan et al. 2001, McLaughlin et al. 2010). More generally, childhood adversity (e.g., abuse, parental loss, negative family environment) is a risk factor for depression, anxiety disorders, personality disorders, and other diseases (Carr et al. 2013, Heim & Nemeroff 2001, Kendler et al. 1992). Therefore, both the presence of certain environmental factors and the time at which they occur are of critical importance in the context of psychiatric risk. Questions about developmental effects are difficult to dissect in an epidemiological context, but as we discuss below, animal models provide a powerful outlet to evaluate the influence of genetic variants throughout development.
Today as well as in the future, it will be important for G × E studies to be standardized to allow for large-scale comparisons. The methods and instruments used by researchers vary dramatically across studies, sometimes making it difficult to compare findings across samples. Variation in the type of scaling used to measure any given environmental factor (e.g., dichotomous response versus Likert scale) also changes how a construct is evaluated and can often inhibit the ability to make adequate comparisons across groups. Collaboration will be crucial for standardization of environmental measures across institutions, and efforts by the PGC have paved the way to facilitate these large-scale coordinated efforts.
Outcome Measures in Genetic Studies
The outcome measure of genetic association studies can be a disease diagnosis, but it can also be an intermediate phenotype or endophenotype, an independent measure that correlates with a disease, such as amygdala reactivity. Just as the definition of the environmental component requires careful design, so too does the definition of the outcome being investigated. Querying disease directly has been the mainstay of genetic association studies; however, intermediate phenotypes such as fear-potentiated startle, stress/anxiety questionnaires, and neuroimaging (such as amygdala activation) are also methods used to understand how genetic variation influences behavioral physiology (Stein et al. 2008). These intermediate phenotypes or endophenotypes define the outcome more narrowly than a potentially broad disease category, and the association of a genetic variant with an endophenotype points to a more specific consequence of that variant. Endophenotypes can be used in both GWAS and G × E studies.
A deeper consideration is the true nosology of a mental health disorder. Definitions from the DSM are explicitly designed to provide the most clinical utility. However, the striking comorbidities of mental health patients raise an important question: At a genetic level, are any two comorbid psychiatric disorders truly distinct, or is there a shared etiology? As we discussed in the introduction, stress, anxiety, and depression can be conceived of as a spectrum; however, this may not be reflected in the DSM criteria that define diseases clinically. In 2013, the Psychiatric Genomics Consortium Cross-Disorder Group endeavored to leverage GWAS data to investigate the shared genetics between major psychiatric disorders for which GWAS have been carried out (Cross-Disord. Group Psychiatr. Genom. Consort. 2013). This study demonstrated that schizophrenia and bipolar disorder, schizophrenia and MDD, attention-deficit/hyperactivity disorder and MDD, and bipolar disorder and MDD share a detectable genetic variation. These findings suggest that certain genetic variants may play a role in the etiology of multiple disorders, and the high comorbidity may be a consequence of this shared susceptibility. On the opposite end of the spectrum, our current disease categories may be too broad, and patients with heterogeneous etiologies are being lumped together in genetic association studies. This is supported by recent work by the CONVERGE consortium, in which two loci were associated with depression, at genomewide significance, in a phenotypically homogeneous cohort of severely depressed Chinese women (CONVERGE consort. 2015).
The primary diseases studied by the PGC possess the most complete GWAS data sets. However, as more diseases are studied and more comprehensive GWAS data sets become available, cross-disorder and subgroup analyses can become more comprehensive and may reveal new outcome measures and genomewide significant loci to be investigated.
GENE-BY-ENVIRONMENT INTERACTIONS
Many studies of G × E interactions use stress, anxiety, or PTSD measures; however, only a few have been robustly replicated. Rather than presenting the full gamut of findings, we lay out what is known about three genes in which variants have been robustly associated with depression-, stress- and anxiety-related G × E interactions: the serotonin transporter promoter (5-HTTLPR) polymorphism, the brain-derived neurotropic factor (BDNF) Val66Met polymorphism, and the FK506 binding protein 5 (FKBP5) polymorphism. We recognize that a great deal of literature exists for mechanistic function of the 5-HTTLPR and the BDNF Val66Met polymorphisms; however, due to space limitations we focus on presenting the epidemiological evidence connecting these two well-established variants to disease, and we emphasize the often conflicting reports that emerge, even for these now well-established associations. For FKBP5 we present a more comprehensive picture of the variant, from discovery to pathophysiological mechanism—a feat achieved from epidemiological research as well as cell culture and animal-model studies.
Serotonin Transporter Promoter Polymorphism (5-HTTLPR)
Several studies and meta-analyses have shown that the 5-HTTLPR (5-HTT gene-linked polymorphic region) variant moderates the relationship between stressful life events and depression. One of the first G × E interactions to be discovered, this variant largely became the testing ground for the concept, with many studies designed to replicate this finding and much debate as to whether this is a true G × E interaction. The 5-HTTLPR polymorphism is a variation in the number of repeats in the promoter region of the serotonin reuptake transporter (SLC6A4); these GC-rich, 20- to 23-bp-long repeat elements are generally studied in the context of the short (S) allele (14 repeats) and the long (L) allele (16 repeats). We refer to individuals with two copies of the short allele as SS, individuals with one copy of the short allele and one copy of the long allele as SL, and individuals with two copies of the long allele as LL. Homozygous S allele carriers, in combination with increased exposure to stressful life events, are predictive of depression (Bogdan et al. 2014, Caspi et al. 2003, Karg et al. 2011). There is also evidence of increased stress reactivity among 5-HTTLPR SS allele carriers. However, these findings remain mixed and suggest that only main stressful life events affect the development of depression (Gillespie et al. 2005, Risch et al. 2009).
Findings are also mixed regarding G × E interactions in predicting risk for PTSD, with some evidence suggesting that the specific population studied may affect the results. Numerous studies have shown SS 5-HTTLPR genotype in combination with trauma exposure of various types to be a risk factor for PTSD across both civilian and veteran populations (Holman et al. 2011, Kolassa et al. 2010, Mercer et al. 2012, Wang et al. 2011, Xie et al. 2009). Moreover, among adult hurricane survivors, Kilpatrick et al. (2007) found that SS allele carriers had significantly greater risk for the development of PTSD following exposure to the hurricane only in the presence of low social support. However, recent evidence in two African American samples showed that the SS 5-HTTLPR allele was associated with lower PTSD re-experiencing and hyperarousal symptoms among individuals exposed to childhood emotional abuse, suggesting that two S alleles could be a protective factor against the development of PTSD symptoms in the presence of childhood abuse (Walsh et al. 2014). This supports evidence from Xie et al. (2012) showing an interaction between childhood maltreatment and one or two copies of the S allele in the 5-HTTLPR genotype in predicting PTSD, but only among European American adults; this G × E interaction was not found in African American adults. Other research with European adults has shown that the LL 5-HTTLPR genotype interacts with trauma exposure to predict increased risk for PTSD (Grabe et al. 2009). Also, in a study of 41 motor vehicle accident survivors, the LL 5-HTTLPR genotype showed an interaction with trauma exposure to predict chronic PTSD (Thakur et al. 2009). These studies thus suggest that the S allele may be protective in certain populations. To complicate things further, some studies have found no evidence of an effect of 5-HTTLPR on risk for PTSD in the face of stressful or traumatic events (Mellman et al. 2009, Sayin et al. 2010). The 5-HTTLPR SS allele was associated with higher likelihood of suicide attempt in African American substance-dependent males, but only in the presence of high levels of reported child abuse (Roy et al. 2007). A recent meta-analysis examined the association between 5-HTTLPR and panic disorder and found no evidence of a relationship (Blaya et al. 2007); however, G × E associations were not examined.
With regard to intermediate phenotypes, within a sample of ethnically diverse college undergraduates, those homozygous for the S allele, who reported higher levels of childhood maltreatment showed significantly higher levels of anxiety sensitivity compared to heterozygotes or homozygous L carriers. Anxiety sensitivity can be seen as an intermediate phenotype for anxiety and depressive disorders (Stein et al. 2008). Alternatively, another study found that the LL genotype interacted with childhood maltreatment to predict increased anxiety sensitivity in a sample of healthy adults (Klauke et al. 2011), again highlighting how mixed the results are in G × E 5-HTTLPR studies. Other evidence from a college sample using daily monitoring techniques to assess daily stress and anxiety levels found that individuals with at least one S 5-HTTLPR allele showed heightened levels of anxious mood in the presence of increased daily stressors (Gunthert et al. 2007).
BDNF Val66Met Polymorphism (V66M)
Brain-derived neurotrophic factor (BDNF) is perhaps the best-studied protein in neuroscience, given its integral role in neural development and function. A wealth of literature describes BDNF’s role at a molecular and behavioral level, but more recently, epidemiological G × E studies have also elucidated interactions between the Met allele at amino acid position 66 and stress in promoting anxiety and depression. Early in the study of G × E, a three-way interaction among V66M, 5-HTTLPR, and maltreatment history was shown to predict depression in a cohort of 196 adult cases and controls (Kaufman et al. 2006). A 2010 study followed up on this finding, investigating whether this three-way interaction could be observed in an adolescent cohort; however, there was no detected association among V66M, 5-HTTLPR, and maltreatment on adolescent depression (Nederhof et al. 2010). In 2012, a study of 780 pairs of Chinese adolescent twins investigated this three-way interaction, finding that V66M, 5-HTTLPR, and maltreatment did indeed associate with adolescent depression symptoms (Chen et al. 2012). These studies of variable outcomes highlight the power of consortium approaches and the increased statistical power they afford, but their results may be confounded by the fact that they were performed in different populations around the world. Endophenotype approaches have found that an interaction between V66M and early life stress is associated with smaller hippocampal and amygdala volumes, elevated heart rate, and reduced working memory (Gatt et al. 2009).
FK506 Binding Protein-5 Polymorphism (FKBP5)
In 2008, an interaction between SNPs in FKBP5 and early childhood trauma (FKBP5 × childhood trauma) was found to influence the severity of adult PTSD symptoms in a population of urban, low-socioeconomic-status African Americans (Binder et al. 2008). A subsequent study replicated this finding in a larger cohort of subjects of African descent (Xie et al. 2010). Another study investigating G × E interactions in chronic pain patients in Pennsylvania demonstrated that the interaction between FKBP5 genotype and total trauma exposure is associated with PTSD (Boscarino et al. 2012). No main effect for FKBP5 genotype and PTSD was detected in any of the preceding studies. Interestingly, adult trauma did not interact with FKBP5 genotype to influence PTSD symptoms, whereas follow-up studies have consistently reported the interaction of childhood trauma and FKBP5 to be significant. This suggests a developmental window in which environmental risk creates a long-lasting molecular alteration in the FKBP5 pathway, which influences the development of PTSD in adulthood. Subsequent molecular analyses of FKBP5 have yielded insight as to how this memory may be maintained by epigenetic mechanisms (Klengel et al. 2013) (see Figure 6 and sidebar Overview of Epigenetics and Chromatin Conformation).
Figure 6.

Development and G × E interactions. There are developmental windows in which trauma and stress have particular influence on epigenetic signaling, thus propagating the effects through an individual’s life span. The HPA axis, in particular, is modified by early life stress, as evidenced by G × E interactions identified in FKBP5. Abbreviations: G × E, gene-by-environment; HPA, hypothalamic-pituitary-adrenal.
OVERVIEW OF EPIGENETICS AND CHROMATIN CONFORMATION.
The regulation of transcription (the production of RNA from DNA) is highly coordinated. Transcription factors (TFs) orchestrate how RNA polymerase II transcribes mRNA. Many TFs bind to specific DNA sequences. The composition and distribution of these sequences hardwire epigenetic and transcription factor binding profiles, and variants in these motifs affect how strongly transcription factors are able to bind. DNA can also be physically modified, changing the physical properties of the DNA and the strength with which TFs can bind to it. 5-methylcytosine (5-mC) results from the addition of a methyl group to the 5-position on a cytosine ring, and 5-mC deposition in some transcription factor motifs has been shown to alter the binding efficacy of transcription factors to those motifs.
The conformation of chromatin refers to the stretches of linear DNA that are in contact with one another. The purpose of the interaction between these distal DNA sequences is to allow enhancer or inhibitory elements, which can increase or decrease, respectively, the rate of Pol II transcription to differentially regulate gene expression. The GR-sensitive intron 2 enhancer in FKBP5 is an example of this.
Klengel et al. (2013) assert that 5-mC reduction in intron 7 of FKBP5 is propagated through childhood, dependent on early life stress. This 5-mC reduction results in increased FKBP5 mRNA expression and a lasting alteration in the homeostatic balance of the HPA axis, which shapes the potential for future posttraumatic stress disorder psychopathology.
The FKBP5 × childhood trauma interaction has also been shown to influence a variety of other psychiatric disorders and traits including depression, schizophrenia, aggression, psychosis, and suicide attempts (Appel et al. 2011, Collip et al. 2013, Dackis et al. 2012, Roy et al. 2010, Zimmermann et al. 2011). These diverse associations can be understood by the fundamental molecular role FKBP5 plays in regulating glucocorticoid signaling in the cell. FKBP5 exerts an inhibitory effect on glucocorticoid receptor (GR)-mediated signaling, acting in an ultrashort feedback loop of the hypothalamic-pituitary-adrenal (HPA) axis. FKBP5 is a co-chaperone in the heat shock protein 90 (Hsp90) steroid receptor complex. FKBP5 binding to the Hsp90 complex results in reduced binding affinity of glucocorticoids to GRs, and overexpression of FKBP5 reduces GR-mediated signaling. Furthermore, FKBP5 is rapidly induced by glucocorticoids in a number of tissues, including brain and peripheral blood. Thus, GR-mediated signaling upregulates FKBP5, a negative regulator of glucocorticoid signaling, resulting in rapid negative feedback of the stress response at the cellular level. Anatomically, FKBP5 expression is strongest in brain regions associated with stress and anxiety responses, including the hippocampus, the amygdala, and the paraventricular nucleus (Zannas & Binder 2014).
The most understood variants in FKBP5 are rs3800373, rs9296158, and rs1360780 (rs designations are used to identify specific SNPs). These SNPs lie in strong linkage disequilibrium within a haplotype that covers the entire gene. Work by Klengel et al. (2013) unraveled the molecular implications of rs1360780. This variant was chosen for its proximity to a GR element (Lee et al. 2013), a short DNA motif that binds GRs, in intron 2 of FKBP5. The risk allele of rs1360780 is A/T, and the protective allele is C/G. Klengel et al. (2013) showed that the A/T allele enhances expression of FKBP5, and that this effect is mediated by differential interaction between intron 2 and the transcription start site (Figure 7c,d). Given that the GRE in intron 2 is responsive to glucocorticoid signaling, the risk allele is thought to enhance the effects of GR signaling on FKBP5 transcription
Figure 7.
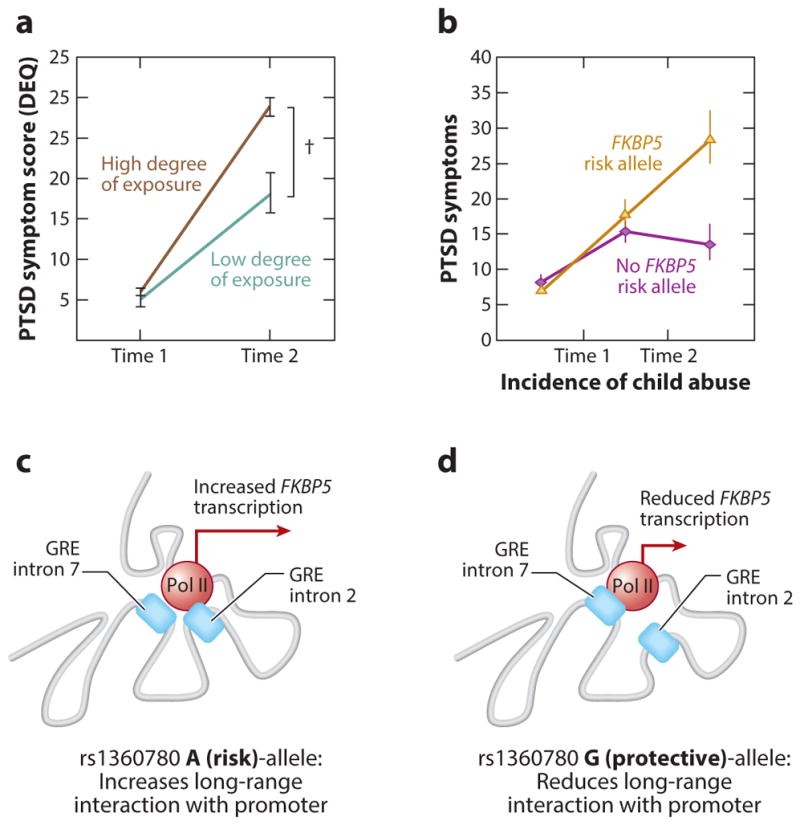
Examples of gene-by-environment (G × E) effects in posttraumatic stress disorder (PTSD). (a) An example of an environmental exposure as a function of time post-trauma, illustrating the severity of PTSD symptoms two to four weeks after a campus shooting by degree of exposure. The graph shows the mean PTSD symptom score [Distressing Event Questionnaire (DEQ)] at time 1 and time 2 (± standard error). A sum ranging from 0 to 2 for positive responses to high-exposure events is classified as a low degree of exposure. (b) An example of G × E exposure as a function of level of childhood trauma exposure, in which an FKBP5 risk allele (tan line) is associated with heightened adult PTSD symptoms following high burdens of childhood trauma compared to the protective allele ( purple line). (c) The risk allele of the rs1360780 SNP in intron 2 of FKBP5 increases the transcriptional output of RNA polymerase II (Pol II) at the transcription start site by increasing the long-distance interactions between the GRE containing intron 2 and the promoter, thus increasing the production of FKBP5 mRNA. (d ) The protective allele reduces the interaction between intron 2 and the promoter, reducing the production of FKBP5 mRNA. by bringing this distal transcriptional enhancer in close proximity to the promoter, therefore curbing the ultrashort HPA axis feedback loop in which FKBP5 participates. This prolonged homeostatic perturbation results in lasting alterations in the neural circuits governing stress and anxiety.
CONCLUSIONS AND FUTURE DIRECTIONS
The robust G × E interactions we have described above are all involved in major signaling systems in development and neural function. However, an important point is that tens, if not hundreds to thousands, of different genes and gene loci are likely involved in genetic heritability, underlying part of the risk for stress- and anxiety-related disorders. These genetic risks are likely interacting with different aspects of the environment, such that some may interact with level of trauma exposure, e.g., 5-HTTLPR and BDNF, whereas others may be particularly sensitive to the time of developmental exposure, as suggested by the finding that HPA-axis genes FKBP5 and CRHR1 interact with childhood trauma, but less so with adult trauma exposure. Still other gene pathways likely interact with sex hormones to provide differential sex effects (e.g., ADCYAP1R1), and others interact with physical toxin exposure (e.g., lead or mercury). The ways in which the environment may differentially integrate with genomic information are extremely varied. Larger-scale studies and studies combining consortia-level GWAS with environmental factors are likely to provide the most important paths to new discovery.
As our understanding of the complexity of genome regulation will continue to expand, so too will our appreciation for a greater understanding of quantitative measurements of exposure, with psychological trauma in particular. Additionally, we must understand which outcome measures best represent the effects of stress and trauma exposure, be they categorical diagnostic outcomes, continuous symptom-level outcomes, comorbid outcomes, and intermediate or endophenotypes outcomes such as physiological or brain imaging–based measures. Despite their complexity, advances in our understanding of G × E interactions will eventually further our knowledge of the biology of the brain and the mind. Such progress will also allow for a better understanding of how the environment—the world around us—creates both mental health and mental illness.
Acknowledgments
This work was primarily supported by the National Institutes of Mental Health (MH071537 and MH096764 to K.J.R.; HD071982 to B.B.; and MH102890 to A.P.). Support was also received from the Emory University and Grady Memorial Hospital General Clinical Research Center, NIH National Centers for Research Resources (M01RR00039), the Howard Hughes Medical Institute (K.J.R.), and the Steven and Alexandra Cohen Foundation (CRM). We are grateful to the staff and participants from the Grady Trauma Project for their time and effort in supporting this research.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review. The contents of this review do not reflect the views of the Department of Veterans Affairs of the United States government.
LITERATURE CITED
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almli LM, Duncan R, Feng H, Ghosh D, Binder EB, et al. Correcting systematic inflation in genetic association tests that consider interaction effects: application to a genome-wide association study of posttraumatic stress disorder. JAMA Psychiatry. 2014;71:1392–99. doi: 10.1001/jamapsychiatry.2014.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almli LM, Stevens J, Smith AK, Kilaru V, Qian M, et al. Genome-wide identified risk variant for PTSD is a methylation quantitative trait locus and confers decreased cortical activation to fearful faces. Am J Med Genet B: Neuropsychiatr Genet. 2015;168:327–36. doi: 10.1002/ajmg.b.32315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Am. Psychiatr. Assoc. Diagnostic and Statistical Manual of Mental Disorders. 5 Washington, DC: Am. Psychiatr. Publ; 2013. [Google Scholar]
- Appel K, Schwahn C, Mahler J, Schulz A, Spitzer C, et al. Moderation of adult depression by a polymorphism in the FKBP5 gene and childhood physical abuse in the general population. Neuropsychopharmacology. 2011;36:1982–91. doi: 10.1038/npp.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. doi: 10.1038/nrg777. [DOI] [PubMed] [Google Scholar]
- Binder E. Molecular mechanisms of gene × environment interaction in stress-related psychiatric disorders. NIH Videocasting Postcasting video. 2014;49:53. Posted Febr. 24, 2014. http://videocast.nih.gov/summary.asp?Live=13688&bhcp=1. [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaya C, Salum GA, Lima MS, Leistner-Segal S, Manfro GG. Lack of association between the serotonin transporter promoter polymorphism (5-HTTLPR) and panic disorder: a systematic review and meta-analysis. Behav Brain Funct. 2007;3:41. doi: 10.1186/1744-9081-3-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan R, Agrawal A, Gaffrey MS, Tillman R, Luby JL. Serotonin transporter-linked polymorphic region (5-HTTLPR) genotype and stressful life events interact to predict preschool-onset depression: a replication and developmental extension. J Child Psychol Psychiatry. 2014;55:448–57. doi: 10.1111/jcpp.12142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscarino JA, Erlich PM, Hoffman SN, Zhang X. Higher FKBP5, COMT, CHRNA5, and CRHR1 allele burdens are associated with PTSD and interact with trauma exposure: implications for neuropsychiatric research and treatment. Neuropsychiatr Dis Treat. 2012;8:131–39. doi: 10.2147/NDT.S29508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady KT, Killeen TK, Brewerton T, Lucerini S. Comorbidity of psychiatric disorders and posttraumatic stress disorder. J Clin Psychiatry. 2000;61(Suppl 7):22–32. [PubMed] [Google Scholar]
- Brewin CR, Andrews B, Valentine JD. Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. J Consult Clin Psychol. 2000;68:748–66. doi: 10.1037//0022-006x.68.5.748. [DOI] [PubMed] [Google Scholar]
- Carr CP, Martins CMS, Stingel AM, Lemgruber VB, Juruena MF. The role of early life stress in adult psychiatric disorders: a systematic review according to childhood trauma subtypes. J Nerv Ment Dis. 2013;201:1007–20. doi: 10.1097/NMD.0000000000000049. [DOI] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–89. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Chen J, Li X, McGue M. Interacting effect of BDNF Val66Met polymorphism and stressful life events on adolescent depression. Genes Brain Behav. 2012;11:958–65. doi: 10.1111/j.1601-183X.2012.00843.x. [DOI] [PubMed] [Google Scholar]
- Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV, et al. Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166:540–56. doi: 10.1176/appi.ajp.2008.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collip D, Myin-Germeys I, Wichers M, Jacobs N, Derom C, et al. FKBP5 as a possible moderator of the psychosis-inducing effects of childhood trauma. Br J Psychiatry. 2013;202:261–68. doi: 10.1192/bjp.bp.112.115972. [DOI] [PubMed] [Google Scholar]
- CONVERGE consort. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis MC, Nugent NR, Amstadter AB, Koenen KC. Genetics of post-traumatic stress disorder: review and recommendations for genome-wide association studies. Curr Psychiatry Rep. 2010;12:313–26. doi: 10.1007/s11920-010-0126-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craske MG, Rauch SL, Ursano R, Prenoveau J, Pine DS, Zinbarg RE. What is an anxiety disorder? Depression Anxiety. 2009;26:1066–85. doi: 10.1002/da.20633. [DOI] [PubMed] [Google Scholar]
- Cross-Disord. Group Psychiatr. Genom. Consort. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–94. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dackis MN, Rogosch FA, Oshri A, Cicchetti D. The role of limbic system irritability in linking history of childhood maltreatment and psychiatric outcomes in low-income, high-risk women: moderation by FK506 binding protein 5 haplotype. Dev Psychopathol. 2012;24:1237–52. doi: 10.1017/S0954579412000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM. Gene-environment interaction in psychological traits and disorders. Annu Rev Clin Psychol. 2011;7:383–409. doi: 10.1146/annurev-clinpsy-032210-104518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn EC, Solovieff N, Lowe SR, Gallagher PJ, Chaponis J, et al. Interaction between genetic variants and exposure to Hurricane Katrina on post-traumatic stress and post-traumatic growth: a prospective analysis of low income adults. J Affect Disord. 2014;152–154:243–49. doi: 10.1016/j.jad.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, et al. Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry. 2009;14:681–95. doi: 10.1038/mp.2008.143. [DOI] [PubMed] [Google Scholar]
- Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, et al. Trauma exposure and stress-related disorders in inner city primary care patients. Gen Hosp Psychiatry. 2009;31:505–14. doi: 10.1016/j.genhosppsych.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie NA, Whitfield JB, Williams B, Heath AC, Martin NG. The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med. 2005;35:101–11. doi: 10.1017/s0033291704002727. [DOI] [PubMed] [Google Scholar]
- Grabe HJ, Spitzer C, Schwahn C, Marcinek A, Frahnow A, et al. Serotonin transporter gene (SLC6A4) promoter polymorphisms and the susceptibility to posttraumatic stress disorder in the general population. Am J Psychiatry. 2009;166:926–33. doi: 10.1176/appi.ajp.2009.08101542. [DOI] [PubMed] [Google Scholar]
- Guffanti G, Galea S, Yan L, Roberts AL, Solovieff N, et al. Genome-wide association study implicates a novel RNA gene, the lincRNA AC068718.1, as a risk factor for post-traumatic stress disorder in women. Psychoneuroendocrinology. 2013;38:3029–38. doi: 10.1016/j.psyneuen.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthert KC, Conner TS, Armeli S, Tennen H, Covault J, Kranzler HR. Serotonin transporter gene polymorphism (5-HTTLPR) and anxiety reactivity in daily life: a daily process approach to gene-environment interaction. Psychosom Med. 2007;69:762–68. doi: 10.1097/PSY.0b013e318157ad42. [DOI] [PubMed] [Google Scholar]
- Halperin E, Stephan DA. SNP imputation in association studies. Nat Biotechnol. 2009;27:349–51. doi: 10.1038/nbt0409-349. [DOI] [PubMed] [Google Scholar]
- Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49:1023–39. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- Hettema JM, Neale MC, Kendler KS. A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry. 2001;158:1568–78. doi: 10.1176/appi.ajp.158.10.1568. [DOI] [PubMed] [Google Scholar]
- Hettema JM, Prescott CA, Myers JM, Neale MC, Kendler KS. The structure of genetic and environmental risk factors for anxiety disorders in men and women. Arch Gen Psychiatry. 2005;62:182–89. doi: 10.1001/archpsyc.62.2.182. [DOI] [PubMed] [Google Scholar]
- Hewitt JK. Editorial policy on candidate gene association and candidate gene-by-environment interaction studies of complex traits. Behav Genet. 2012;42:1–2. doi: 10.1007/s10519-011-9504-z. [DOI] [PubMed] [Google Scholar]
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Spengler D. DNA memories of early social life. Neuroscience. 2014;264:64–75. doi: 10.1016/j.neuroscience.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Holman EA, Lucas-Thompson RG, Lu T. Social constraints, genetic vulnerability, and mental health following collective stress. J Trauma Stress. 2011;24:497–505. doi: 10.1002/jts.20671. [DOI] [PubMed] [Google Scholar]
- Int. HapMap Consort. The International HapMap Project. Nature. 2003;426:789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Karg K, Burmeister M, Shedden K, Sen S. The serotonin transporter promoter variant (5-HTTLPR), stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch Gen Psychiatry. 2011;68:444–54. doi: 10.1001/archgenpsychiatry.2010.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman J, Yang BZ, Douglas-Palumberi H, Grasso D, Lipschitz D, et al. Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children. Biol Psychiatry. 2006;59:673–80. doi: 10.1016/j.biopsych.2005.10.026. [DOI] [PubMed] [Google Scholar]
- Keller MC. Gene × environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol Psychiatry. 2014;75:18–24. doi: 10.1016/j.biopsych.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. Childhood parental loss and adult psychopathology in women: a twin study perspective. Arch Gen Psychiatry. 1992;49:109–16. doi: 10.1001/archpsyc.1992.01820020029004. [DOI] [PubMed] [Google Scholar]
- Kilpatrick DG, Koenen KC, Ruggiero KJ, Acierno R, Galea S, et al. The serotonin transporter genotype and social support and moderation of posttraumatic stress disorder and depression in hurricane-exposed adults. Am J Psychiatry. 2007;164:1693–99. doi: 10.1176/appi.ajp.2007.06122007. [DOI] [PubMed] [Google Scholar]
- Kilpatrick DG, Ruggiero KJ, Acierno R, Saunders BE, Resnick HS, Best CL. Violence and risk of PTSD, major depression, substance abuse/dependence, and comorbidity: results from the National Survey of Adolescents. J Consult Clin Psychol. 2003;71:692–700. doi: 10.1037/0022-006x.71.4.692. [DOI] [PubMed] [Google Scholar]
- Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, et al. MAOA, maltreatment, and gene–environment interaction predicting children’s mental health: new evidence and a meta-analysis. Mol Psychiatry. 2006;11:903–13. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- Klauke B, Deckert J, Reif A, Pauli P, Zwanzger P, et al. Serotonin transporter gene and childhood trauma—a G × E effect on anxiety sensitivity. Depression Anxiety. 2011;28:1048–57. doi: 10.1002/da.20840. [DOI] [PubMed] [Google Scholar]
- Klengel T, Binder EB. FKBP5 allele-specific epigenetic modification in gene by environment interaction. Neuropsychopharmacology. 2015;40:244–46. doi: 10.1038/npp.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, et al. Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat Neurosci. 2013;16:33–41. doi: 10.1038/nn.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolassa I, Ertl V, Eckart C, Glöckner F, Kolassa S, et al. Association study of trauma load and SLC6A4 promoter polymorphism in posttraumatic stress disorder: evidence from survivors of the Rwandan genocide. J Clin Psychiatry. 2010;71:543–47. doi: 10.4088/JCP.08m04787blu. [DOI] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–94. doi: 10.1038/ng.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue MW, Amstadter AB, Baker DG, Duncan L, Koenen KC, et al. The Psychiatric Genomics Consortium Posttraumatic Stress Disorder Workgroup: Posttraumatic stress disorder enters the age of large-scale genomic collaboration. Neuropsychopharmacology. 2015;40:2287–97. doi: 10.1038/npp.2015.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue MW, Baldwin C, Guffanti G, Melista E, Wolf EJ, et al. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol Psychiatry. 2013;18(8):937–42. doi: 10.1038/mp.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMillan HL, Fleming JE, Streiner DL, Lin E, Boyle MH, et al. Childhood abuse and lifetime psychopathology in a community sample. Childhood. 2001;158(11):1878–83. doi: 10.1176/appi.ajp.158.11.1878. [DOI] [PubMed] [Google Scholar]
- Major Depressive Disord. Work. Group Psychiatr. GWAS Consort. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18(4):497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarroll SA. Extending genome-wide association studies to copy-number variation. Hum Mol Genet. 2008;17:R135–42. doi: 10.1093/hmg/ddn282. [DOI] [PubMed] [Google Scholar]
- McLaughlin KA, Conron KJ, Koenen KC, Gilman SE. Childhood adversity, adult stressful life events, and risk of past-year psychiatric disorder: a test of the stress sensitization hypothesis in a population-based sample of adults. Psychol Med. 2010;40:1647–58. doi: 10.1017/S0033291709992121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman TA, Alim T, Brown DD, Gorodetsky E, Buzas B, et al. Serotonin polymorphisms and post-traumatic stress disorder in a trauma exposed African American population. Depression Anxiety. 2009;26:993–97. doi: 10.1002/da.20627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer KB, Orcutt HK, Quinn JF, Fitzgerald CA, Conneely KN, et al. Acute and posttraumatic stress symptoms in a prospective gene × environment study of a university campus shooting. Arch Gen Psychiatry. 2012;69:89–97. doi: 10.1001/archgenpsychiatry.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nederhof E, Bouma EM, Oldehinkel AJ, Ormel J. Interaction between childhood adversity, brain-derived neurotrophic factor val/met and serotonin transporter promoter polymorphism on depression: the TRAILS study. Biol Psychiatry. 2010;68:209–12. doi: 10.1016/j.biopsych.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Nievergelt CM, Maihofer AX, Mustapic M, Yurgil KA, Schork NJ, et al. Genomic predictors of combat stress vulnerability and resilience in U.S. Marines: A genome-wide association study across multiple ancestries implicates PRTFDC1 as a potential PTSD gene. Psychoneuroendocrinology. 2015;51:459–71. doi: 10.1016/j.psyneuen.2014.10.017. [DOI] [PubMed] [Google Scholar]
- Psychiatr. GWAS Consort. Steer. Comm. A framework for interpreting genome-wide association studies of psychiatric disorders. Mol Psychiatry. 2009;14:10–17. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Herrell R, Lehner T, Liang K-Y, Eaves L, et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA. 2009;301:2462–71. doi: 10.1001/jama.2009.878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Gorodetsky E, Yuan Q, Goldman D, Enoch MA. Interaction of FKBP5, a stress-related gene, with childhood trauma increases the risk for attempting suicide. Neuropsychopharmacology. 2010;35:1674–83. doi: 10.1038/npp.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Hu X-Z, Janal MN, Goldman D. Interaction between childhood trauma and serotonin transporter gene variation in suicide. Neuropsychopharmacology. 2007;32:2046–52. doi: 10.1038/sj.npp.1301331. [DOI] [PubMed] [Google Scholar]
- Sayin A, Kucukyildirim S, Akar T, Bakkaloglu Z, Demircan A, et al. A prospective study of serotonin transporter gene promoter (5-HTT gene linked polymorphic region) and intron 2 (variable number of tandem repeats) polymorphisms as predictors of trauma response to mild physical injury. DNA Cell Biol. 2010;29:71–77. doi: 10.1089/dna.2009.0936. [DOI] [PubMed] [Google Scholar]
- Schizophr. Work. Group Psychiatr. Genom. Consort. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–27. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nat Rev Genet. 2014;15:335–46. doi: 10.1038/nrg3706. [DOI] [PubMed] [Google Scholar]
- Stein MB, Schork NJ, Gelernter J. Gene-by-environment (serotonin transporter and childhood maltreatment) interaction for anxiety sensitivity, an intermediate phenotype for anxiety disorders. Neuropsychopharmacology. 2008;33:312–19. doi: 10.1038/sj.npp.1301422. [DOI] [PubMed] [Google Scholar]
- Thakur GA, Joober R, Brunet A. Development and persistence of posttraumatic stress disorder and the 5-HTTLPR polymorphism. J Trauma Stress. 2009;22:240–43. doi: 10.1002/jts.20405. [DOI] [PubMed] [Google Scholar]
- Walsh K, Uddin M, Soliven R, Wildman DE, Bradley B. Associations between the SS variant of 5-HTTLPR and PTSD among adults with histories of childhood emotional abuse: results from two African American independent samples. J Affect Disord. 2014;161:91–96. doi: 10.1016/j.jad.2014.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Baker DG, Harrer J, Hamner M, Price M, Amstadter A. The relationship between combat-related posttraumatic stress disorder and the 5-HTTLPR/rs25531 polymorphism. Depression Anxiety. 2011;28:1067–73. doi: 10.1002/da.20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Farrer L, Gelernter J. Serotonin transporter 5-HTTLPR genotype moderates the effects of childhood adversity on posttraumatic stress disorder risk: a replication study. Am J Med Genet B: Neuropsychiatr Genet. 2012;159:644–52. doi: 10.1002/ajmg.b.32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, et al. Interactive effect of stressful life events and the serotonin transporter 5-HTTLPR genotype on posttraumatic stress disorder diagnosis in 2 independent populations. Arch Gen Psychiatry. 2009;66:1201–9. doi: 10.1001/archgenpsychiatry.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Poling J, Stein MB, Anton RF, et al. Interaction of FKBP5 with childhood adversity on risk for post-traumatic stress disorder. Neuropsychopharmacology. 2010;35:1684–92. doi: 10.1038/npp.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie P, Kranzler HR, Yang C, Zhao H, Farrer LA, Gelernter J. Genome-wide association study identifies new susceptibility loci for posttraumatic stress disorder. Biol Psychiatry. 2013;74:656–63. doi: 10.1016/j.biopsych.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannas AS, Binder EB. Gene-environment interactions at the FKBP5 locus: sensitive periods, mechanisms and pleiotropism. Genes Brain Behav. 2014;13:25–37. doi: 10.1111/gbb.12104. [DOI] [PubMed] [Google Scholar]
- Zhang H. Statistical analysis in genetic studies of mental illnesses. Stat Sci. 2011;26:116–29. doi: 10.1214/11-STS353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann P, Bruckl T, Nocon A, Pfister H, Binder EB, et al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry. 2011;168:1107–16. doi: 10.1176/appi.ajp.2011.10111577. [DOI] [PMC free article] [PubMed] [Google Scholar]