

Abstract
It is almost four decades since N-(2-hydroxypropyl)methacrylamide (HPMA) – based copolymers arose as drug carriers. Although fundamentals have been established and significant advantages have been proved, the commercialization of this platform technology was hampered due to modest outcome of clinical trial initiated with PK1, the symbol of first generation polymer-drug conjugates. In this review, we illustrate the exciting progress and approaches offered by more effective 2nd generation HPMA-based polymer-drug conjugates in cancer treatment. For example, a new synthetic strategy endorses inert HPMA polymer with biodegradability, which permitted to prepare high molecular weight HPMA-drug conjugates with simple linear architecture while maintaining good biocompatibility. As expected, extended long-circulating pharmacokinetics and enhanced antitumor activities were achieved in several preclinical investigations. In addition, greater inhibition of tumor growth in combination regimes exhibits the remarkable capability and flexibility of HPMA-based macromolecular therapeutics. The review also discusses the main challenges and strategies for further translation development of 2nd generation HPMA-based polymer-drug conjugates.
Keywords: HPMA (N-(2-hydroxypropyl)methacrylamide), polymer-drug conjugates, cancer, backbone degradable polymers, combination therapy
Graphical abstract
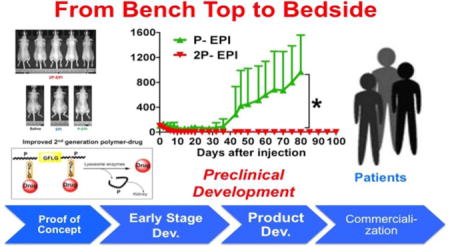
1. A Brief Retrospective: Invention and early advances of HPMA copolymer in nanomedicine
The concept of polymer-drug conjugates was developed to address the lack of specificity of low molecular weight drugs for cancer cells [1,2]. In a typical HPMA-based polymer drug conjugate, drug is bound to HPMA polymer backbone via a tetrapeptide sequence (GFLG) that is stable in the blood stream but susceptible to cleavage by lysosomal enzymes in cancer cells. Optionally, a targeting moiety can be attached to HPMA backbone to enhance the selectivity of polymeric prodrugs. This approach was based on the work of de Duve, who discovered that the endocytic pathway is suitable for lysosomotropic drug delivery [3].
As one of the earliest developed drug-delivery systems, HPMA polymer–drug conjugates have been extensively studied [4–8, reviewed in 9] (Fig. 1). Early-stage studies focused on structure-activity relationship, such as optimization of oligopeptide spacer structure and the effect of polymer molecular weight (Mw) on cell uptake and intracellular degradation [reviewed in 9]; these data constitute a firm foundation for the design of advanced conjugated therapeutics.
Figure 1.
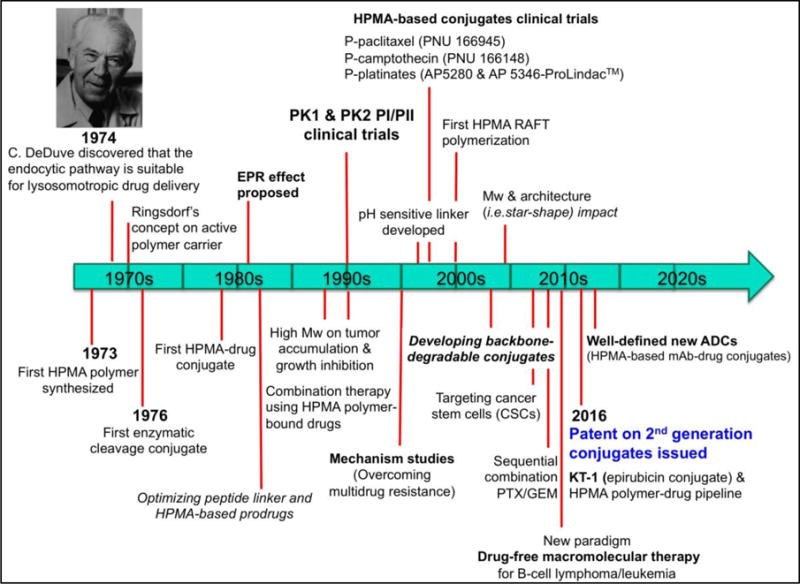
Historical timeline of major developments in HPMA-based conjugates for cancer treatment.
In the mid of 1990’s, the first synthetic polymer-doxorubicin (DOX) conjugate, named PK1, entered clinical trials [10,11], followed by PK2 [12]. This landmark of development greatly advanced the field of macromolecular therapeutics. From late 1990’s to the beginning of the 21st century, HPMA polymer-bound drugs containing paclitaxel (PTX) [13], camptothecin (CPT) [14,15], and platinates [16–19] have reached Phase I and/or Phase II clinical evaluations. Nevertheless, none of these investigations moved into routine clinical use. There are numerous reviews documenting the evolution of HPMA-based polymer-drug conjugates spanning from ‘the dawning era’ to ‘the end of the beginning’ [20–22]. Some other reviews judiciously discussed the design rationale, synthetic approaches, comprehensive state-of-the-art of the field and future directions [23–28]. Herein we do not intend to repeat these opinions, but describe briefly the intensive research efforts on the development of 2nd generation conjugates, and identify major challenges hindering their translation into clinical application.
2. The development of the second generation of HPMA copolymer-drug conjugates
As a water-soluble polymer, pHPMA is cleared from the body mainly by renal excretion. Conjugation of a drug to HPMA polymer backbone improves the drug solubility and stability, increasing its half-life in blood circulation. It was found that when the Mw of an HPMA conjugate is above renal threshold (~50 kDa), the conjugate accumulates in tumor tissue at much higher concentrations than in normal tissue/organs. Maeda defined this phenomenon as the enhanced permeability and retention (EPR) effect [29,30], which is caused by the leaky walls of immature growing vasculature and less developed lymphatic drainage system in tumor. Therefore, EPR effect is usually proportional to the rate of tumor growth, and works well in aggressive tumor models. A more detailed discussion of EPR is in section 3.1. Because the HPMA copolymer backbone is non-degradable and possesses a wide molecular weight distribution inherent to conventional free-radical polymerization used for synthesis, the conjugates that stepped into clinical trials were suboptimal (<40 kDa) to guarantee eventual renal elimination (Fig. 2).
Figure 2.
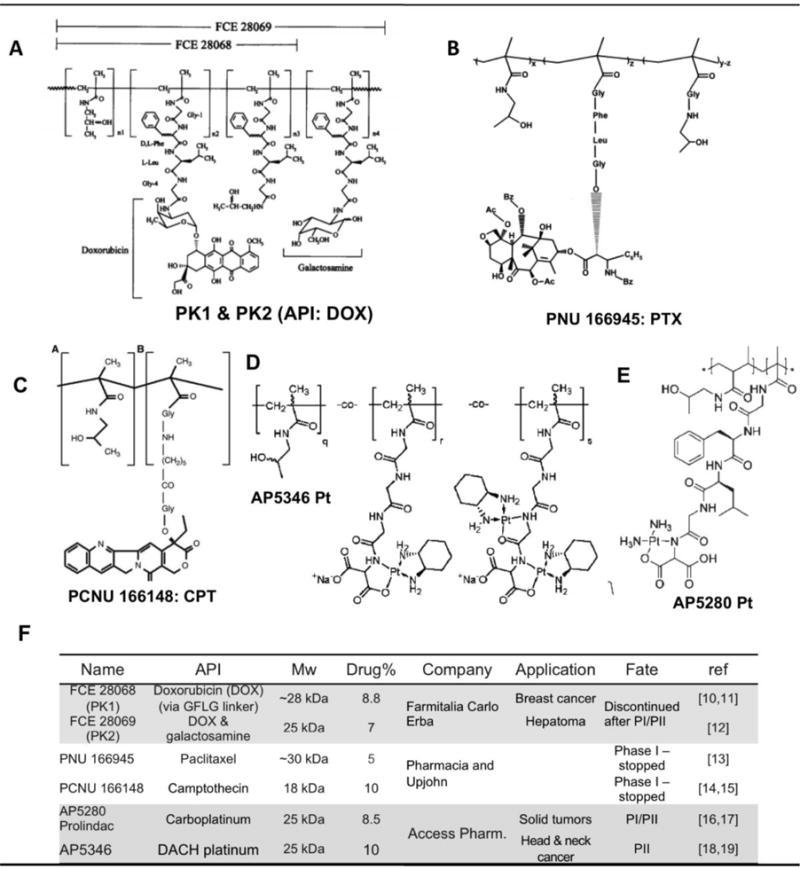
First-generation HPMA polymer–anticancer drug conjugates used in clinical trials
Herein we present the previous translation attempts of the first generation HPMA-based polymer-drug conjugates, along with commentary regarding what we believe to be the primary cause for their lack of success. We will then demonstrate that each of these shortcomings are not an inherent limitation of the HPMA copolymer platform itself and discuss recent developments that have overcome these early “growing pains” with modern advances in polymer chemistry and improved understanding of cancer biology.
2.1. Lessons learnt from 1st generation conjugates
2.1.1 PK1 & PK2
PK1 was initially co-developed at the Institute of Macromolecular Chemistry (Kopeček’s former laboratory), Prague, and University of Keele (Duncan’s group), Keele (UK). A polymer precursor containing 4-nitrophenyl ester groups was first prepared followed by attachment of DOX via aminolysis. After purification, usually there is a tiny amount of free DOX left. Prior to use in humans, the mechanism of action and anticancer efficacy of PK1 were intensively investigated in vitro and in vivo. Preclinical studies demonstrated PK1 enters the cell via a different pathway and has completely dissimilar pharmacokinetics compared with free DOX. A plasma distribution half-life was extended from 5 min to 1 h, and initial peak level in the heart was reduced 100-fold. PK1 showed great activity in both solid tumors and leukemia [31]. PK1 is not a substrate for P-glycoprotein (Pgp) and multidrug resistant protein (MRP) drug efflux pumps. As a result, P-DOX has potential to overcome multidrug resistance. Indeed, P-DOX was observed to be effective in both sensitive and resistant tumor models, whereas free drug was ineffective in the tumor derived from resistant cells [32].
In March 1994, PK1 was licensed to Pharmacia (formerly Farmitalia Carlo Erba and later Pfizer) for co-development [33]. The Phase I study was conducted in United Kingdom; 36 patients (20 males and 16 females) enrolled, and a total 100 cycles of PK1 were administered with dose frequency as once every 3 weeks. Similar to preclinical study, PK1 demonstrated significant reduction of nonspecific toxicity. Maximum tolerated dose (MTD) of PK1 in humans was 320 mg/m2 of DOX equivalent, whereas for free (unbound) DOX in humans is 60–80 mg/m2. The enhanced MTD of the polymer-bound DOX (PK1) is primarily attributed to low uptake in heart tissue. Importantly, PK1 showed activity in chemotherapy-refractory patients. Two partial responses (PR) in non-small-cell lung cancer (NSCLC) patients and two minor responses, one in a metastatic colorectal cancer patient and one in a breast cancer patient were observed. Dose-limiting toxicities were neutropenia and mucositis. The recommended Phase II dose was 280 mg/m2 every 3 weeks.
Following PK1 Phase I study, a sister compound PK2 containing galactosamine as a targeting ligand for liver hepatocytes was developed and also entered clinical trials. However, although targeted conjugate indeed effectively accumulated in the liver, the level of the conjugate in liver tumor was substantially lower than that in healthy tissue (3±6% vs 17±4% of the injected dose). Antitumor activity was observed in a few patients with advanced liver carcinoma. MTD was found as 160 mg/m2, which is lower that that of PK1, and dose-limiting side effect was comparable to PK1, and recommended dose for Phase II was 120 mg/m2 [12].
Phase II trial with PK1 was conducted with 17 breast cancer patients (all anthracycline-naïve), 29 non-small cell lung cancer (NSCLC) and 16 patients with colorectal cancer). Up to 8 courses of PK1 (280 mg/m2 doxorubicin-equivalent) were i.v. administered. Overall toxicities were tolerable, with grade 3 neutropenia more prominent in patients with breast cancer (4/17, 23.5% compared with 5/62, 8.1% overall). Of 14 evaluable patients with breast cancer 3 patients had partial responses (PR). In 26 evaluable patients with NSCLC, 3 chemotherapy-naïve patients had PR. In contrast, none of the 16 evaluable patients with colorectal cancer responded [11]. The 6/62 PR results support the concept that polymer-drug conjugates can have modified toxicity and improved efficacy. However, clinical studies discontinued in 2008 due to marginal improvement of anticancer activities [34]. Apparently, conjugates efficient in humans need to possess longer circulating time in blood stream in order to produce a sustained concentration gradient between the vasculature and solid tumor. Consequently, HPMA conjugates with high Mw and degradability need to be developed.
2.1.2. HPMA copolymer-paclitaxel conjugate
(PNU166945) was developed in Pharmacia and Upjohn. Acute and subchronic toxicity studies in mice, rats, and dogs indicated that dose-limiting toxicity was myelotoxicity with no signs of hypersensitivity, neurotoxicity or cardiovascular toxicity. It was evaluated just in Phase I clinical trial and 12 patients were enrolled [13]. The highest dose level was 196 mg/m2, at which no dose-limiting toxicities have been observed. Hematologic toxicity was mild and dose dependent. One patient developed Grade 3 neurotoxicity (no premedication was given). For reasons unclear, additional 13-week toxicity study in rats was performed with doses 60 and 90 mg/kg (corresponds to 440 and 600 mg/m2 in human!!). Serious neurotoxicity found in rats directly caused discontinuation of the Phase I study. It is known that polymer-PTX conjugate is incapable of crossing the brain-blood barrier. The neurotoxicity found in PNU166945 clinical trial was apparently from free PTX (peripheral neuropathy is a common adverse effect of free PTX). In PNU166945, PTX is bound via ester bond. So PTX is partially released from polymer carrier while circulating in the blood stream. Moreover, the conjugate was prepared by a polymeranalogous reaction (attachment of drug to polymer precursor); consequently, free drug may have been part of the formulation. The toxicity in rats was most probably a result of these two factors and of extremely high doses of PNU166945 administered.
2.1.3. HPMA copolymer-camptothecin (CPT) conjugate
(MAG-CPT, PCNU166148) was also developed in Pharmacia and Upjohn. Compared to DOX, CPT has lower aqueous solubility but higher toxicity. Instead of using GFLG linker, covalent attachment of CPT to HPMA polymer precursor had two steps: first, a glycine residue was used to modify 20-OH of CPT via ester bond formation; in the next step, the modified CPT was conjugated to the polymer. The release of free camptothecin into the blood stream was then dependent on the rate of pH mediated esterolytic cleavage. MAG-CPT was evaluated in several Phase I trials [14,15]. However, serious cumulative bladder toxicity was observed. In addition, MAG-CPT did not show any clinical evidence of antitumor activity. Therefore the Phase I trial was stopped. This failure can be attributed to two reasons: first, as mentioned above, conjugation of CPT to hydrophilic HPMA polymer altered its pharmacokinetic profile/biodistribution, resulted in elimination of CPT from body mainly via kidney. Considering the Mw was only 18 kDa, a fast body clearance resulted in loss of antitumor activity. Second, inappropriate rate of drug-release led to complete elimination of activity and to off-target toxicity.
2.1.4. HPMA copolymer-platinates
In late 1990s when PK1 underwent clinical trials, Duncan et al. initiated development of HPMA copolymer-platinates following the same rationale as PK1 [17]. The technology was licensed to Access Pharmaceuticals, who eventually brought two conjugates, AP5280 (carboplatinum analogue) and AP5346 (oxaliplatin analogue), to clinical trials [19]. The synthesis of the conjugates consists of a series reactions employing elegant chemistry. It was reported that a co-development contract was signed with Korea and China, respectively in early 2008. These conjugates were expected to be the first HPMA copolymer conjugate to come to the market, but there has been no updated report on these conjugates available after the review article in 2009 [19]. Because of extremely high dose (i.e. 3300 mg Pt/m2 for AP5280 Phase II studies), we consider it may not be suitable for drug delivery strategy.
2.1.5. Other promising HPMA-based conjugates in clinical studies
In addition to the classic linker GFLG, an acid-sensitive hydrazone linkage has been utilized alone or in combination with GFLG in a number of HPMA-based polymer drug conjugates [35]. Unlike GFLG, this pH-sensitive linker enables hydrolytically controlled release of drug molecules from HPMA polymer carrier—it is relatively stable in blood circulation (pH 7.4), but is unstable at intratumor and intracellular (pH ~ 5–6) environment. In this case, the activity of cathepsin B is not essential for biological activity of the conjugate. One such conjugate consisting of hydrazone linker and drug pirarubicin (P-THP) has been applied together with proton beam radiation to a patient with stage IV prostate cancer and extensive metastases, resulting in complete remission and no evidence of relapse was observed after 20 months [36].
Říhová and coworkers treated six patients with HPMA copolymer – drug (DOX, EPI) – IgG conjugates [37,38]. Patient No. 1 diagnosed with metastatic angiosarcoma was treated with HPMA copolymer-epirubicin conjugated to autologous IgG, and 5 metastatic breast cancer patients were treated with HPMA copolymer-doxorubicin conjugate either with autologous (patient No. 2) or normal IgG (patients Nos. 3–6). In four (out of six) patients a positive clinical response was recorded lasting 6 to 18 months. Numerous biochemical, immunological, and hematological parameters were recorded from patients’ blood up to 10 months following the last application. No serious side effects were recorded and elevated number of immunocompetent cells indicated that HPMA copolymer-drug conjugates are able to stimulate anticancer immunity. This is an important outcome that might indicate that administration of polymeric prodrugs may result in double attack on cancer cells – by cytotoxicity and via immunostimulation [39,40].
In summary, all these studies except the camptothecin conjugate provide a clinical ‘proof of principle’ and demonstrated polymer-drug conjugates could reach human tumors, internalize and be cleaved to exert bioactive function. HPMA copolymer as drug carrier has been administered to more than 250 patients but no polymer-related toxicity has been reported.
Recently an article [41] evaluated hemodynamic effects of HPMA copolymer-DOX conjugate in rats. While the biological techniques were up-to-date, the results have no validity due to inaccurate polymer structure employed. They used an HPMA copolymer from Polymer Laboratories as control, which contains reactive p-nitrophenyl ester groups that could initiate numerous side reactions if used directly. The samples must be years old and are not characterized correctly. Even though the p-nitrophenyl ester groups may have been randomly hydrolyzed as time goes by, the resultant polymer contains ionizable groups at side chain termini whose in vitro cytotoxicity has been reported [42]. Moreover, reactions of the p-nitrophenyl group with the polymer carrier and redox changes of their structure may have occurred.
2.2 Backbone degradable long-circulating macromolecular therapeutics
In order to directly address the limitations imposed by the low molecular weight and rapid clearance of previous clinical trial candidates, many efforts have been made to enlarge molecular weight and/or architecture of non-degradable HPMA conjugates without impairing the biocompatibility [43–46].
Enzyme cleavable oligopeptide linkers were used to prepare conjugates with different architectures (Fig. 3A). When these conjugates were incubated with cathepsin B, in addition to drug release, all conjugates were gradually degraded into composing fragments that were small enough to be eliminated from body. When Mw of the conjugate increased from 22 to 160 kDa, the drug accumulation in tumor dramatically increased, with concomitant enhancement of tumor growth inhibition [46] (Fig. 3B). However, the major drawback is the complexity of structure and lack of batch-to-batch synthetic reproducibility [45].
Figure 3.
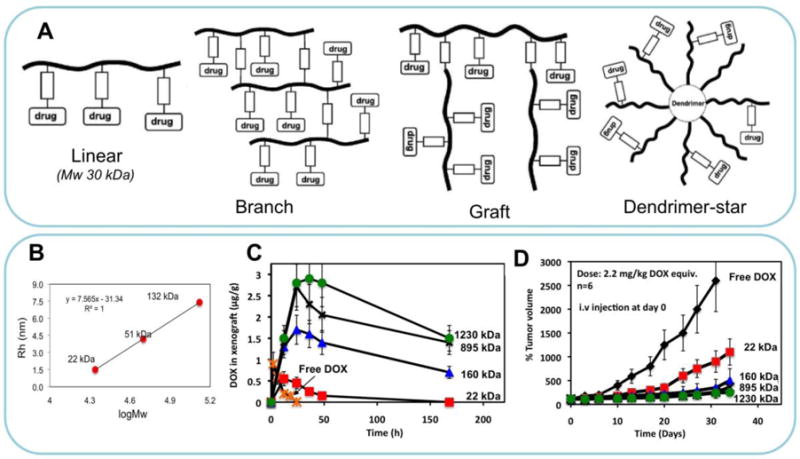
(A) Different architectures of degradable HPMA copolymer conjugates. Adapted form ref. [27]. (B). Correlation of molecular weight with hydrodynamic radius of HPMA homopolymer standards. (C) Concentration of DOX in OVCAR-3 carcinoma xenografts in nu/nu mice after i.v. bolus of free DOX or P-DOX of different MW. (D) Growth inhibition of s.c. human ovarian OVCAR-3 carcinoma xenografts in nu/nu mice by long-circulating P-DOX conjugates. The mice received i.v. injection of 2.2 mg/kg DOX equivalent dose as P-DOX of different Mw [45,46].
2.2.1 Breakthrough long-circulating HPMA polymer-drug conjugates
Advances in polymer chemistry have provided a powerful tool to synthesize well-defined, end-functionalized HPMA polymer conjugates [47]. A new generation of linear, backbone-degradable HPMA copolymers was synthesized via reversible addition-fragmentation chain transfer (RAFT) polymerization and subsequent polymer coupling (Fig. 4A) [48–50]. In particular, a bifunctional chain transfer agent containing an enzymatically degradable sequence (Peptide2CTA) was designed [50] (Fig. 4B). During RAFT polymerization, HPMA and comonomers incorporate at both dithiobenzoate groups of the Peptide2CTA with identical efficiency. When the conjugate was incubated with papain, a thiol proteinase with similar specificity as lysosomal proteinases, the molecular weight decreased to half of the original value (Fig. 4C). Thus Peptide2CTA permits to synthesize degradable diblock copolymers in one reproducible, industrially scalable step. Further expansion of molecular weight can be achieved by click reactions, producing multiblock copolymers. These polymers are based on the same biocompatible HPMA chemistry; they are linear block copolymers where synthetic segments alternate with degradable ones. Such conjugates will be able to circulate systemically in the blood for long periods of time and take full advantage of the EPR effect while maintaining the ability to be eliminated via renal filtration following enzymatic degradation. For example, the exposure of a multiblock HPMA copolymer-DOX conjugate to lysosomal cathepsin B led to gradual release of DOX as well as complete degradation of the polymer backbone (Fig. 4 D & E).
Figure 4.
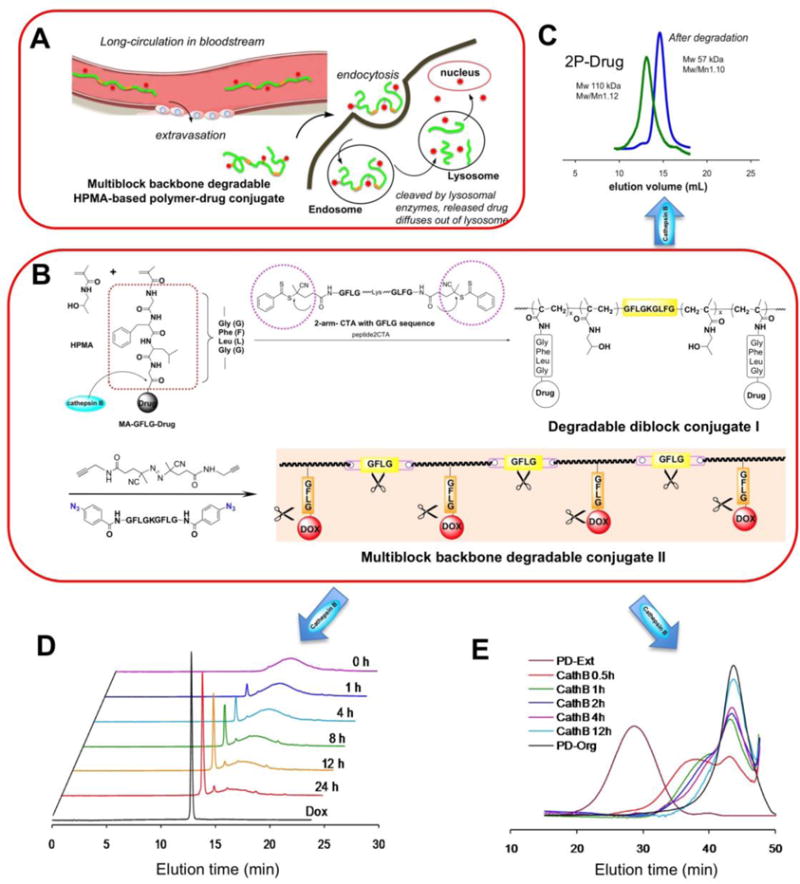
Design of backbone degradable long-circulating HPMA copolymer – drug conjugates. A) Illustration of extravasation, cell uptake followed by cleavage by lysosomal enzymes of 2nd generation conjugate. B) Scheme of conjugate synthesis. Two dithiobenzoate chain transfer agents were linked with lysosomal enzyme cleavable peptide GFLG-K-GLFG resulting in a biodegradable RAFT agent, peptide2CTA. This permits one-step synthesis of diblock copolymers. Post-polymerization click reaction produces multiblock HPMA copolymer-drug conjugates with different chain lengths. C) The diblock HPMA copolymer-drug conjugates degraded into half of their initial Mw, indicating the potential to employ diblock conjugates with 100 kDa Mw without impairing their biocompatibility (the degradation products are below the renal threshold); D) and E) using High-performance liquid chromatography and size-exclusion chromatography to monitor drug release and degradation of polymer backbone in the presence of cathepsin B [50].
Using this strategy, a series of drug (gemcitabine (GEM) and paclitaxel (PTX)) conjugates were synthesized and evaluated in a preclinical tumor model bearing s.c. human ovarian A2780 xenografts [51]. Each group consisted of 5 female mice. Treatment with HPMA copolymer-GEM conjugates revealed a clear advantage of macromolecular therapeutics, as all conjugates (P-GEM; 2P-GEM; mP-GEM200; and mP-GEM300) were considerably more active when compared with free GEM (Fig. 5A).
Figure 5.
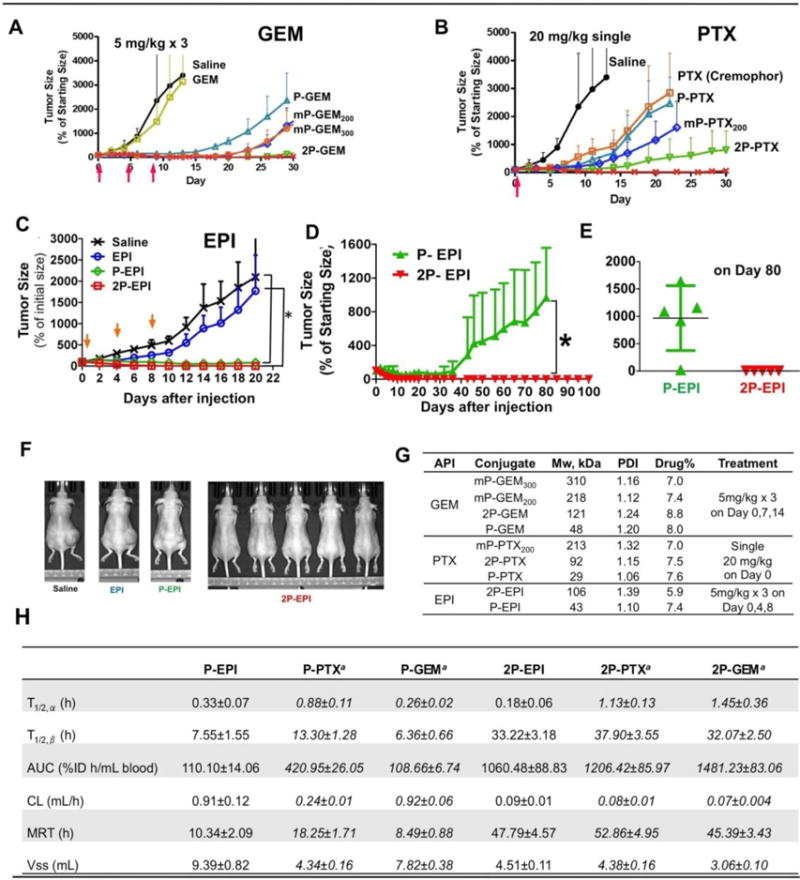
In vivo antitumor efficacy of HPMA copolymer-drug conjugates against A2780 human ovarian carcinoma xenografts. The dose for each injection is expressed as a dose equivalent to free drug (gemcitabine or epirubicin 5 mg/kg, paclitaxel 20 mg/kg). Tumor growth was inhibited by (A): Gemcitabine monotherapy. (B) Paclitaxel monotherapy. (C) Epirubicin monotherapy. Free drug and untreated groups were stopped on day 20 due to large size of tumors. (D) Long-term monitoring of tumor growth in conjugate treatments. (E) The tumor size on day 80 of the polymer conjugates. (F) End point photographs of tumor-bearing mice from variable treatments. (G) Characterization of the conjugates. (H) Comparison of pharmacokinetic parameters for 125I-labeled conjugates in mice. Adapted from refs. [51,54].
Gemcitabine has very short intravascular half-life (15.7 min) in mouse due to fast metabolism and clearance. No significant toxicity was found following i.v. injection of doses as high as 150 mg/kg [unpublished data]. In a treatment experiment of human ovarian carcinoma A2780 xenografts the dose of GEM/GEM equivalent was only 5 mg/kg. As expected (at such a low dose) free GEM showed similar activity to saline. The 1st generation (Mw ~ 40 kDa) conjugate P-GEM possessed the lowest activity among all polymer conjugates indicating the importance of the molecular weight of the carrier on activity. Interestingly, diblock conjugate 2P-GEM (Mw ~ 100 kDa) inhibited tumor growth more effectively than multiblock conjugates mP-GEM200 (tetrablock) and mP-GEM300 (hexablock). A similar trend was found in PTX conjugates except free PTX. Due to poor solubility, PTX has to be formulated prior to administration. In this study, 30 mg PTX was emulsified with 5 mL vehicle (Cremophor EL/ethanol 1:1). PTX was gradually released from the vehicle, therefore showed similar tumor inhibition ability to P-PTX, the first generation conjugate. Unexpectedly, mP-PTX200 demonstrated similar tumor as P-PTX in tumor inhibition. To elucidate this phenomenon, 131I-labeled PTX conjugates were prepared and their pharmacokinetics/biodistribution in mice were determined. mP-PTX200 was found to have the same pattern of ‘Blood radioactivity-time’ profile as P-PTX but much higher uptake in spleen and liver (data not shown), which suggested higher Mw conjugate mP-PTX200 tend to form larger size inter-/intra- chain associates and was taken up by macrophages, resulting in fast clearance from bloodstream after intravascular injection [52]. This data are in agreement with the study of the relationship between molecular weight of backbone degradable HPMA copolymer-doxorubicin conjugates and their antitumor efficacy toward resistant human ovarian carcinoma A2780/AD xenografts [53].
To confirm that diblock structure is sufficient to dramatically enhance efficacy, HPMA copolymer-epirubicin conjugates (including 1st generation conjugate P-EPI and diblock conjugate 2P-EPI) were synthesized [54]. Epirubicin, the 4′-epimer of the anthracycline doxorubicin (DOX), has been regarded as one of the most active drugs for the patients with cancer, particularly with metastatic disease. It has shown equivalent cytotoxic effects to DOX in human ovarian cancer cells, but decreased cardiotoxicity and myelotoxicity than DOX at equimolar doses. Thus, epirubicin is thought to have a better therapeutic index than DOX [55].
The therapeutic potential of the conjugates was evaluated in female nude mice bearing A2780 human ovarian carcinoma xenografts, the same animal model as mentioned above [54]. The mice were intravenously injected with three doses of 5 mg/kg EPI equivalent on days 0, 4, and 8. Free drug EPI was also administered for comparison. The advantage of polymer-drug conjugates over free drug on tumor inhibition was clearly demonstrated: At day 20, complete tumor regression was achieved in the five mice treated with conjugate 2P-EPI; the tumors treated with P-EPI also shrank to 80±37% of the initial size. In contrast, free drug EPI at equivalent doses only slightly delayed tumor growth when compared with saline, and mice had to be sacrificed on day 20 due to large tumor burden.
To assess whether there is significant difference between treatment with 2P-EPI and P-EPI, long-term observation was conducted. In P-EPI group, tumor started regrowth from day 35 and in four mice (out of 5) the tumors grew back to ~1200% at day 80 (p<0.01) (Fig. 5D & E). In contrast, no observable tumor was detected in the mice treated with 2P-EPI at day 100. The complete tumor regression and long-term inhibition of tumorigenesis are most likely due to long circulation time and sufficient extravasation of the conjugates at the tumor site by the EPR effect (Fig. 5E & F).
For safety concern, body weight of the mice was closely recorded during and after treatment. At initial administration with multiple dosages, the body weights of the mice decreased less than 10%, then recovered gradually and remained stable, which suggests the doses used were tolerable.
To highlight the molecular weight effect on conjugate plasma concentration and circulation time, the blood radioactivity-time profiles were determined and calculated pharmacokinetic parameters of the conjugates are listed in Fig. 5H.
Although individual drugs (EPI, PTX, and GEM) have different metabolism and clearance time, the second generation conjugates 2P-EPI/2P-PTX/2P-GEM (when compared to first generation conjugates, P-EPI/P-PTX/P-GEM) showed significantly improved pharmacokinetics parameters such as terminal half life, total body clearance, and steady-state volume of distribution, which indicates conjugation of drug to polymer carrier can improve its stability in plasma, and the elimination rate of the conjugates is primarily determined by the polymer carrier.
Thus it was confirmed that diblock backbone degradable HPMA-drug conjugates possess superior advantages over current marketed chemotherapeutical agents as well as the 1st generation conjugates (Mw<50 kDa). Furthermore, the backbone degradable diblock conjugates are a platform – a list of conjugates containing current chemotherapy agents or new inhibitors that are still in clinical trials (i.e., GDC0980) presents a pipeline for translation development. Among them, KT-1 (also known as 2P-EPI) as an exciting candidate lies at the top of this Polymer-Drug Conjugate Platform for Solid Tumors (breast, non-small cell lung (NSCL), pancreatic, and ovarian cancers).
In addition to abovementioned features, the second-generation HPMA polymer-drug conjugates have a competitive advantage with simplicity of structure, proven safety of the polymer carrier, and utilization of current effective drugs. The involved synthesis procedures are suitable for scale-up and industrial production. This technology provides a platform for the preparation of a large variety of polymer-drug conjugates with tailor-made properties, such as predetermined circulation time and composition. Up to now a list of polymer-drug conjugates have been prepared; their anti-tumor activities either used alone or as combination therapy were explored intensively in order to expand the array of potential applications.
2.3. Combination therapy
Cancer is a disease of abnormal and uncontrolled cell growth; attacking one pathway may not be enough. Due to complexity of human body and heterogeneity of tumor cells and tissues, when two or more drugs with different mechanism of action are used simultaneously or sequentially, there is benefit from avoiding development of drug-resistance. However, such cocktail treatment usually is limited by the cumulative toxic effects of the free drugs. Polymer-drug conjugates have advantages on maximizing synergy in the therapeutic effects due to long-circulating pharmacokinetics and reduced non-specific toxicity [56].
The first combination therapy using polymer bound drugs for chemo- and photodynamic therapy on two cancer models, Neuro 2A neuroblastoma and human ovarian carcinoma heterotransplanted in the nude mice, resulted in synergistic response [57]. Incorporation of targeting moiety further increased the therapeutic efficacy [58].
There are many factors influencing combination therapeutic effects: drug selection and their ratio; administration regime, i.e., sequential or simultaneous; and the formulation: such as binding two drugs to the same polymer chain, or preparation of two individual polymer-drug conjugates for co-administration. It has been frequently reported that two or more drugs entrapped in one nanoparticle can maintain the synergistic ratio of the drugs thus delivering the drugs to the same cell to achieve synchronization in tumor treatment. However, the major disadvantage of such system is the loss of flexibility in adjusting each drug dose independently. Once two or more agents are fixed in the final drug product, it becomes ‘one-size fits all’. This inflexibility can be of particular concern for a patient who may not tolerate one of the drugs in the combination product; thereby reducing amount of the drug or changing the order of administration is impossible.
Below we discuss three interesting examples of combination treatment utilizing the HPMA copolymer delivery platform.
2.3.1 Combination of PTX and GEM—a regimen from clinics
PTX and GEM are one the most common combination regimens in clinics. They possess distinct mechanisms of anticancer effect: PTX interacts with tubulin in cell cytoplasm, whereas GEM triphosphate replaces cytidine during DNA replication and causes replication arrest and apoptosis. There is synergistic potential in PTX and GEM combination.
For conjugates combination, formulation selection is the first step. Three conjugates were prepared and their interactions with cells were investigated by in vitro combination index (CI) study [59]. When both GEM and PTX were conjugated to the same copolymer backbone, moderate antagonism (CI 1.3–1.6) was observed. Synergistic combined effects (CI < 0.7) were observed when A2780 cells were treated with mixture of two individual conjugates. Therefore, in all next studies, combination was conducted using HPMA polymer-PTX and HPMA polymer-GEM conjugates.
CI study was also used to optimize combination regimen. It was demonstrated that the combination of PTX and GEM is schedule-dependent; treatment with PTX followed by GEM has the strongest synergism [60] (Fig. 6.I). One possible mechanism for the synergism is that the amount of microtubulin rapidly decreased in the tumor cells exposed to such sequential treatment.
Figure 6.
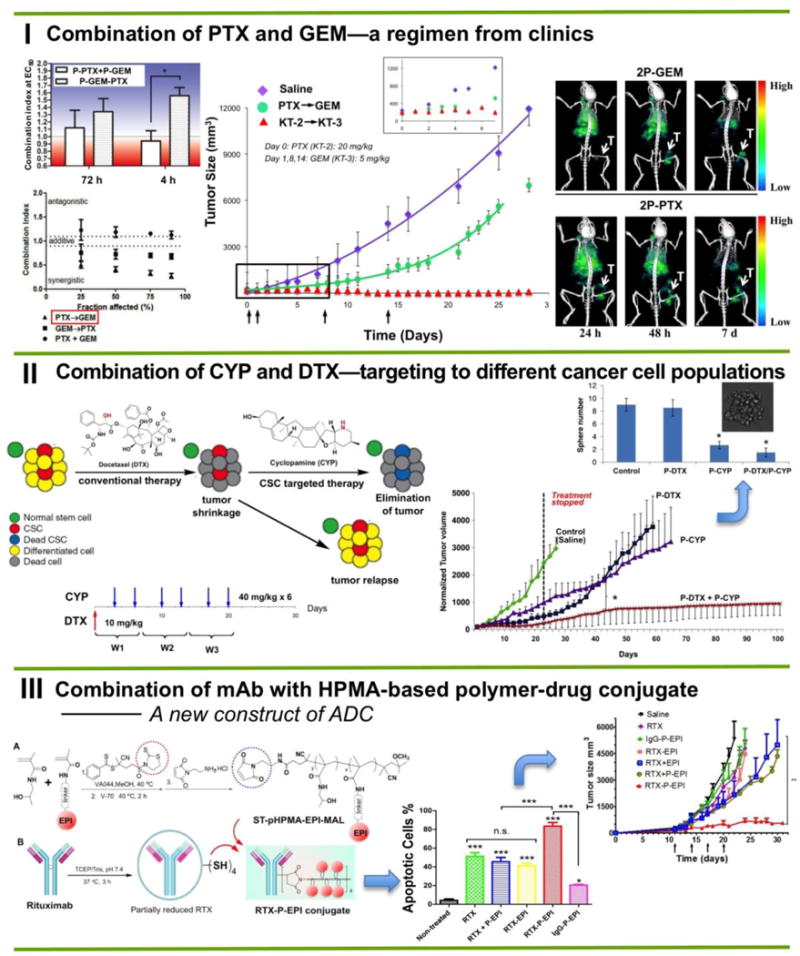
Three examples of HPMA-based macromolecular combination therapeutics. (I) Long-circulating backbone degradable combination chemotherapy based on clinical chemotherapeutic agents (gemcitabine and paclitaxel). (II) Combination therapy targeting different cell types: cancer cells and cancer stem cells using chemoagent docetaxel and hedgehog pathway inhibitor cyclopamine. Such strategy produced long-term survivors in an animal model of prostate cancer. (III) A new construct of antibody-polymer-drug conjugate. Well-defined semitelechelic HPMA polymer-epirubicin conjugate was selectively attached to freshly reduced Rituximab. In vitro Ramos cell apoptosis induction and in vivo antitumor efficacy were evaluated. Adapted from refs. [60,62,66].
Combination of gemcitabine with paclitaxel was evaluated on nu/nu mice bearing s.c. A2780 human ovarian carcinoma xenografts [60]. Notably, long retention time of drugs in the circulation is crucial to cell-cycle-specific drugs such as PTX and GEM, which can exert effective actions on cells only in a specific phase of cell cycle. Besides, as previously mentioned, GEM is rapidly metabolized into inactive 2-deoxy-2,2-difluorodeoxyuridine (dFdU) by cytidine deaminase present in the blood, liver, and other tissues. Attaching GEM to HPMA copolymer dramatically decreases GEM’s metabolism rate. Instead of using multiple injections with a dose of 100 mg/kg [61], GEM was administered still at 5 mg/kg on Day 1, 8 and 14.
Overwhelming results were observed: the differences of tumor size between free drug and conjugates were statistically significant. Two mice were tumor-free and the other three mice had tumor at ∼3% of initial size. Such success is attributed to enhanced bioavailability, prolonged circulation time, increased intratumoral drug concentration, and combination strategy. As SPECT imaging indicated, conjugate accumulated in tumor tissue and can be detected even one week after i.v. administration (Fig. 6.I).
2.3.2 Combination of cyclopamine and docetaxel—targeting to different cancer cell populations
In this example, we chose to target two different cell populations: cancer stem cells and differentiated cells. Docetaxel (DTX) is a traditional first-line chemotherapeutic agent for advanced prostate cancer that shows effective anti-tumor effect during treatment period. However, the progression free survival of patients is not satisfactory. One main possible reason is that DTX alone provokes an enlarged and more resistant CSC pool. Accumulating evidence indicates the significance of cyclopamine (CYP) as a potential stem-cell inhibitor. CYP inhibits the hedgehog (Hh) signaling pathway by directly binding to SMO heptahelical bundle and suppressing the SMO receptor. However, CYP has very poor water-solubility. In preclinical studies/clinical trial, the administration approach is oral or i.p. with high dose and frequency (i.e. 40 mg/kg daily). Therefore, we conjugated CYP to HPMA polymer carrier to improve its solubility and pharmacokinetics. By combination of P-CYP/P-DTX to eradicate differentiated cancer cells as well as the cancer stem cell (CSC) population we have achieved long-term survivors [62] (Fig. 6.II).
This promising result implies potential improvement of pancreatic cancers treatment. Pancreatic cancer contains numerous tumorigenic cancer stem cells (CSCs), which are highly resistant to chemotherapy, resulting in a relative increase in CSC numbers during chemotherapy. Combination of P-CYP with P-GEM could represent a novel treatment strategy.
2.3.3 Combination of Rituximab (RTX) with HPMA copolymer-epirubicin conjugate – a novel construct of antibody-drug conjugates
As the first FDA approved monoclonal antibody, RTX has provided a revolutionary contribution to the treatment of B-cell non-Hodgkin’s lymphomas (NHL) over the past two decades. However, the major barrier for RTX clinical application is the development of resistance—RTX is less effective in patients with relapsed lymphoma. Although combination of RTX with chemotherapy (i.e. the most common regimen R-CHOP) was reported to provide better outcomes, RTX conjugated with doxorubicin (DOX) [63], liposomal DOX [64], and HPMA copolymer-DOX conjugates [65] did not significantly enhance treatment activities.
Recently we have developed a new strategy to generate therapeutically efficient HPMA-based RTX-epirubicin conjugates [66]. Taking advantage of living/controlled free radical polymerization technology, a well-defined semitelechelic (ST) pHPMA-EPI conjugate with maleimide end-group was first prepared, which was conjugated to partially reduced RTX according to the scheme illustrated in Fig. 6.III. This approach allows the introduction of a large payload of drug to antibody without adding attachment sites. The cytotoxicity of resultant RTX-P-EPI against Ramos cells was determined. Interestingly, a two-fold increase in apoptosis level was observed in cells treated with RTX-P-EPI compared with the equivalent mixture of RTX and the first generation HPMA copolymer-epirubicin conjugate P-EPI. Treatment of male SCID mice bearing subcutaneous Ramos B-cell lymphoma tumors also demonstrated the superior efficacy of RTX-P-EPI when compared to the combination of RTX with free EPI (RTX + EPI) and P-EPI (RTX + P-EPI) (Fig. 6.III). The results indicate the synergism of immunotherapy combined with established macromolecular therapy.
This study has profound influence on ADCs development: Instead of using extremely potent toxins, conventional chemo-agents could be utilized to generate highly effective ADCs. Consequently, the risk of off-target binding will be lowered, and the tolerability greatly improved.
The aforementioned examples demonstrated the great versatility and high potency of 2nd generation HPMA polymer-drug conjugates.
3. Main challenges and strategies
It has been a long journey since the first HPMA paper was published in 1973 [4]. The discontinuation of clinical trials from 1st generation conjugates have resulted in seriously reduced interest (both in industry and academia) in HPMA-based polymer-drug conjugates as an attractive therapeutic option. ‘HPMA’ is wrongly considered outdated. Based on a recent publication on cancer nanomedicine [67], nanoparticles and nanomedicine are often used interchangablely, whereas the polymer-drug conjugates are no longer one of mainstays. In this review, we have analyzed the factors for the failure of early conjugates clinical trials, and the emergence and progress of the 2nd generation of HPMA polymer-drug conjugates. Nonetheless, to make the project attractive and to move it into clinical development, many challenges still lay ahead.
3.1 EPR effect: Yes or No?
Researchers have fought over this question for many years because of the significant different outcome between preclinical animal studies and clinical trials. The culprit behind this dispute is tumor heterogeneity.
The concept of EPR effect was proposed by Meada in the process of developing a macromolecular therapeutics poly(Styrene-co-Maleic Acid) – NeoCarzinoStatin (SMANCS), a polymer conjugate (12 kDa protein + two 1.5 kDa polymer chains) that non-covalently binds albumin in the circulation to reach a molecular weight of around 80 kDa [68]. It was originally based on the observation that the macromolecules preferentially accumulated in tumor tissue. As cancer biology evolved, this preferentially biodistribution was attributed to the fenestration in the imperfect blood vessels and the poor lymphatic drainage in the tissue. Therefore EPR effect has become a design principle and cornerstone of nanomedicine. After ‘So many papers and so few drugs’ [69] EPR has been swung from over-interpretation [70] to declining belief. There is skepticism about whether EPR exists in human tumors [71].
However, recent findings from CRLX101 clinical trial demonstrate EPR is at work in humans [72]. CRLX101 is also a prodrug consisting of a cyclodextrin-containing polymer-conjugate of camptothecin (CPT), which self-assemble into nanoparticles with 20–30 nm diameter and 10 wt% CPT. In Phase II study, CPT was detected in tumor samples of 5/9 gastric cancer patients 24–48 after administration. Although biological activity from the delivered CPT was reported, there is no efficacy outcome yet, which is dependent on the concentration of the nanoparticle in tumor and the cleavage of CPT from the carrier.
Additionally, EPR effect merely explains or predicts extravasation and biodistribution of a drug delivery system at the tissue level. Cancer treatment is a much more complex biological process, and reaching the tumor tissue is only one step to a successful treatment. A drug carrier has to overcome tumor penetration, cell uptake and drug release, etc. Each step presents additional challenges.
Molecular weight needs to be optimized for particular applications. Higher molecular weight conjugates possess a longer intravascular half-life resulting in efficient extravasation into solid tumors; lower molecular weight conjugates may diffuse better into the solid tumor interior. While low molecular weight conjugates show high efficiency in animal models [73] they have difficulties in clinical settings [10] due to a short intravascular half-life. In a human body where the ratio of tumor to whole body is considerably smaller than in animals a sustained concentration gradient between vasculature and tumor is needed to achieve tumor accumulation.
Ultimately, for a successful clinical study, a good drug delivery system is only one piece of the puzzle. It also needs a right treatment regime and selection of a right patient. Indeed patient selection is becoming an important factor. As cancers often differ depending on their tissue of origin and the stage of the tumor. But even two cases of one type of cancer at the same clinical and pathological stage might differ substantially. Miller et al. [74] suggested an approach for predicting the extent of the EPR effect by tracking the accumulation of a clinically approved tracer nanoparticle, Based on the result, it may have potential to identify the extent of the EPR effect and select a patient.
3.2 The gap between animal model and the complexity of human body
Significant advances have been made in developing various cancer therapies, but the majority of promising new findings failed in clinical trials due to lack of efficacy. There has been a robust discussion on the predictive power of preclinical murine models. To minimize such high-cost-but-low-efficacy development activity, more clinical relevant animal models should be used such as patient-derived tumor explant (PDX) model and genetically engineered mouse model (GEMMs) that more faithfully reflect the morphology, complexity, and heterogeneity of clinical tumors [75]. In addition, for tumors not limited to a specific sex, both male and female animal should be tested, and different type of tumor models may be used to validate the results.
3.3. Lack of a clear commercialization plan
As Flynn pointed out [76], the science is important, and no products will come to market without it; but how that will happen should not be underestimated. It is all about the commercialization.
Unfortunately, there is no defined path with necessary steps for everyone to follow. It is important for successful technology translation to build up a good commercialization plan and set up milestones when translating from bench top to bedside.
For example, to develop KT-1, the most potent 2nd generation HPMA polymer-epirubicin conjugate, to clinical trials, both technique milestones and business milestones are listed below.
To implement this project, however, the first step in current status is to bridge laboratory synthesis and large-scale production.
Bringing new nanomedicines to the market is still a challenge for manufacturers in the pharmaceutical industry due to escalating complexity in the chemistry, manufacturing and controls (CMC) and good manufacturing practice (GMP) requirements. It is worth noting that KT-1 is produced by copolymerization of HPMA with polymerizable EPI derivative (MA-GFLG-EPI) using RAFT polymerization technique, which allows the preparation of well-defined, backbone-degradable diblock HPMA copolymer-drug conjugates in one step. We have tested synthesis of polymeric carriers from 1 mmol up to 50 mmol scale; this process proved scalable with excellent reproducibility. However, to incorporate the active ingredient, the existing conditions and operation parameters need to be modified to fit the solubility requirement. Combination of online monitoring and continuous flow polymerization has great potential to significant improvement of scale-up synthesis [77].
Another vital step is establishing new methodologies for characterization of the conjugates to meet the requirements for regulatory approval. Resources such as the Nanotechnology Characterization Laboratory (NCL) can play an essential role in this process and should be better utilized by investigators.
3.4 Final thoughts
Nanomedicine is the convergence of several technologies, and no one person has all the necessary skills to bring a product to market. Therefore building a team and finding suitable partners is the key to success.
The last but not the least, the investment environment is a critical factor that usually limits the activity. To conduct preclinical evaluations and move the project forward, financial need is a major obstacle to overcome. Publicly grants are limited and become more competing. Therefore various levels of fundraising are imperative and necessary.
Conclusions
Water-soluble HPMA polymer has been widely used as a biomedical material due to its good biocompatibility and non-immunogenicity. However, the great potential of HPMA polymer-drug conjugates as a therapeutic modality for cancer treatment has not been fully exploited. Although early clinical trials confirmed the improved tolerance of these nanomedicines in humans, the lack of degradability limited the molecular weight of these candidates to sub-optimal sizes. This, in turn, curtailed their clinical effectiveness and prevented the system from achieving its full potential.
However, the recent development of second-generation HPMA polymer-drug conjugate technology has profoundly changed the conjugate circulation time in blood and significantly enhanced the treatment efficacy in preclinical animal models. This innovative approach has created amazing flexibility and greater synthetic diversity than previously possible. As such, this platform technology may initiate a new wave of HPMA-based polymer-drug conjugate development. We eagerly await future endeavors exploring the clinical benefit of this new modality.
Figure 7.
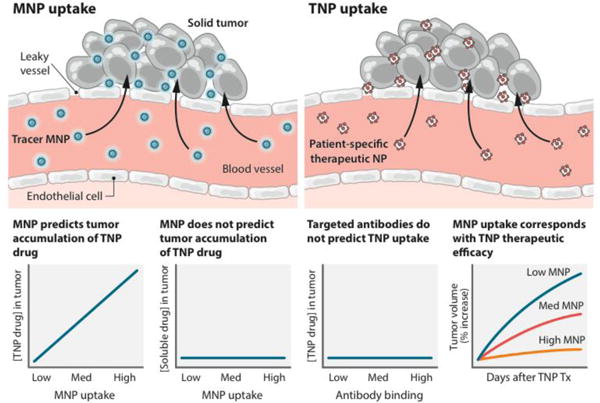
Tumor-specific profiling of the accumulation of nanomedicines using companion nanoparticles. Companion magnetic nanoparticles (MNP) accumulate within tumors, and can be imaged to predict the accumulation of therapeutic nanoparticles (TNP)—a physical effect that is specific to the nanomedicine drugs. The MNPs did not predict free drug accumulation nor did tumor-specific antibodies predict TNP accumulation. Reprinted with permission from [57].
Figure 8.
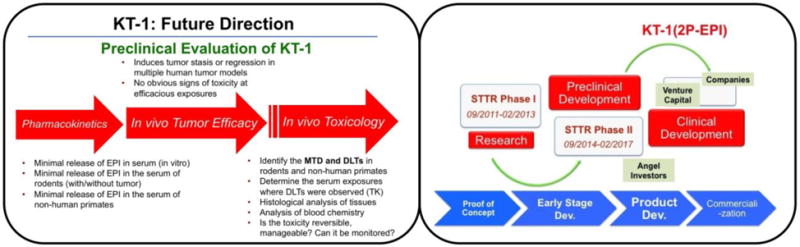
Table 1.
HPMA-based 2nd generation polymer-drug pipeline
| Name | Drug | Application |
|---|---|---|
| KT-1 | Epirubicin (EPI) | Wide spectra for various solid tumor chemotherapy |
| KT-2 | Paclitaxel (PTX) | |
| KT-3 | Gemcitabine (GEM) | |
| KT-4 | Doxorubicin (DOX) | |
| KZ-1 | Docetaxel (DTX) | Hedgehog signaling pathway inhibitor PI3K/mTOR dual inhibitor hematologic malignancies |
| * KZ-2 | Cyclopamine (CYP) | |
| * KZ-3 | GDC0980 (GDC) | |
| KS-1 | Cytarabine | |
|
| ||
| **K-0231 | Rituximab/EPI | B-cell leukemia/lymphoma |
Drug is still in clinical trial.
Acknowledgments
The research was supported in part by NIH grant NIH grant R42 CA156933 from the National Cancer Institute, by Department of Defense Grant W81XWH-13-1-0160 and by the University of Utah Research Foundation. Thanks to D.C. Radford for carefully reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kopeček J. Soluble biomedical polymers. Polim Med. 1977;7:191–221. [PubMed] [Google Scholar]
- 2.Yang J, Kopeček J. Encyclopedia of Polymeric Nanomaterials. Springer; Berlin Heidelberg: 2014. Polymeric drugs; pp. 1–9. [Google Scholar]
- 3.De Duve C, De Barsy T, Poole B, Trouet A, Tulkens P, van Hoof F. Lysosomotropic agents. Biochem Pharmacol. 1974;23:2495–531. doi: 10.1016/0006-2952(74)90174-9. [DOI] [PubMed] [Google Scholar]
- 4.Kopeček J, Bažilová H. Poly[N-(2-hydroxypropyl)methacrylamide]. 1. Radical polymerization and copolymerization. Eur Polym J. 1973;9:7–14. [Google Scholar]
- 5.Kopeček J, Cífková I, Rejmanová P, Strohalm J, Obereigner B, Ulbrich K. Polymers containing enzymatically degradable bonds. 4. Preliminary experiments in vivo. Makromol Chem. 1981;182:2941–49. [Google Scholar]
- 6.Rejmanová P, Pohl J, Baudyš M, Kostka V, Kopeček J. Polymers containing enzymatically degradable bonds. 8. Degradation of oligopeptide sequences in N-(2-hydroxypropyl)methacrylamide copolymers by bovine spleen cathepsin B. Makromol Chem. 1983;184:2009–20. [Google Scholar]
- 7.Říhová B, Kopečková P, Strohalm J, Rossmann P, Větvička V, Kopeček J. Antibody directed affinity therapy applied to the immune system: In vivo effectiveness and limited toxicity of daunomycin conjugates to HPMA copolymers and targeting antibody. Clin Immunol Immunopathol. 1988;46:100–14. doi: 10.1016/0090-1229(88)90010-4. [DOI] [PubMed] [Google Scholar]
- 8.Duncan R, Kopečková P, Strohalm J, Hume IC, Lloyd JB, Kopeček J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. 2. Evaluation of daunomycin conjugates in vivo against L1210 leukaemia. Brit J Cancer. 1988;57:47–156. doi: 10.1038/bjc.1988.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **9.Kopeček J, Kopečková P. HPMA copolymers: Origins, early developments, present, and future. Adv Drug Delivery Rev. 2010;62:122–49. doi: 10.1016/j.addr.2009.10.004. A seminal review from pioneering work to the development of HPMA polymeric carriers that have since been used in multiple biomedical applications such as polymer-drug conjugates and hybrid hydrogels. It also described a biologically inspired new strategy-drug-free macromolecular therapeutics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Vasey PA, Kaye SB, Morrison R, Twelves C, Wilson P, Duncan R, et al. Phase I clinical and pharmacokinetic study of PK1 [N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first member of a new class of chemotherapeutic agents-drug-polymer conjugates Cancer Research Campaign Phase I/II Committee. Clin Cancer Res. 1999;5:83–94. First-in-human testing of a polymeric prodrug for cancer chemotherapy, demonstrating the greatly improved tolerance of drug-delivery system compared with free drug. [PubMed] [Google Scholar]
- 11.Seymour LW, Ferry DR, Kerr DJ, Rea D, Whitlock M, Poyner R, et al. Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the treatment of breast, lung and colorectal cancer. Int J Oncol. 2009;34:1629–36. doi: 10.3892/ijo_00000293. [DOI] [PubMed] [Google Scholar]
- 12.Seymour LW, Ferry DR, Anderson D, Hesslewood S, Julyan PJ, Poyner R, et al. (2002) Hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J Clin Oncol. 2002;20:1668–76. doi: 10.1200/JCO.2002.20.6.1668. [DOI] [PubMed] [Google Scholar]
- 13.Meerum Terwogt JM, Bokkel Huinink WW, Schellens JH, Schot M, Mandjes IA, Zurlo MG, et al. Phase I clinical and pharmacokinetic study of PNU166945, a novel water-soluble polymer-conjugated prodrug of paclitaxel. Anticancer Drugs. 2001;12:315–23. doi: 10.1097/00001813-200104000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Schoemaker NE, van Kesteren C, Rosing H, Jansen S, Swart M, Lieverst J, et al. A phase I and pharmacokinetic study of MAG-CPT, a water-soluble polymer conjugate of camptothecin. Br J Cancer. 2002;87:608–14. doi: 10.1038/sj.bjc.6600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bissett D, Cassidy J, de Bono JS, Muirhead F, Main M, Robson L, et al. Phase I and pharmacokinetic (PK) study of MAG-CPT (PNU 166148): A polymeric derivative of camptothecin (CPT) Br J Cancer. 2004;91:50–55. doi: 10.1038/sj.bjc.6601922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rademaker-Lakhai JM, Terret C, Howell SB, Baud CM, De Boer RF, Pluim D, et al. A Phase I and pharmacological study of the platinum polymer AP5280 given as an intravenous infusion once every 3 weeks in patients with solid tumors. Clin Cancer Res. 2004;10:3386–95. doi: 10.1158/1078-0432.CCR-03-0315. [DOI] [PubMed] [Google Scholar]
- 17.Gianasi E, Wasil M, Evagorou EG, Keddle A, Wilson G, Duncan R. HPMA copolymer platinates as novel antitumour agents: in-vitro properties, pharmacokinetics and antitumour activity in vivo. Eur J Cancer. 1999;35:994–1002. doi: 10.1016/s0959-8049(99)00030-1. [DOI] [PubMed] [Google Scholar]
- 18.Campone M, Rademaker-Lakhai JM, Bennouna J, Howell SB, Nowotnik DP, Beijnen JH, et al. Phase I and pharmacokinetic trial of AP5346, a DACH-platinum-polymer conjugate, administered weekly for three out of every 4 weeks to advanced solid tumor patients. Cancer Chemother Pharmacol. 2007;60:523–33. doi: 10.1007/s00280-006-0397-0. [DOI] [PubMed] [Google Scholar]
- 19.Nowotnik DP, Cvitkovic E. ProLindac (AP5346): A review of the development of an HPMA DACH platinum polymer therapeutic. Adv Drug Deliv Rev. 2009;61:1214–19. doi: 10.1016/j.addr.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 20.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–60. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 21.Vicent MJ, Ringsdorf H, Duncan R. Polymer therapeutics: clinical applications and challenges for development. Adv Drug Deliv Rev. 2009;61:1117–20. doi: 10.1016/j.addr.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Duncan R, Vicent MJ. Polymer therapeutics – prospects for 21st century: the end of the beginning. Adv Drug Deliv Rev. 2013;65:60–70. doi: 10.1016/j.addr.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Haag R, Kratz F. Polymer therapeutics: Concepts and applications. Angew Chem Int Ed. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- 24.Bildstein L, Dubernet C, Couvreur P. Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev. 2011;63:3–23. doi: 10.1016/j.addr.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 25.Zhou Y, Kopeček J. Biological rationale for the design of polymeric anti-cancer nanomedicines. J Drug Targeting. 2013;21:1–26. doi: 10.3109/1061186X.2012.723213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kopeček J. Polymer – drug conjugates: Origins, progress to date and future directions. Adv Drug Delivery Rev. 2013;65:49–59. doi: 10.1016/j.addr.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ulbrich K, Šubr V. Structural and chemical aspects of HPMA copolymers as drug carriers. Adv Drug Deliv Rev. 2010;62:150–66. doi: 10.1016/j.addr.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Kopeček J. Design of smart HPMA copolymer-based nanomedicines. J Controlled Release. 2016;240:9–23. doi: 10.1016/j.jconrel.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. doi: 10.1016/j.addr.2015.01.002. [DOI] [PubMed] [Google Scholar]
- *30.Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H. Early Phase Tumor Accumulation of Macromolecules: A Great Difference in Clearance Rate between Tumor and Normal Tissues. Jpn J Cancer Res. 1998;89:307–14. doi: 10.1111/j.1349-7006.1998.tb00563.x. Excellent study that highlights the effect of molecular weight of HPMA polymer-drug conjugates on extravasation and accumulation in tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seymour LW, Ulbrich K, Steyger PS, Brereton M, Šubr V, Strohalm J, et al. Tumour tropism and anti-cancer efficacy of polymer-based doxorubicin prodrugs in the treatment of subcutaneous murine B16F10 melanoma. Brit J Cancer. 1994;70:636–41. doi: 10.1038/bjc.1994.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minko T, Kopečková P, Kopeček J. Efficacy of chemotherapeutic action of HPMA copolymer-bound doxorubicin in a solid tumor model of ovarian carcinoma. Int J Cancer. 2000;86:108–17. doi: 10.1002/(sici)1097-0215(20000401)86:1<108::aid-ijc17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Bilim V. Technology evaluation: PK1, Pfizer/Cancer Research UK. Curr Opinion Molecular Ther. 2003;5:326–30. [PubMed] [Google Scholar]
- 34.Williams R. Discontinued drugs in 2008: Oncology drugs. Exp Opinion Invest Drugs. 2009;18:1581–94. doi: 10.1517/13543780903151806. [DOI] [PubMed] [Google Scholar]
- 35.Etrych T, Chytil P, Jelínková M, Říhová B, Ulbrich K. Synthesis of HPMA copolymers containing doxorubicin bound via a hydrazone linkage. Effect of spacer on drug release and in vitro cytotoxicity. Macromol Biosci. 2002;2:43–52. [Google Scholar]
- 36.Dozono H, Yanazume S, Nakamura H, Etrych T, Chytil P, Ulbrich K, et al. HPMA copolymer-conjugated pirarubicin in multimodal treatment of a patient with stage IV prostate cancer and extensive lung and bone metastases. Target Oncol. 2016;11:101–6. doi: 10.1007/s11523-015-0379-4. [DOI] [PubMed] [Google Scholar]
- 37.Říhová B, Kubáčková M. Clinical implications of N-(2-hydroxypropyl)-methacrylamide copolymers. Curr Pharmaceutical Biotechnol. 2003;4:311–22. doi: 10.2174/1389201033489711. [DOI] [PubMed] [Google Scholar]
- 38.Říhová B. Clinical experience with anthracycline antibiotics-HPMA copolymer-human immunoglobulin conjugates. Adv Drug Deliv Rev. 2009;61:1149–58. doi: 10.1016/j.addr.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 39.Říhová B, Strohalm J, Kubáčková M, Jelínková M, Hovorka O, Kovář M, et al. Acquired and specific immunological mechanisms co-resposible for efficacy of polymer-bound drugs. J Control Release. 2002;78:97–104. doi: 10.1016/s0168-3659(01)00489-8. [DOI] [PubMed] [Google Scholar]
- 40.Říhová B, Kovář L, Kovář M, Hovorka O. Cytotoxicity and immunostimulation: double attack on cancer cells with polymeric therapeutics. Trends Biotechnol. 2009;27:11–17. doi: 10.1016/j.tibtech.2008.10.006. Excellent example how HPMA copolymers are able to not only directly destroy cancer cells but also stimulate systemic tumor-specific response. [DOI] [PubMed] [Google Scholar]
- 41.Cheah HY, Šarenac O, Arroyo JJ, Vasić M, Lozić M, Glumac S, et al. Hemodynamic effects of HPMA copolymer based doxorubicin conjugate: A randomized controlled and comparative spectral study in conscious rats. Nanotoxicology. 2017 doi: 10.1080/17435390.2017.1285071. http://dx.doi.org/10.1080/17435390.2017.1285071. [DOI] [PMC free article] [PubMed]
- 42.Říhová B, Strohalm J, Hovorka O, Šubr V, Etrych T, Chytil P, Pola R, Plocová D, Bouček J, Ulbrich K. Doxorubicin release is not a prerequisite for the in vitro cytotoxicity of HPMA-based pharmaceuticals: In vitro effect of extra drug-free GlyPheLeuGly sequences. J Controlled Release. 2008;127:110–20. doi: 10.1016/j.jconrel.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Wang D, Kopečková P, Minko T, Nanayakkara V, Kopeček J. Synthesis of star-like N-(2-hydroxypropyl)methacrylamide copolymers – potential drug carriers. Biomacromolecules. 2000;1:313–9. doi: 10.1021/bm0000236. [DOI] [PubMed] [Google Scholar]
- 44.Chytil P, Koziolová E, Janoušková O, Kostka L, Ulbrich K, Etrych T. Synthesis and properties of star HPMA copolymer nanocarriers synthesized by RAFT polymerization designed for selective anticancer drug delivery and imaging. Macromol Biosci. 2015;15:839–50. doi: 10.1002/mabi.201400510. [DOI] [PubMed] [Google Scholar]
- 45.Dvořák M, Kopečková P, Kopeček J. High-molecular weight HPMA copolymer-adriamycin conjugates. J Control Release. 1999;60:321–32. doi: 10.1016/s0168-3659(99)00087-5. [DOI] [PubMed] [Google Scholar]
- *46.Shiah JG, Dvořák M, Kopečková P, Sun Y, Peterson CM, Kopeček J. Biodistribution and antitumor efficacy of long-circulating N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates in nude mice. Eur J Cancer. 2001;37:131–39. doi: 10.1016/s0959-8049(00)00374-9. This excellent study demonstrated the higher the molecular weight of the conjugate, the more accumulation of anticancer drug into tumors, providing a proof for the use of large-size, long-circulating therapeutic agents in cancer treatment. [DOI] [PubMed] [Google Scholar]
- *47.Scales CV, Vasilieva YA, Convertine AJ, Lowe AB, McCormick CL. Direct, controlled synthesis of the nonimmunogenic, hydrophilic polymer, poly(N-(2-hydroxypropyl)methacrylamide) via RAFT in aqueous media. Biomacromolecules. 2005;6:1846–50. doi: 10.1021/bm0503017. A seminal work on the controlled living polymerization of HPMA that essentially laid the foundation for the use of RAFT polymerization technique to synthesize well-defined HPMA copolymers with narrow polydispersity and end-functional group(s) [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Luo K, Pan H, Kopečková P, Kopeček J. Synthesis of biodegradable multiblock copolymers by click coupling of RAFT-generated heterotelechelic polyHPMA conjugates. Reactive Functional Polym. 2011;71:294–302. doi: 10.1016/j.reactfunctpolym.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luo K, Yang J, Kopečková P, Kopeček J. Biodegradable multiblock N-(2-hydroxypropyl)methacrylamide copolymers via reversible addition-fragmentation chain transfer polymerization and click chemistry. Macromolecules. 2011;44:2481–88. doi: 10.1021/ma102574e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *50.Pan H, Yang J, Kopečková P, Kopeček J. Backbone degradable multiblock N-(2-hydroxypropyl)methacrylamide copolymer conjugates via reversible addition-fragmentation chain transfer polymerization and thiol-ene coupling reaction. Biomacromolecules. 2011;12:247–52. doi: 10.1021/bm101254e. A highly innovative approach to synthesis of diblock biodegradable HPMA polymer-drug conjugates in one step, and its potential for use in preparation of multiblock biodegradable long-circulating HPMA-based conjugates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Zhang R, Pan H, Li Y, Fang Y, Zhang L, et al. Backbone degradable HPMA copolymer conjugates with gemcitabine and paclitaxel: Impact of molecular weight on activity toward human ovarian carcinoma xenografts. Mol Pharmaceutics. 2017;14:1384–94. doi: 10.1021/acs.molpharmaceut.6b01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fox ME, Szoka FC, Fréchet JM. Soluble polymer carriers for the treatment of cancer: the importance of molecular architecture. Acc Chem Res. 2009;42:1141–51. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pan H, Sima M, Yang J, Kopeček J. Synthesis of long-circulating backbone degradable HPMA copolymer-doxorubicin conjugates and evaluation of molecular weight dependent antitumor efficacy. Macromol Biosci. 2013;13:155–60. doi: 10.1002/mabi.201200353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang J, Zhang R, Radford DC, Kopeček J. FRET-Trackable Biodegradable HPMA Copolymer-Epirubicin Conjugates for Ovarian Carcinoma Therapy. J Controlled Release. 2015;218:36–44. doi: 10.1016/j.jconrel.2015.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plosker GL, Faulds D. Epirubicin: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in cancer chemotherapy. Drugs. 1993;45:788–856. doi: 10.2165/00003495-199345050-00011. [DOI] [PubMed] [Google Scholar]
- 56.Yang J, Kopeček J. Polymeric biomaterials and nanomedicines. J Drug Deliv Sci Technol. 2015;30:318–30. doi: 10.1016/j.jddst.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krinick NL, Sun Y, Joyner D, Spikes JD, Straight RC, Kopeček J. A polymeric drug delivery system for the simultaneous delivery of drugs activatable by enzymes and/or light. J Biomat Sci Polym Ed. 1994;5:303–24. doi: 10.1163/156856294x00040. [DOI] [PubMed] [Google Scholar]
- 58.Shiah JG, Sun Y, Kopečková P, Peterson CM, Straight RC, Kopeček J. Combination chemotherapy and photodynamic therapy of targetable N-(2-hydroxypropyl)-methacrylamide copolymer – doxorubicin/mesochlorin e6 – OV- TL16 antibody immunoconjugates. J Control Release. 2001;74:249–53. doi: 10.1016/s0168-3659(01)00325-x. [DOI] [PubMed] [Google Scholar]
- 59.Larson N, Yang J, Ray A, Cheney DL, Ghandehari H, Kopeček J. Biodegradable multiblock poly(N-2-hydroxypropyl)methacrylamide gemcitabine and paclitaxel conjugates for ovarian cancer cell combination treatment. Int J Pharmaceutics. 2013;454:435–43. doi: 10.1016/j.ijpharm.2013.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang R, Yang J, Sima M, Zhou Y, Kopeček J. Sequential combination therapy of ovarian cancer with backbone degradable HPMA copolymer paclitaxel and gemcitabine conjugates. Proc Natl Acad Sci USA. 2014;111:12181–6. doi: 10.1073/pnas.1406233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meng H, Wang M, Liu H, Liu X, Situ A, Wu B, et al. Use of lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano. 2015;9:3540–7. doi: 10.1021/acsnano.5b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y, Yang J, Rhim J, Kopeček J. HPMA copolymer-based combination therapy toxic to both prostate cancer stem/progenitor cells and differentiated cells induces durable anti-tumor effects. J Control Release. 2013;172:946–53. doi: 10.1016/j.jconrel.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Law CL, Cerveny CG, Gordon KA, Klussman K, Mixan BJ, Chace DF, Meyer DL, Doronina SO, Siegall CB, Francisco JA, Senter PD, Wahl AF. Efficient elimination of B-lineage lymphomas by anti-CD20 – auristatin conjugates. Clin Cancer Res. 2004;10:7842–7851. doi: 10.1158/1078-0432.CCR-04-1028. [DOI] [PubMed] [Google Scholar]
- 64.Sapra P, Allen TM. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody targeted liposomal drugs. Cancer Res. 2002;62:7190–94. [PubMed] [Google Scholar]
- 65.Lidický O, Janoušková O, Strohalm J, Alam M, Klener P, Etrych T. Anti-lymphoma efficacy comparison of anti-CD20 monoclonal antibody-targeted and non-targeted star-shaped polymer-prodrug conjugates. Molecules. 2015;20:19849–64. doi: 10.3390/molecules201119664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang L, Fang Y, Kopeček J, Yang J. A new construct of antibody-drug conjugates for treatment of non-Hodgkin’s lymphoma. Eur J Pharm Sci. 2017;103:36–46. doi: 10.1016/j.ejps.2017.02.034. [DOI] [PubMed] [Google Scholar]
- 67.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: Progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maeda H. SMANCS and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev. 2001;46:169–85. doi: 10.1016/s0169-409x(00)00134-4. [DOI] [PubMed] [Google Scholar]
- *69.Venditto VJ, Szóka FC. Cancer nanomedicines: so many papers and so few drugs! Adv Drug Deliv Rev. 2013;65:80–8. doi: 10.1016/j.addr.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *70.Lammers T, Kiessling F, Hennink W, Storm G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175–87. doi: 10.1016/j.jconrel.2011.09.063. One of the most comprehensive reviews on the subject of drug targeting to tumors. [DOI] [PubMed] [Google Scholar]
- 71.Danhier F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J Control Release. 2016;244:108–21. doi: 10.1016/j.jconrel.2016.11.015. [DOI] [PubMed] [Google Scholar]
- 72.Clark AJ, Wiley DT, Zuckerman JE, Webster P, Chao J, Lin J, et al. CRLX101 nanoparticles localize in human tumors and not in adjacent, nonneoplastic tissue after intravenous dosing. Proc Natl Acad Sci USA. 2016;113:3850–4. doi: 10.1073/pnas.1603018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Camacho KM, Menegatti S, Mitragotri S. Low-molecular-weight polymer-drug conjugates for synergistic anticancer activity of camptothecin and doxorubicin combinations. Nanomedicine (Lond) 2016;11:1139–51. doi: 10.2217/nnm.16.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miller MA, Gadde S, Pörschke C, Engblom C, Sprachman MM, Kohler RH, et al. Predicting therapeutic nanomedicine efficacy using a companion MR imaging nanoparticle. Sci Transl Med. 2015;7:314ra183. doi: 10.1126/scitranslmed.aac6522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Day C-P, Merlino G, Dyke TV. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163:39–53. doi: 10.1016/j.cell.2015.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Flynn T, Wei C. The pathway to commercialization for nanomedicine. Nanomed Nanotechnol Biol Med. 2005;1:47–51. doi: 10.1016/j.nano.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 77.Junkers T. Precision polymer design in microstructured flow reactors: Improved control and first upscale at once. Macromol Chem Phys. 2017;218:1600421. [Google Scholar]