

Myofibroblasts are key effector cells in tissue stiffening and fibrosis progression, but the contribution of cells undergoing epithelial–mesenchymal transition (EMT) is unclear. Unlike EMT, myofibroblasts contribute to tissue stiffening through their contractility and expression of fibrillar collagens, which is associated with aberrant FAK (Y397) hyperactivation.
Abstract
The contribution of epithelial-to-mesenchymal transition (EMT) to the profibrotic stiff microenvironment and myofibroblast accumulation in pulmonary fibrosis remains unclear. We examined EMT-competent lung epithelial cells and lung fibroblasts from control (fibrosis-free) donors or patients with idiopathic pulmonary fibrosis (IPF), which is a very aggressive fibrotic disorder. Cells were cultured on profibrotic conditions including stiff substrata and TGF-β1, and analyzed in terms of morphology, stiffness, and expression of EMT/myofibroblast markers and fibrillar collagens. All fibroblasts acquired a robust myofibroblast phenotype on TGF-β1 stimulation. Yet IPF myofibroblasts exhibited higher stiffness and expression of fibrillar collagens than control fibroblasts, concomitantly with enhanced FAKY397 activity. FAK inhibition was sufficient to decrease fibroblast stiffness and collagen expression, supporting that FAKY397 hyperactivation may underlie the aberrant mechanobiology of IPF fibroblasts. In contrast, cells undergoing EMT failed to reach the values exhibited by IPF myofibroblasts in all parameters examined. Likewise, EMT could be distinguished from nonactivated control fibroblasts, suggesting that EMT does not elicit myofibroblast precursors either. Our data suggest that EMT does not contribute directly to the myofibroblast population, and may contribute to the stiff fibrotic microenvironment through their own stiffness but not their collagen expression. Our results also support that targeting FAKY397 may rescue normal mechanobiology in IPF.
INTRODUCTION
Physiologic tissue stiffness supports tissue-specific functions and is maintained in normal homeostatic conditions. In contrast, normal tissue stiffness becomes chronically increased in a variety of prevalent diseases, including fibrosis, sclerosis, and cancer, where it is increasingly pointed to as a major driving force of disease progression (Ingber, 2003; Butcher et al., 2009; Ho et al., 2014). In the context of pulmonary physiopathology, it is well known that lungs are soft and elastic organs, which enables the dramatic cyclic volume changes required for breathing. In contrast, normal pulmonary stiffness can be increased up to 30-fold in pulmonary fibrotic disorders concomitantly with a loss of normal pulmonary function and architecture (Liu et al., 2010; Hinz, 2012). Of note, such tissue stiffening remains currently irreversible in idiophatic pulmonary fibrosis (IPF), which is a very aggressive disease characterized by the progressive and chronic replacement of normal pulmonary tissue by a stiff dysfunctional scar in the absence of any known cause (King et al., 2011; Hinz, 2012). Intriguingly, unlike other pulmonary fibrotic disorders, IPF remains frequently lethal due to suboptimal therapeutic options, with a median survival of only ∼2–5 yr from time of diagnosis (Blackwell et al., 2014). Moreover, it is worth noting that IPF is a rare disease that affects ∼5 million patients worldwide, yet it has a prevalence of ∼1/5000 people that continues to rise (King et al., 2011). Accordingly, there is an urgent need in understanding the critical mechanobiological alterations that drive this devastating disease.
IPF progression is associated with the appearance of areas of active fibrosis referred to as fibroblast foci, which are rich in activated fibroblasts/myofibroblasts in the background of abundant fibrillar collagens (Willis et al., 2006; Scotton and Chambers, 2007; King et al., 2011). Myofibroblasts have been identified as major drivers of the progression of IPF and other fibrotic disorders (Hinz, 2012; Ho et al., 2014). Likewise, myofibroblasts have been pointed to as major effector cells of tissue stiffening, since they combine the extracellular matrix (ECM)-synthesizing features of fibroblasts with the cytoskeletal characteristics of contractile smooth muscle cells. Indeed, the most common myofibroblast markers are the abundant extracellular deposition of fibrillar collagens and the intracellular overexpression of alpha-smooth muscle actin (α-SMA) (King et al., 2011; Hinz et al., 2012). Although the processes underlying the aberrant accumulation of myofibroblasts in IPF remain poorly understood, it has been reported that transforming growth factor beta 1 (TGF-β1), which is the most potent fibroblast activator cytokine characterized to date, is frequently upregulated in fibrotic lung diseases (Willis et al., 2006; Scotton and Chambers, 2007; King et al., 2011). Moreover, studies using hydrogel-based culture substrata with tunable elasticity have revealed that TGF-β1 can induce α-SMA expression in stiff but not in soft substrata (Arora et al., 1999; Balestrini et al., 2012; Hinz, 2012), thereby underscoring the prominent role of tissue stiffening in organ fibrosis.
In addition to α-SMA up-regulation, there is growing evidence that matrix stiffening alone can trigger the expression of collagens and other profibrotic factors independently of TGF-β1 in fibroblasts (Ebihara et al., 2000; Liu et al., 2010; Thannickal et al., 2014). Moreover, we recently showed that fibrosis-like extracellular rigidities alone can enhance the growth and survival of lung fibroblasts through β1 integrin and pFAKY397 signaling (Puig et al., 2015). Conversely, it was reported that extracellular stiffening strongly suppressed fibroblast expression of cyclooxygenase-2 (COX-2) and synthesis of prostaglandin E2 (PGE2), which is an autocrine inhibitor of fibrogenesis (Liu et al., 2010). More recently it was described that gradients of matrix stiffness reminiscent of those occuring in fibrotic tissues can attract fibroblasts through durotaxis (Liu et al., 2010; Lagares et al., 2016). Collectively, these previous in vitro studies support that tissue stiffening per se provides a strong profibrotic microenvironment. In agreement with this interpretation, a panel of basic, translational, and clinical IPF researchers gathered in 2014 by the National Heart, Lung and Blood Institute in the United States highlighted the need to “elucidate the mechanisms of matrix stiffening and cell responses to increased stiffness” as an important future direction in IPF research (Blackwell et al., 2014).
Given the prominent role of fibrogenic myofibroblasts in tissue stiffening and overall disease progression in IPF and other fibrotic disorders, there is increasing interest in defining their cellular source. It remains widely accepted that lung myofibroblasts arise from the proliferation and migration of resident fibroblasts. In addition, alveolar epithelial cells undergoing epithelial-to-mesenchymal transition (EMT) have been pointed to as potential contributors to the myofibroblast population (Willis et al., 2006; Scotton and Chambers, 2007; Hinz, 2012). During EMT, epithelial cells undergo a dramatic phenotypic change that includes loosening cell–cell adhesions, down-regulating epithelial markers, up-regulating mesenchymal markers and pro-EMT transcription factors, and acquiring an elongated fibroblast-like morphology (Willis et al., 2006; Scotton and Chambers, 2007; Moreno-Bueno et al., 2009). In support of the EMT source of myofibroblasts, studies in vitro have revealed that EMT may be induced by TGF-β1 (Willis et al., 2006; Hinz, 2012; O’Connor and Gomez, 2014). Moreover, studies using in vivo models of pulmonary fibrosis support that a significant fraction of fibroblasts in fibrotic lesions may be epithelial derived (Kim et al., 2006; Wu et al., 2007; Kim et al., 2009; Degryse et al., 2010). Likewise, cells coexpressing epithelial and mesenchymal markers have been reported in IPF biopsies (Willis et al., 2005; Kim et al., 2006; Yamaguchi et al., 2017). However, recent lineage tracing experiments have failed to unambiguously provide evidence of EMT in in vivo models of lung fibrosis (Rock et al., 2011; Ho et al., 2014), and technical concerns on previous EMT analyses of IPF biopsies have been raised (Rock et al., 2011). Similar controversial data have been reported in liver and kidney fibrosis (Humphreys et al., 2010; Bartis et al., 2014; Giannandrea and Parks, 2014; Ho et al., 2014). On the other hand, it should be mentioned that previous EMT studies have focused on the expression of epithelial and mesenchymal markers, whereas functional mechanical measurements have been largely absent. Accordingly, it remains unclear what the contribution of EMT is to the profibrotic stiff microenvironment and overall myofibroblast accumulation in IPF.
To shed light on these questions, we gathered a panel of EMT-competent lung epithelial cells and primary lung fibroblasts derived from either fibrosis-free donors (used as control cell models for resident fibroblasts) or IPF patients. To mimic a profibrotic microenvironment, we cultured cells on stiff culture substrata in the presence of exogenous TGF-β1 at concentrations capable of inducing both EMT and fibroblast activation. The phenotypic effects of the profibrotic microenvironment were analyzed in terms of morphology and expression of both EMT and myofibroblast markers. To assess the potential impact of pro-fibrotic stimuli on tissue mechanics, we analyzed cellular stiffness through nanoindentation measurements by atomic force microscopy (AFM) as well as the expression of fibrillar collagens.
Results
TGF-β1 induces EMT in selected lung epithelial cell lines but fails to match the vimentin expression levels and morphological features of IPF-derived fibroblasts
Unlike fibroblasts, isolating and maintaining human lung epithelial cells remains rather challenging and poorly efficient (Shannon et al., 1987; Yu et al., 2007). To overcome these limitations, we selected a panel of lung epithelial cell lines previously reported to express levels of epithelial and mesenchymal markers indicative of either a high (H441, PC9) or a moderate/low (A549, H1975) epithelial phenotype in basal conditions, which has been associated with poor or strong EMT competence, respectively (Thomson et al., 2005; Yauch et al., 2005; Zhang et al., 2011; Jolly et al., 2016). Although these four cell lines were originally derived from lung adenocarcinomas, they were selected because they are accepted models of either poor or strong EMT, and there is extensive evidence that major EMT hallmarks are shared between normal epithelial cells during development and selected transformed cells (Moreno-Bueno et al., 2009). Furthermore, the A549 cell line in particular is a widely used model of alveolar epithelial-like behavior in noncancer-oriented studies (Lieber et al., 1976). To provide cells a profibrotic microenvironment, both epithelial cell lines and primary fibroblasts were cultured on tissue culture plastic for 24 h in culture medium containing 10% fetal bovine serum (FBS), thereby facilitating cell attachment to a rigid substrata (Paszek et al., 2005). Next, cell cultures were washed, maintained in serum-free medium for 24 h to growth arrest and synchronize them, and, finally, stimulated for 3 d with serum-free medium (used as negative control) or 5 ng/ml TGF-β1, which is a master regulator of fibrosis in the lung and other organs (Willis et al., 2006; Scotton and Chambers, 2007). The latter conditions have been extensively reported to be sufficient to induce both fibroblast activation as well as EMT in A549 and other epithelial cell lines (Zhang et al., 2011; Gabasa et al., 2013; Vizoso et al., 2015; Yamaguchi et al., 2017).
As expected, immunofluorescence analysis revealed that TGF-β1 stimulation induced EMT selectively in some of the epithelial cell lines (Figure 1A and Supplemental Figure S1). Specifically, TGF-β1 increased the average expression of the mesenchymal marker vimentin by ∼50–100% in A549 and H1975 epithelial cells, whereas vimentin remained virtually unaltered in H441 and PC9 cell lines (Figure 1B). Likewise, vimentin levels did not exhibit a significant fold increase in primary fibroblasts derived from different randomly selected control donors or IPF patients on TGF-β1 treatment (Figure 1B), in agreement with previous observations (Goldberg et al., 2007). Conversely, TGF-β1 reduced the expression of the epithelial marker E-cadherin further in A549 and H1975, whereas H441 and PC9 cells retained high/moderate E-cadherin expression levels (Figure 1, A and B, and Supplemental Figure S1). The down-regulation of E-cadherin by TGF-β1 in A549 cells was further confirmed by Western blotting (Supplemental Figure S1). In contrast, primary myofibroblasts were negative for E-cadherin. On the other hand, the absolute intensity values of vimentin fluorescence after TGF-β1 treatment shown in Figure 1C were somewhat cell line (in epithelial cells) and patient (in fibroblasts) dependent, which may be associated with the particular genetic alterations of each cell line and the intrinsic patient variability, respectively, as well as with potential limitations in our image processing. Yet, despite the strong EMT phenotype in A549 and H1975 cells supported by the large fold induction of vimentin (Figure 1B), their corresponding absolute vimentin levels in the presence of TGF-β1 were less than half of those found in IPF fibroblasts and control fibroblasts activated with TGF-β1 as shown in Figure 1C. In addition, TGF-β1 treatment held the number of primary fibroblasts; likewise, it maintained or decreased the population of epithelial cells in a cell-line–dependent manner (Supplemental Figure S2), in agreement with previous results (Wang et al., 2015). These observations are consistent with the universal cytostatic effects and selective apoptotic function of TGF-β1 in epithelial cells (Siegel and Massague, 2003). All these results on epithelial cells are consistent with previous observations (Yauch et al., 2005; Yamaguchi et al., 2017) and support that TGF-β1 elicits a strong EMT phenotype in A549 and H1975 cells but not in H441 and PC9 cells. Accordingly, these cell lines were used as models of strong (A549, H1975) and weak (H441, PC9) EMT thereafter.
FIGURE 1:

Immunofluorescence analysis of epithelial (E-cadherin) and mesenchymal (vimentin) markers in EMT-competent lung epithelial cells and primary lung fibroblasts. (A) Representative immunofluorescence images of E-cadherin (left) and vimentin (right) in epithelial cell lines (H441, A549) and primary fibroblasts obtained from randomly selected control (fibrosis-free) or IPF patients (referred by the three-digit number following # symbol) cultured for 3 d in the absence or presence of 5 ng/ml TGF-β1. Images were obtained with a ×20 objective (seven images/condition). Blue indicates DAPI staining here and thereafter. Scale bar here, 50 μm. Representative images from other epithelial cell lines (PC9, H1975) and patients are shown in Supplemental Figure S1. (B) Ratio of the average vimentin fluorescence intensity per cell obtained in the presence or absence of TGF-β1 in a panel of epithelial cell lines and primary fibroblasts from randomly selected control and IPF patients. Horizontal gray dashed line corresponds to no fold change (ratio = 1), whereas horizontal solid blue lines correspond to the average values for each cellular model (i.e., epithelial cells exhibiting weak or strong EMT, n = 2; and primary fibroblasts from control or IPF patients, n = 3). (C) Absolute average vimentin fluorescence intensity per cell obtained on stimulation with TGF-β1 in the same cell models shown in B. *p ≤ 0.05 with respect to 1 were determined by Student’s t test (here and hereafter).
A common feature of EMT is a morphological change in which epithelial cells exhibit an elongated fibroblast-like shape (Moreno-Bueno et al., 2009). To assess to what extent the shape of epithelial cell lines stimulated with TGF-β1 resembled that of actual lung fibroblasts, we conducted a morphometric analysis of phase contrast images of cells on a 3-d treatment with TGF-β1. In the absence of exogenous TGF-β1, both H441 and A549 cells exhibited cell–cell adhesions and a roundish shape with limited spreading, whereas all primary (nonactivated) fibroblasts exhibited the usual spindle shape and marked elongation (Figure 2A and Supplemental Figure S3). On TGF-β1 stimulation, A549 cells scattered and exhibited a lower value of circularity (adimensional index constrained between 0 and 1, where 1 corresponds to a perfect circle) and a higher elongation (ratio between cell length and width) and spreading (cell projected area) than H441 cells (Figure 2, B–D). The shape changes elicited by TGF-β1 in A549 are hallmarks of EMT (Moreno-Bueno et al., 2009; O’Connor and Gomez, 2014) and were also observed in H1975 but not in PC9. Spreading analysis revealed a progressive increase in the average cell area in cells undergoing EMT (∼1500 μm2), control myofibroblasts (∼2500 μm2), and IPF myofibroblasts (∼3000 μm2) (Figure 2D). The latter differences in spreading attained statistical significance between each EMT model compared with both sources of primary myofibroblasts but not between IPF and control myofibroblasts (Figure 2D).
FIGURE 2:

Morphometric analysis of EMT-competent lung epithelial cells and primary lung fibroblasts stimulated with the profibrotic cytokine TGF-β1 for 3 d. (A) Representative phase contrast images of epithelial cells (H441, A549) and primary fibroblasts obtained from randomly selected control or IPF patients in the absence or presence of TGF-β1. Images were obtained with a ×20 objective (two to three images/condition). Scale bar, 50 μm. Representative images from other epithelial cell lines (PC9, H1975) and patients are shown in Supplemental Figure S3. (B–D) Average cell values of the circularity (B), elongation (C), and spreading (D) assessed on stimulation with TGF-β1 in epithelial cells undergoing weak or strong EMT and in primary fibroblasts from control or IPF patients. On average, 30 cells were analyzed per experimental condition. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.005 with respect to IPF fibroblasts; #p ≤ 0.05, ##p ≤ 0.01, and ###p ≤ 0.005 with respect to control fibroblasts. All comparisons were determined by Student’s t test.
Likewise, all morphological parameters were statistically different between weak and strong EMT models (p < 0.05, Student’s t test) but not between control and IPF fibroblasts. However, to address our main goal (i.e., compare different EMT models with fibroblasts), and for the sake of simplicity, we reduced two-group comparisons to those between different EMT models and fibroblasts only. Of note, the morphological differences between cells undergoing EMT and primary fibroblasts stimulated with TGF-β1 were even more dramatic and statistically significant in terms of elongation and circularity than of spreading. Collectively, these results indicate that TGF-β1 induces a mesenchymal-like phenotype in EMT-competent cells but fails to match the vimentin expression and morphological features of activated primary fibroblasts.
TGF-β1 induces a robust assembly of α-SMA into stress fibers in primary fibroblasts but not in cells undergoing EMT
The primary marker of the myofibroblast phenotype in vitro is the expression of α-SMA and its assembly into a cytoskeleton rich in stress fibers (Tomasek et al., 2002). In addition, some authors have described an intermediate phenotype between a nonactivated fibroblast and a myofibroblast characterized by the prominent presence of stress fibers containing little or no α-SMA and have referred to it as proto-myofibroblast (Tomasek et al., 2002; Hinz, 2012). To visualize simultaneously both the actin cytoskeleton and α-SMA, we immunostained cells cultured as in the previous experiments for both F-actin and α-SMA, as shown in Figure 3 and Supplemental Figure S4, and performed two complementary colocalization analyses with ImageJ (Abramoff et al., 2004). First, we computed the Manders colocalization coefficient M(α-SMA), which measures the fraction of α-SMA co-occurring with F-actin staining (Manders et al., 1993). This coefficient is constraint between 0 (absence) and 1 (complete colocalization) and is considered the most biologically relevant quantitative assessment of colocalization (Dunn et al., 2011). As shown in Figure 4A, the average M(α-SMA) values on TGF-β1 stimulation were similarly very high in both IPF fibroblasts and control fibroblasts (>0.95) compared with epithelial cells exhibiting either strong (∼0.8) or weak (∼0.7) EMT. The M(α-SMA) values of each EMT model (but not in control fibroblasts) were statistically different than those of IPF fibroblasts. These results reveal that EMT failed to match the colocalization found in activated primary fibroblasts.
FIGURE 3:

Visualization of the actin cytoskeleton, α-SMA, and their colocalization on stimulation with TGF-β1 for 3 d. Selected regions from representative immunofluorescence images of F-actin (left), α-SMA (middle), and their corresponding colocalization (right) in epithelial cells (H441, A549) and primary fibroblasts obtained from randomly selected control or IPF patients cultured in the absence or presence of 5 ng/ml TGF-β1. The colocalization of F-actin and α-SMA was labeled in white. Images were obtained with a ×20 objective (seven images/condition). Scale bar, 50 μm. Full images as well as additional representative images from other epithelial cell lines (PC9, H1975) and patients are shown in Supplemental Figure S4.
FIGURE 4:
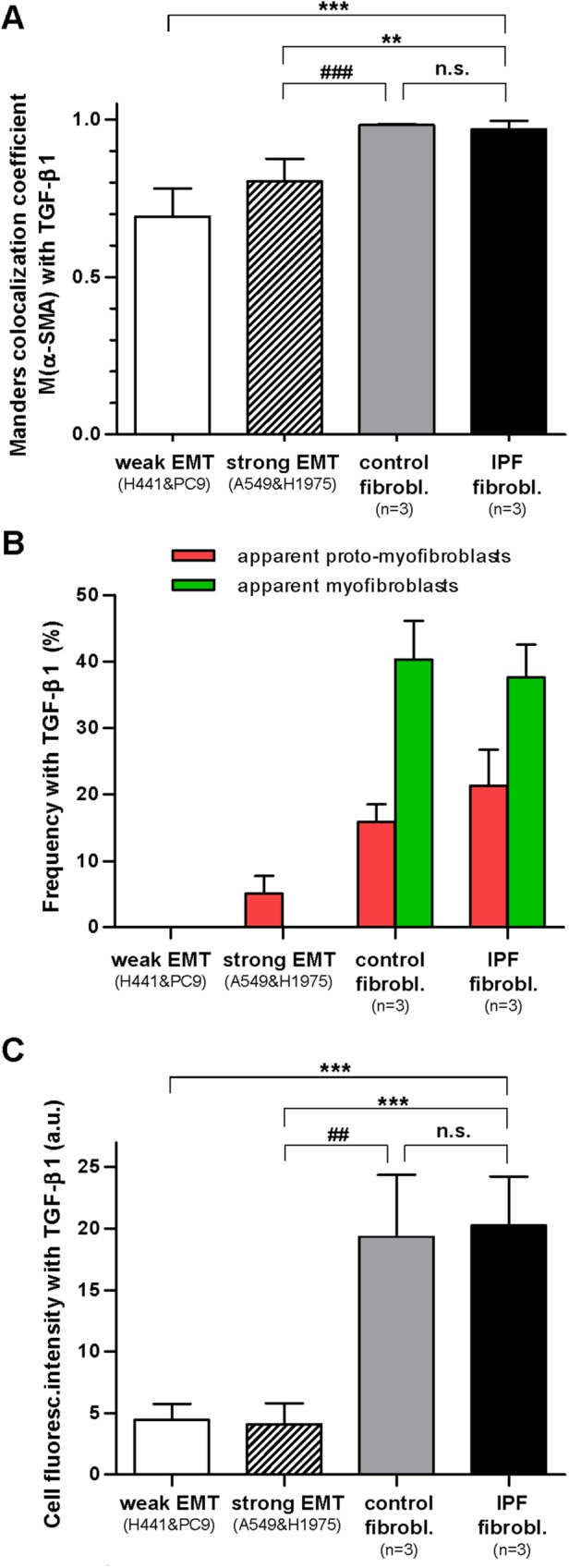
Assembly of α-SMA into stress fibers in EMT-competent lung epithelial cells and primary lung fibroblasts on stimulation with TGF-β1 for 3 d. (A) Quantitative colocalization analysis of the images in Figure 3 through the Manders colocalization coefficient M(α-SMA), which corresponds to the fraction (between 0 and 1) of α-SMA co-occurring with F-actin. (B) Colocalization analysis of the images in Figure 3 in terms of the percentage of cells stimulated with TGF-β1 that exhibited at least three straight and continuous white lines (green bars) or three straight and discontinuous (but not speckled) white lines (red bars), which were interpreted as apparent (well-differentiated) myofibroblasts or proto-myofibroblasts, respectively. (C) Absolute average F-actin fluorescence intensity per cell obtained on stimulation with TGF-β1 in epithelial cells undergoing weak or strong EMT and in primary fibroblasts from either control or IPF patients. Statistical analysis as in Figure 2.
Since M(α-SMA) values capture global rather than specific colocalization of α-SMA into stress fibers, we conducted a second (qualitative) visual colocalization analysis with ImageJ based on highlighting in white overlapping F-actin and α-SMA stainings as shown in Figure 3 and Supplemental Figure S4. This visual analysis revealed abundant white straight lines suggestive of α-SMA aligned into actin stress fibers in both control and IPF fibroblasts on TGF-β1 stimulation, whereas such white straight lines were much less abundant or largely absent in epithelial cells exhibiting strong or weak EMT, respectively. In an attempt to relate these colocalization images with the appearance of the myofibroblast phenotype, those cells exhibiting at least three straight and continous white (colocalization) straight lines were taken as apparent well-differentiated myofibroblasts, whereas cells that did not fulfill the latter criteria but exhibited at least three straight but discontinuous white lines (not speckled/noisy looking) were counted as proto-myofibroblast–like cells. The latter colocalization analysis revealed a progressive increase in the percentage of apparent proto-myofibroblasts in cells undergoing EMT (∼5%), control fibroblasts (∼15%), and IPF fibroblasts (∼20%) on 3-d stimulation with TGF-β1. In contrast, apparent well-differentiated myofibroblasts were observed in both control and IPF fibroblasts to a similar extent (∼40%), but they were largely absent in EMT models (Figure 4B). Adding the percentages of apparent proto-myofibroblasts and well-differentiated myofibroblasts elicited an average ∼60% value in both control and IPF fibroblasts, which is consistent with the percentages of α-SMA positive cells reported in primary lung fibroblasts obtained from the same type of IPF and control (fibrosis-free) patients elsewhere (Gabasa et al., 2013).
Given the importance of F-actin in cell mechanics (Alcaraz et al., 2017), we also analyzed quantitatively F-actin fluorescence stainings. Unlike vimentin stainings, we observed a similar average F-actin intensity in all EMT models (i.e., weak and strong) in response to 5 ng/ml TGF-β1 that was approximately fourfold lower than the F-actin intensity obtained in both control and IPF myofibroblasts (Figure 4C). Collectively these observations reveal that EMT-competent lung cells do not exhibit standard cytoskeletal markers indicative of well differentiated myofibroblasts when stimulated with TGF-β1 in vitro.
Potential relative contribution of epithelial cells and fibroblasts to the stiff microenvironment in IPF
The stiff microenvironment that characterizes IPF and other fibrotic disorders has been largely associated with the enhanced contractile properties of myofibroblasts and their excessive deposition of fibrillar collagens (King et al., 2011; Hinz, 2012; Ho et al., 2014). To assess the relative contribution of cells undergoing EMT to such stiff microenvironment in the context of IPF at the cellular level, we first measured cell stiffening in response to TGF-β1 by nanoindentation measurements with AFM, which enables computing the local Young’s modulus (E) of single cells (Alcaraz et al., 2003). E is a standard physical parameter that describes the resistance of a sample to the deformation imposed by a low/moderate external loading force, and is widely used to characterize the mechanical properties of cells and other biological samples (Butcher et al., 2009; Giménez et al., 2017b). In addition, we previously reported that E measurements are good indicators of cellular contractility (Roca-Cusachs et al., 2008). During each nanonindentation measurement, a cantilever force sensor provided with a micrometer sized pyramidal blunted tip—exhibiting a radius of curvature at the apex of ∼20 nm—was brought into contact with the cell surface (Figure 5A) and used to apply an increasing loading force up to ∼1 nN while monitoring cell resistance to deformation. These conditions elicited a maximum tip-cell contact area (∼0.1–0.2 μm2), which is approximately three orders of magnitude lower than cell spreading data (Figure 2D), thereby probing the local mechanical properties of the cell. Caution was taken to perform nanoindentation measurements at the perinuclear region and at low/moderate relative indentations to probe the mechanics of the cytoskeleton while minimizing contributions of the nucleus and the underlying tissue culture plastic (Roca-Cusachs et al., 2008; Giménez et al., 2017b). E data for each cell line and patient are shown in Supplemental Figure S5. To assess cell stiffening, we computed the ratio of E measured in the presence or absence of TGF-β1. As shown in Figure 5B, TGF-β1 stiffened all epithelial cell models in a cell-line–dependent manner but PC9, which may be associated with the particular genetic alterations of this cell line (see the Supplemental Material for an extended discussion). However, the largest cell stiffening elicited by TGF-β1 was consistenly observed in IPF fibroblasts (∼3-fold on average), whereas TGF-β1 induced only a modest cell stiffening in control fibroblasts (∼1.5-fold) (Figure 5B).
FIGURE 5:
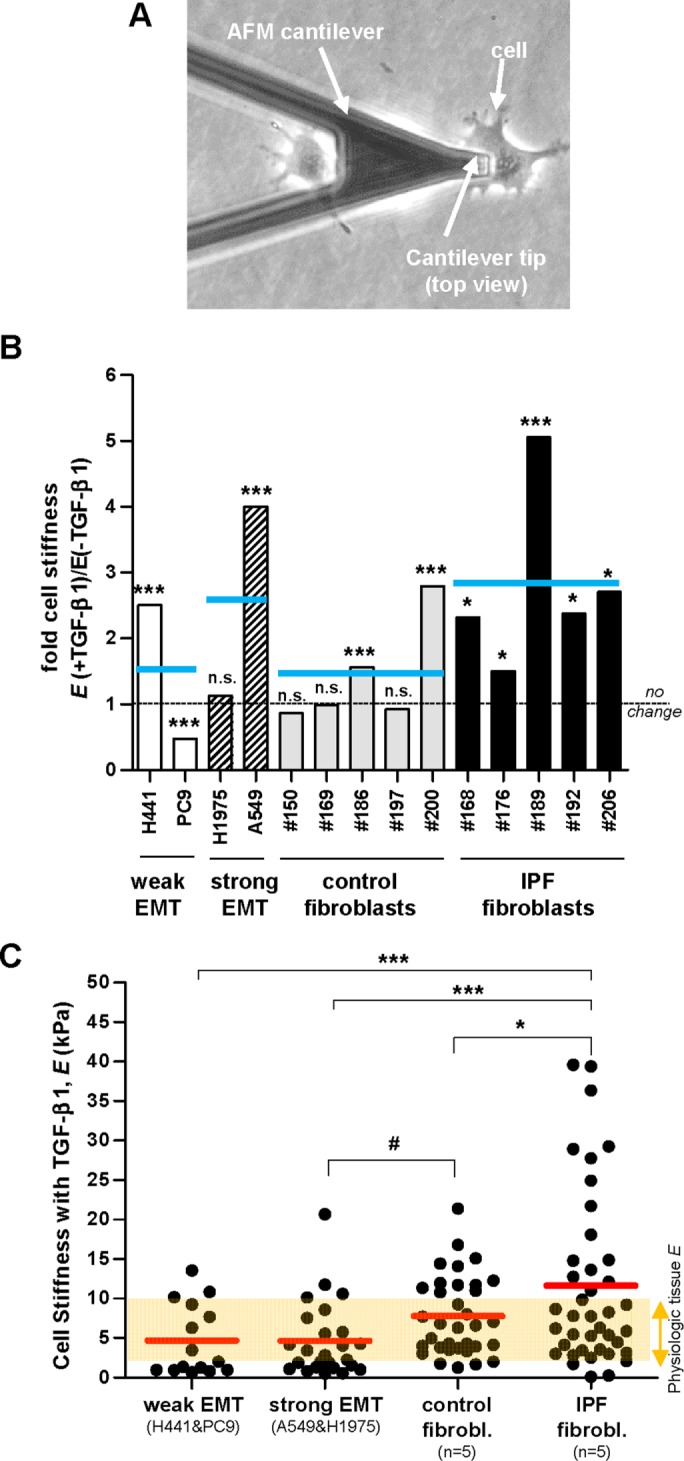
Nanoindentation elasticity measurements of single cells carried out with AFM on lung epithelial cells and primary lung fibroblasts. (A) Representative phase contrast image of an AFM force sensor (cantilever) on top of a single lung epithelial cell (A549). AFM force sensors were used to indent locally the cell surface and assess the corresponding opposing force to compute the Young’s modulus (E) as described in Material and Methods, using three technical replicates. (B) Ratio of the average E values obtained on single cells cultured in the presence or absence of TGF-β1 in a panel of epithelial cell lines (n = 4) and primary fibroblasts from randomly selected control (n = 5) and IPF (n = 5) patients. On average 10 cells were measured per experimental condition (two independent experiments). Horizontal gray dashed line corresponds to no fold change (ratio = 1), whereas horizontal solid blue lines correspond to the average values for each cellular model (i.e., epithelial cells exhibiting weak or strong EMT, and primary fibroblasts from control or IPF patients) as in Figure 1B. The E data for each cell line and patient are shown in Supplemental Figure S5. Statistical analysis as in Figure 1B. (C) E values obtained on single cells on TGF-β1 stimulation from each cellular model. Red horizontal lines correspond to the average E values for each model as in Figure 1C. The semi-transparent orange horizontal band corresponds to the E values assessed on normal pulmonary tissue elsewhere (see the main text for references) and was added to illustrate the aberrant mechanical behavior of IPF fibroblasts. Statistical analysis as in Figure 2.
To better compare the potential contribution of EMT cells and myofibroblasts to tissue stiffness, we plotted the absolute E values of single cells recorded after 3 d of TGF-β1 stimulation concomitantly with a horizontal band spanning the physiologic range of E data from pulmonary tissue (∼2–10 kPa) reported elsewhere (Ebihara et al., 2000; Liu et al., 2010; Brown et al., 2013) in Figure 5C. Although we observed a progressive increase in E in cells undergoing EMT (∼5 kPa), control myofibroblasts (∼8 kPa) and IPF myofibroblasts (∼13 kPa), their average E values were beyond the physiologic range in IPF myofibroblasts only (Figure 5C). Interestingly, the difference in E values observed in control and IPF myofibroblasts was statistically significant, supporting our initial observation of larger cell stiffening in myofibroblasts from IPF patients compared with those from control donors (Figure 5B). Likewise, E values of epithelial cells undergoing EMT were also different from E values obtained in either control or IPF fibroblasts with statistical significance. In addition we noticed that the differences in the average E values of cells undergoing EMT and primary myofibroblasts were much lower than the differences between other parameters examined in this study that had been previously pointed to as regulators of cell stiffness, including spreading (Figure 2D), F-actin (Figure 4C), and apparent myofibroblast enrichment (Figure 4, A and B) (Roca-Cusachs et al., 2008; Hinz, 2012). These observations suggest that the mechanical differences between epithelial cells undergoing EMT and myofibroblasts are due to other unidentified factors.
We next analyzed the expression of type I and type III collagens, which are the most abundant fibrillar collagens that are also frequently used as myofibroblast and EMT markers, respectively (Forino et al., 2006; Hinz et al., 2012). For this purpose, we assessed the mRNA levels of COL1A1 and COL3A1 genes by quantitative reverse transcription PCR (qRT-PCR) in cells stimulated with TGF-β1 for 3 d as in the previous experiments. In agreement with cell mechanical data, the expression of both COL1A1 and COL3A1 was moderately higher (35 and 25%, respectively) in myofibroblasts from IPF patients compared with those from control donors, and this difference attained statistical significance (Figure 6, A and B). In contrast, the expression of these genes in epithelial cells undergoing EMT was 3 orders of magnitude lower than in myofibroblasts. Interestingly, the expression of COL3A1 (but not COL1A1) in epithelial cells exhibiting a weak EMT was 3 orders of magnitude lower than those exhibiting strong EMT, providing additional support that COL3A1 is a suitable EMT marker (Forino et al., 2006). To validate the differences in the mRNA levels of fibrillar collagens between myofibroblasts from IPF and control patients at the protein level, we assessed the total amount of secreted collagen (types I–V) in the culture medium of primary fibroblasts stimulated with TGF-β1 for 3 d using the Sircol collagen assay as shown in Figure 6C. Total secreted collagen was ∼25% higher in myofibroblasts from IPF compared with control patients, which is very close to the ∼35% and ∼25% increases in COL1A1 and COL3A1 mRNA expression in the former cells, respectively.
FIGURE 6:
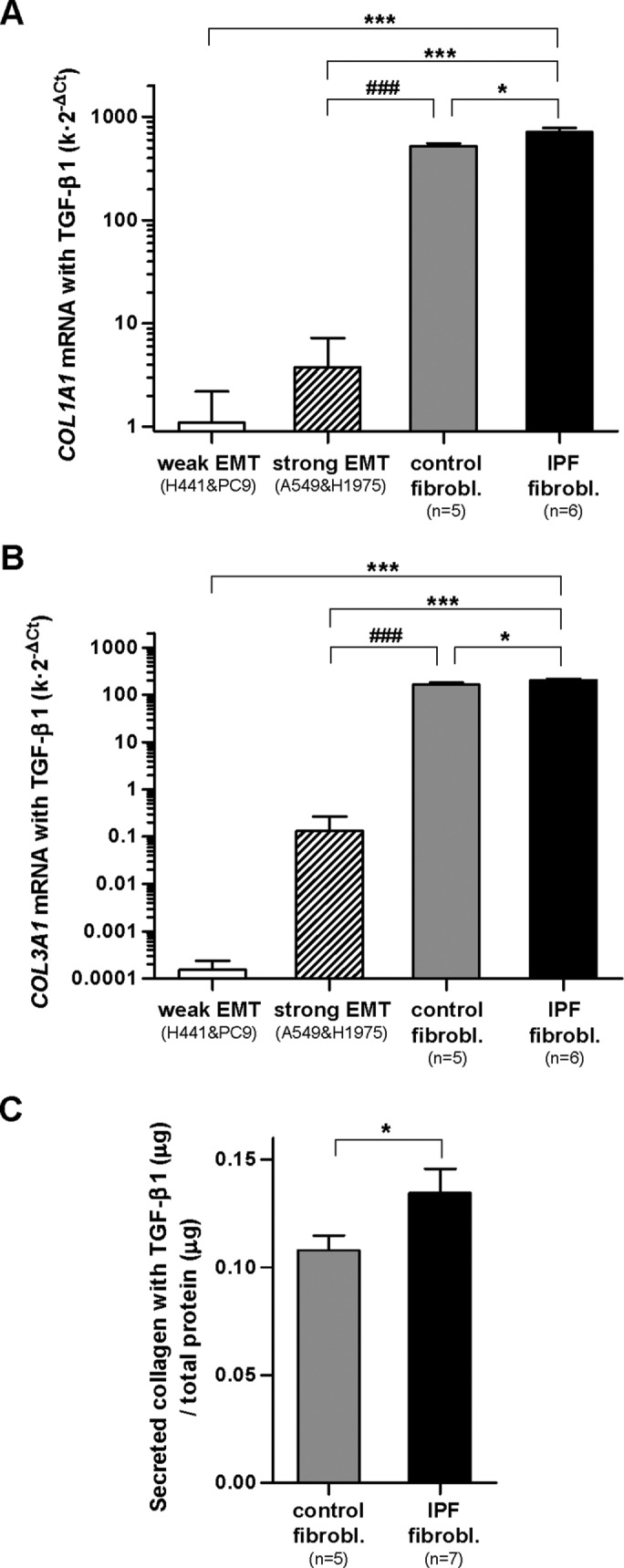
Expression of fibrillar collagens in EMT-competent lung epithelial cells and primary lung fibroblasts on stimulation with TGF-β1 for 3 d. (A, B) mRNA levels of COL1A1 (A) and COL3A1 (B) assessed by qRT-PCR using three technical replicates. POLR2A was used as endogenous gene. (C) Total secreted collagens of primary myofibroblasts assessed with the Sircol assay. Statistical analysis as in Figure 2. Note the vertical log scale in A and B.
Combining the mechanical and collagen expression data supports that myofibroblasts are the larger contributors to the stiff microenvironment in terms of both cell stiffness and expression of fibrillar collagens. Moreover, to the best of our knowledge, our results show for the first time that the contribution of epithelial cells undergoing EMT to the stiff microenvironment may be relevant in terms of cell stiffness but not of expression of fibrillar collagens. In addition, our measurements reveal that myofibroblasts derived from IPF patients exhibit aberrant mechanobiology responses to TGF-β1 in terms of both cell stiffness and expression of fibrillar collagens, which may be a critical contributor of the profibrotic tissue stiffening in IPF.
Once elucidated that EMT fails to induce a myofibroblast-like phenotype in our cell models in vitro, we wondered whether EMT may induce bona fide traits of nonactivated fibroblasts. For this purpose, we compared the average values of all the parameters examined throughout this work in both EMT-competent cells stimulated with TGF-β1 and in (nonactivated) fibroblasts from control donors cultured in the absence of TGF-β1. As shown in Figure 7, cells undergoing EMT could be distinguished from nonactivated primary fibroblasts in all parameters but spreading, exhibiting the largest differences in terms of vimentin (Figure 7A), morphology (elongation, circularity) (Figure 7, E and F), and expression of both COL1A1 and COL3A1 (Figure 7, H and I).
FIGURE 7:

Comprehensive comparison of the phenotype of lung cells undergoing EMT on stimulation with TGF-β1 and that of nonactivated primary fibroblasts from control donors cultured in the absence of TGF-β1 in terms of vimentin (A), F-actin (B), % of apparent proto-myofibroblasts/myofibroblasts (C), spreading (D), circularity (E), elongation (F), stiffness (G), and expression of COL1A1 (H) and COL3A1 (I). Statistical analysis as in Figure 2.
FAKY397 activity is enhanced in IPF fibroblasts compared with control fibroblasts stimulated with TGF-β1, which may underlie the larger stiffness and collagen expression of the former
Prompted by the enhanced stiffness and expression of fibrillar collagens of IPF fibroblasts compared with control fibroblasts on TGF-β1 stimulation, we began to examine potential underlying mechanisms. Focal adhesion kinase (FAK) is an important regulator of the bidirectional cell-ECM mechanical interactions in fibroblasts and other cell types through its phosphorylation at Y397, which is also upregulated by TGF-β1 (Thannickal et al., 2003; Mitra et al., 2005; Ho et al., 2014). Interestingly, previous in vivo studies revealed that FAKY397 activity is enhanced within fibroblast foci in IPF patients (Cai et al., 2010; Lagares et al., 2012; Ding et al., 2013), and its forced down-regulation protected against bleomycin-induced lung fibrosis in mice (Lagares et al., 2012; Zhao et al., 2016). To examine FAKY397 activity in our in vitro models, we first identified a suitable timing on TGF-β1 stimulation by conducting a time-course experiment in control fibroblasts (Figure 8A). Western blot analysis revealed a peak in FAKY397 at ∼6 h, which is in agreement with previous studies (Thannickal et al., 2003) and was used thereafter. Of note, FAKY397 activity was increased approximately twofold in IPF fibroblasts compared with control fibroblasts following TGF-β1 treatment (Figure 8B). To assess whether enhanced FAK activity is sufficient to increase fibroblast stiffness and/or expression of fibrillar collagens, we took advantage of a well-established FAK cell model based on mouse embryonic fibroblasts that are either wild type (FAK+/+) or FAK null (FAK–/–) (Lagares et al., 2012). As expected, TGF-β1 increased FAKY397 activity in FAK+/+ fibroblasts, whereas the expression of both total FAK and FAKY397 were largely absent in FAK–/– fibroblasts (Figure 8C). Likewise, AFM analysis reported ∼60% larger average E values in FAK+/+ compared with FAK–/– fibroblasts (Figure 8D), which was associated with a larger percentage of α-SMA colocalized with F-actin in the former as revealed by immunofluorescence (Figure 8E). In agreement with AFM data, qRT-PCR measurements showed a strikingly higher expression of both Col1a1 and Col3a1 mRNA levels in FAK+/+ compared with FAK–/– fibroblasts on TGF-β1 stimulation (Figure 8, F and G). Taken together, and to the best of our knowledge, these observations report for the first time a larger FAKY397 activity in TGF-β1-activated fibroblasts derived from IPF patients compared with control patients, and strongly support that such FAKY397 hyperactivation may be a driving mechanism underlying the aberrant mechanobiology of IPF fibroblasts, at least in terms of cell stiffness and expression of fibrillar collagens.
FIGURE 8:

Analysis of FAKY397 activity in primary fibroblasts on TGF-β1 stimulation, and effect of loss of FAK activity in the stiffness, cytoskeletal organization, and collagen expression of fibroblasts. (A) Representative Western blot showing the time-course expression of FAKY397, total FAK, and β-actin in primary control fibroblasts stimulated with TGF-β1. (B) Representative Western blot showing FAKpY397 and total FAK of control fibroblasts and IPF fibroblasts stimulated with TGF-β1 for 6 h; right: corresponding densitometry analysis of FAKpY397/FAK in control (n = 4) and IPF fibroblasts (n = 4). (C) Representative Western blot showing FAKpY397, total FAK and α-tubulin of FAK wild-type (FAK+/+), or FAK null (FAK–/–) mouse embryonic fibroblasts stimulated with TGF-β1 for 6 h; right: corresponding densitometry analysis of FAKpY397/α-tubulin (n = 3). (D) E of FAK+/+ and FAK–/– fibroblasts stimulated with TGF-β1 for 3 d assessed by AFM as in Figure 5. (E) Selected regions from representative immunofluorescence images of F-actin (left), α-SMA (middle), and their corresponding colocalization (right) of FAK+/+ and FAK–/– fibroblasts stimulated with TGF-β1 for 3 d. Images were obtained as in Figure 3. Scale bar, 50 μm. (F, G) mRNA levels of Col1a1 (F) and Col3a1 (G) of FAK+/+ and FAK–/– fibroblasts stimulated with TGF-β1 for 3 d assessed as in Figure 6. Statistical analysis as in Figure 2 (n = 2, unless otherwise indicated).
DISCUSSION
It is increasingly acknowledged that the chronic stiffening of the tissue microenvironment observed in IPF and other fibrotic disorders provides profibrotic cues that are essential for the progression of organ fibrosis (Hinz, 2012; Ho et al., 2014). Myofibroblasts are pointed to as critical effector cells in both tissue stiffening and fibrosis progression in the lung and other organs (King et al., 2011; Hinz, 2012; Ho et al., 2014). Yet it remains controversial what the contribution of epithelial cells undergoing EMT is to tissue stiffening and overall myofibroblast population (Bartis et al., 2014; O’Connor and Gomez, 2014). By examining a panel of commonly used lung EMT models, we found evidence supporting that lung cells undergoing EMT may contribute to the profibrotic stiff microenvironment through their enhanced endogenous stiffness but not through their expression of fibrillar collagens. In addition, we found that EMT renders a phenotype that is clearly different from that of myofibroblasts derived from IPF patients in terms of mechanics, collagen expression, morphology, and cytoskeletal organization. Indeed, cells undergoing EMT could be easily distinguished even from nonactivated fibroblasts, thereby challenging the ability of EMT to provide myofibroblast precursors further, unlike previously suspected (Kim et al., 2006; Willis et al., 2006; Degryse et al., 2010; Yamaguchi et al., 2017). Conversely, the responses of fibroblasts derived from control (fibrosis-free) donors—which were used to model resident fibroblasts—to the profibrotic cytokine TGF-β1 were similar to those of myofibroblasts derived from IPF patients in all the parameters examined. Accordingly, our data support that myofibroblasts in IPF are directly populated largely from resident fibroblasts (Rock et al., 2011; Giannandrea and Parks, 2014). In contrast, EMT may contribute indirectly to the myofibroblast population in IPF by perturbing normal parenchymal architecture (through loss of cell–cell adhesions required for alveolar integrity) and by providing profibrotic cytokines, in agreement with previous observations in mouse models of pulmonary fibrosis or renal fibrosis on induction of partial EMT (Wu et al., 2007; Kim et al., 2009; Grande et al., 2015).
Previous AFM mechanical measurements in the context of pulmonary fibrosis have been limited to whole and decellularized tissue sections (Liu et al., 2010; Melo et al., 2014; Alcaraz et al., 2017), whereas no studies have been conducted on IPF fibroblasts. Likewise, most previous EMT studies have focused on the expression of epithelial and mesenchymal markers, whereas studies examining the corresponding mechanical alterations had been scarce (Moreno-Bueno et al., 2009; Buckley et al., 2012; Bartis et al., 2014). Remarkably, our AFM measurements revealed that the average cell stiffness (E) of IPF myofibroblasts was beyond the physiologic range of pulmonary tissue stiffness, whereas that of myofibroblasts from control donors was not, underscoring the aberrant mechanical behavior of IPF myofibroblasts. Moreover, we found that a panel of lung cells undergoing EMT stiffened on average up to fourfold in response to TGF-β1, although their average E was ∼50% lower than that of control fibroblasts. Such a range of cell stiffening is in agreement with the twofold increase in E of A549 cells measured by AFM on treatment with 5 ng/ml TGF-β1 for 48 h elsewhere (Buckley et al., 2012) and with the fivefold stiffening reported in mouse mammary epithelial cells stimulated with 10 ng/ml TGF-β1 for 48 h (Schneider et al., 2013). To the best of our knowledge, our observations provide the first direct evidence of the mechanical diffences between cells undergoing EMT and myofibroblasts probed by AFM, and support that the contribution of the former to the tissue stiffening in IPF cannot be neglected, even though it is lower than that of myofibroblasts.
Most of the few previous attempts to examine the responses of normal lung epithelial cells to TGF-β have used type II alveolar epithelial cells (AECs) derived from rodents (Zhang et al., 2004; Willis et al., 2005; Bhaskaran et al., 2007; Zhao et al., 2013). These studies have consistently reported anti-proliferative effects of TGF-β1, which were also observed in the four epithelial cell lines used here. Likewise, a previous study of EMT induction by TGF-β1 on rat AECs reported common EMT hallmarks (i.e., down-regulation of epithelial markers, a fibroblast-like morphology, and an up-regulation of mesenchymal markers, including vimentin, type I collagen, and α-SMA) (Willis et al., 2005) that are reminiscent of our observed EMT induction elicited by TGF-β1 in selected cell lines (A549 and H1975). The latter similarity is unsurprising considering that EMT hallmarks have been reported in both normal epithelial cells during development and in pathologic conditions including organ fibrosis and cancer (Moreno-Bueno et al., 2009). Thus, even though the molecular underpinnings underlying TGF-β1 responses may be altered during malignant transformation (particularly in PC9 cells), the consistently similar responses to TGF-β1 in terms of cytostasis, stiffening, and expression of EMT markers compared with normal rodent epithelial cells reported elsewhere support that the former were a reasonable experimental model to address the aims of this study.
The enhanced cellular stiffening of both IPF myofibroblasts and selected epithelial cells undergoing EMT may have important profibrotic consequences. First, it may increase directly tissue rigidity through the expected enhanced traction forces and ECM adhesion strength of both cell types (Roca-Cusachs et al., 2008; Acerbi et al., 2012; Vizoso et al., 2015), which is in agreement with the critical mechanical roles of fibroblasts during the final contraction of a wound closure (Singer and Clark, 1999) and on the mechanical strength of fibroblast-populated three-dimensional collagen gels (Wakatsuki et al., 2003; Gehler et al., 2009). Moreover, cell stiffening may contribute indirectly to tissue stiffening by increasing the activation of extracellular TGF-β1, for there is growing evidence that such activation requires intracellular tension (Hinz, 2012; O’Connor and Gomez, 2014). Since matrix stiffness and TGF-β1 can cooperate to activate fibroblasts and promote EMT, all these observations support that cell stiffening contributes to a vicious cycle in which matrix stiffening activates fibroblasts and promotes EMT, which in turn increase tissue stiffness further, thereby amplifying fibrosis (Liu et al., 2010; Ho et al., 2014).
We previously showed that increasing the concentration of type I collagen or collagen cross-linking in acellular hydrogels was sufficient to effectively increase gel stiffness as measured by AFM (Alcaraz et al., 2011; Vicens-Zygmunt et al., 2015; Gimenez et al., 2017a). Based on the latter evidence and on the reported abundant deposition of fibrillar collagens and collagen crosslinkers concomitantly with tissue stiffening in organ fibrosis (Barry-Hamilton et al., 2010; Liu et al., 2010; King et al., 2011; Hinz, 2012; Ho et al., 2014), it is conceivable that the excessive and sustained expression of fibrillar collagens in fibrogenic myofibroblasts (but not in epithelial cells undergoing EMT) may directly contribute to the tissue stiffening in IPF. In addition, such excessive collagen expression may contribute to IPF progression through other processes, including disrupting normal alveolar architecture (Zeisberg et al., 2001), providing a profibrotic hypoxic microenvironment by hindering oxygen diffusion (Moreno-Bueno et al., 2009; Colom et al., 2014; Ho et al., 2014) and even hindering the diffusion of molecular therapeutics (Egeblad et al., 2010; Galgoczy et al., 2014). Conversely, all these observations strongly suggest that restoring normal collagen homeostasis may be a useful therapeutic strategy in organ fibrosis. In support of this hypothesis, we and others have reported that nintedanib—a recently approved drug to treat IPF—markedly reduces collagen expression by fibroblasts in vitro and in vivo (Hostettler et al., 2014; Wollin et al., 2014; Gabasa et al., 2017).
Remarkably, our in vitro analysis revealed modest yet significantly larger responses to TGF-β1 in fibroblasts derived from IPF patients compared with control donors in terms of both cell stiffness and expression of fibrillar collagens. Likewise, TGF-β1 elicited larger FAKY397 activity in IPF fibroblasts compared with control fibroblasts, thereby expanding previous histologic observations of enhanced FAKY397 expression within fibroblast foci in IPF samples (Cai et al., 2010; Lagares et al., 2012; Ding et al., 2013). On the other hand, we found that FAK depletion in fibroblasts was sufficient to markedly reduce both the stiffness and expression of fibrillar collagens in response to TGF-β1, concomitantly with a decrease in α-SMA colocalized with actin stress fibers. In agreement with our findings, a previous study on the FAK-related nonkinase protein FRNK, which is an independently expressed protein that negatively regulates FAK, reported its down-regulation in IPF samples; moreover, genetic down-regulation of FRNK in mouse fibroblasts was sufficient to increase FAKY397 concomitantly with collagen expression, contractility, and α-SMA in response to TGF-β1 (Ding et al., 2013). Unlike cell stiffness and collagen expression, we could not detect statistically significant differences between IPF fibroblasts and control fibroblasts in terms of F-actin content and its colocalization with α-SMA. The latter negative observations may reflect either lack of sufficient resolution in our immunofluorescence quantitative approaches or that the aberrant mechanical features of IPF myofibroblasts are due to other cytoskeletal associated proteins. Taken together, these novel observations support that FAKY397 hyperactivation in IPF fibroblasts may be a driving mechanism of the aberrant mechanobiology of these cells. Moreover, they support that targeting FAKY397 could be a suitable therapeutic approach aiming to restore normal fibroblast mechanobiology in IPF. In support of these interpretations, both pharmacologic and genetic inhibition of FAK activity protected against bleomycin-induced lung fibrosis in mice (Lagares et al., 2012; Zhao et al., 2016).
In summary, our study sheds light on the controversial contribution of EMT to the increased myofibroblast population in IPF. In addition, our work provides evidence supporting the direct pathologic contribution of IPF myofibroblasts to the stiff microenvironment in IPF through their own aberrant stiffness and excessive expression of fibrillar collagens, and reveals that such aberrant mechanobiology is associated with FAKY397 hyperactivation. Moreover, our results support that EMT may contribute to tissue stiffening by increasing epithelial cell stiffness, although to a lesser extent than myofibroblasts, but not through collagen deposition. Our results underscore also the remarkable potential of TGF-β1 to increase tissue stiffness through its direct impact on cell stiffness and collagen expression in the context of an already stiff microenvironment. These novel insights may help in designing future therapeutic studies aiming to restore normal mechanobiology in IPF (Ho et al., 2014).
MATERIAL AND METHODS
Isolation of adult human lung fibroblasts
Primary fibroblasts were isolated in sterile conditions by outgrowth of tissue explants from pulmonary tissue as described elsewhere (Gabasa et al., 2013) and frozen stored in liquid nitrogen at passages 2 and 3 until use. This procedure was approved by the Ethics Committees of the Hospital Clínic de Barcelona and the Universitat de Barcelona. Histologically normal parenchymal tissue was obtained from patients undergoing surgical pleurodesis to treat recurrent spontaneous pneumothorax (n = 9). IPF tissue was obtained from lung biopsies of patients exhibiting the histopathological pattern of usual interstitial pneumonia (n = 8). Patients gave written informed consent.
Cell culture and treatments
Before experiments, fibroblasts were fast thawed and grown in fibroblast culture media containing DMEM supplemented with 10% FBS (FBS Gold, PAA Laboratories), 100 IU/ml penicillin, 100 mg/ml streptomycin (Invitrogen), and 2 mg/ml amphotericin B (Sigma). All fibroblasts were used up to five to six passages to prevent replicative senescence and guarantee their phenotypic stability (Chang et al., 2002; Lugo et al., 2016). H441 and A549 lung adenocarcinoma cell lines were obtained from the American Type Culture Collection (ATCC). PC9 and H1975 lung adenocarcinoma cell lines were kindly provided by Miquel Tarón (USP Dexeus University Institute, Barcelona, Spain), and their identity was confirmed in terms of accepted EGFR mutations (Blanco et al., 2009; van der Wekken et al., 2016) as described elsewhere (Molina-Vila et al., 2008; Simonetti et al., 2010). Epithelial cells were maintained in culture medium consisting of HEPES (Sigma)-buffered RPMI 1640 medium supplemented with 10% FBS, 1 mM l-glutamine (Invitrogen), and the same antibiotics as in fibroblasts. Mouse embryonic fibroblasts from wild-type (FAK+/+) or FAK-deficient (FAK–/–) mice (ATCC) were cultured in DMEM as previously described (Lagares et al., 2012). All cultures were kept in a 5% CO2 humidified incubator at 37°C. For experiments on tissue culture plastic or glass substrata, cells were seeded in the corresponding culture media until they reached ∼80% confluency (∼1 to 2 d), washed three times in phosphate-buffered saline (PBS), growth arrested in serum-free culture media (SFM) for 24 h, and treated for 3 d with SFM alone (used as control) or supplemented with 5 ng/ml human TGF-β1 (R&D Systems).
Bright-field optical microscopy
To assess major morphological features, cells were examined under an inverted optical microscope (Eclipse TE2000; Nikon) placed on a vibration isolation table (Isostation; Newport), using a ×20 objective and phase contrast optics. Phase contrast images were acquired with a cooled charge-coupled device (CCD) camera (Orca AG; Hamamatsu Photonics). Cell spreading (A), elongation, perimeter (P), and circularity (C, computed as C = 4·π·A/P2) were assessed by manually tracing the boundary of single cells by ImageJ (Abramoff et al., 2004) as reported elsewhere (Roca-Cusachs et al., 2008; Lugo et al., 2016).
Fluorescence microscopy and image quantification
Cells were cultured on four-well chamber slides (BD Falcon) to analyze the expression of EMT and myofibroblast markers by immunofluorescence. Slides were washed with PBS, fixed with cold 4% paraformaldehyde (Sigma) for 15 min, permeabilized with 0.5% Triton X-100 for 30 min, and blocked with 1% bovine serum albumin (BSA) in PBS for 1 h. Chamber slides were incubated for 1 h at 37°C with primary antibodies against either α-SMA (1:500), vimentin (1:100) (Invitrogen), or E-cadherin (1:100) (BD Biosciences). To detect each primary antibody, chamber slides were washed with PBS and incubated with a suitable fluorescent secondary antibody for 1 h at 37°C. F-actin was detected by incubating with tetramethylrhodamine (TRITC)-phalloidin (Sigma) for 1 h. Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole; 1:10,000; Sigma). Slides were mounted with Prolong gold antifade reagent (Invitrogen). To quantify the fluorescence signal from a specifically labeled protein, slides were mounted on the motorized stage of the Eclipse TE2000 microscope (Nikon), and images were captured at seven randomized nonoverlapping locations with a ×20 objective and a cooled CCD camera (Orca AG) using Metamorph software (Molecular Devices). For each location, multiplex imaging was performed by acquiring images corresponding to DAPI and protein staining using the same exposure time in subsaturation conditions. All quantitative image measurements were conducted with ImageJ. DAPI images were used to assess the number of cells per image. After background substraction, the average protein fluorescence intensity per cell was assessed by dividing the total intensity per image by the corresponding cell number and by averaging the values obtained in all locations. Nuclear counting of DAPI stainings were also used to assess the average cell number in the presence or absence of TGF-β1. Colocalization analysis of F-actin and α-SMA stainings was performed by using both the Manders coefficients and the colocalization highlighter plug-ins (ImageJ 1.41n).
qRT-PCR
Cells were plated in triplicate, and the corresponding total RNA was isolated using the RNeasy Mini kit (QIAGEN). A total of 1 µg of RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit and RNase inhibitor (Applied Biosystems). Real-time PCRs were performed on 40 ng of each cDNA sample using TaqMan Gene Expression Master Mix and TaqMan gene-specific primer pairs and probes for human genes encoding collagen type I, alpha 1 (COL1A1) (Hs00164004_m1), collagen type III, alpha 1 (COL3A1) (Hs00164103_m1), and the RNA polymerase II polypeptide A (POLR2A, used as a reference gene) (Hs00172187_m1), and for mouse genes encoding Col1a1 (Mm00801666_g1), Col3a1 (Mm01254476_m1), and Polr2a (Mm00839502_m1, used as a reference gene) (Applied Biosystems). Reactions were carried out for 40 cycles (95°C for 21 s and 60°C for 20 s) in a 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative gene expression was assessed using the comparative Ct method as described elsewhere (Livak and Schmittgen, 2001). In brief, a common threshold was defined in the exponential phase of the PCR amplification curves for each gene, and the corresponding threshold cycles (Ct) were obtained and averaged over each replicate. Relative gene expression with respect to POLR2A was assessed as k⋅2–ΔCt, where k is the ratio of the threshold numbers used for the target and the endogenous genes, respectively, and ΔCt is the difference between the average Ct of the target and the reference (endogenous) genes.
Sircol collagen assay
Total secreted soluble collagen (types I–V) in the culture medium of fibroblasts was assessed using the Sircol collagen assay as reported elsewhere (Gabasa et al., 2013). Briefly, culture medium was collected for each experimental condition, concentrated, and stained with Sirius red dye to form a collagen-dye complex, which was precipitated and solubilized. The corresponding absorbance was measured at 540 nm with a microplate enzyme-linked immunosorbent assay (ELISA) reader, converted into collagen concentration through a calibrated standard curve, and multiplied by each sample volume to assess the total secreted collagen in micrograms. Results were normalized to total protein content, which was measured with the Lowry assay. Final results are shown as secreted collagen (micrograms)/total protein (micrograms).
Western blotting
Western blotting analysis of FAK activity was conducted as described elsewhere (Puig et al., 2015). In brief, fibroblasts were lysed in extraction buffer supplemented with phosphatase and protease inhibitors. Equal protein amounts were separated on 10% Mini-PROTEAN TGX precast gels (Bio-Rad), transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare), blocked, and incubated overnight with suitable primary antibodies including anti-FAK (3285; Cell Signalling), anti-FAKpY397 (700255; Invitrogen), anti–α-tubulin (5174 and 2144, Cell Signalling), or anti–β-actin (Sigma). The latter two markers were used as loading controls. Protein bands were labeled and visualized by chemiluminescence in a digital imaging system (ImageQuant LAS 4000; GE Healthcare). Protein band intensities were quantified with ImageJ and normalized to the corresponding loading control.
Single cell stiffness assessed from nanoindentation measurements with AFM
Cells were cultured on glass-bottomed 35-mm Petri dishes (MatTek) and placed on the stage of an inverted optical microscope (Olympus IX70-S8F2) equipped with a microinbucator (HCMIS ALA Science) to hold the temperature at 37°C. A commercial stand-alone AFM (Bioscope, Veeco) adapted to the optical microscope and provided with a low-spring constant (0.03 nN/nm) AFM cantilever (Veeco) was used to assess the Young’s modulus (E) of single cells, following a protocol described in detail elsewhere (Alcaraz et al., 2003, 2011). The spring constant of the cantilever was calibrated using the thermal fluctuations method (Roca-Cusachs et al., 2008). In brief, three standard force versus displacement curves (F vs. z) were recorded on the perinuclear region of each cell at moderate loading force (∼1 nN) and low speed (∼5 µm/s). The E of a single cell was computed by fitting a suitable contact elastic model to each F-versus-z curve and averaging it over the three recordings. The final E for a cell population in each experimental condition was calculated from at least nine measurements for each independent experiment (n ≥ 2).
Statistical analysis
Paired comparisons of data obtained from fibroblasts from the same patients were carried out using paired Student’s t test. Comparisons between each different cell types and IPF fibroblasts, between selected EMT models (A549, H1975) and control fibroblasts, or between FAK+/+ and FAK–/– fibroblasts were assessed by unpaired Student’s t test. Statistical analyses were performed with either SigmaStat (Systat Software) or Graphpad Prism (Graphpad). Statistical significance was assumed at p < 0.05. Unless otherwise stated, data are given as mean ± SE for all independent experiments (n ≥ 2).
Supplementary Material
Acknowledgments
We thank Maria Dolores Royo and Virginia Amador (Institut d’Investigacions Biomèdiques August Pi i Sunyer [IDIBAPS]), Rocío Nieto, Irene Acerbi, Juanjo Uriarte, and Roland Galgoczy (Universitat de Barcelona [UB]); Miquel Tarón (Dexeus) for technical assistance; and César Picado, Noemí Reguart (IDIBAPS-Clínic), Daniel Navajas, and Ramon Farré (UB) for support. This work was further supported by grants from the Ministerio de Economía y Competitividad (SAF2009-13243, PI13/02368, and SAF2016-79527-R to J.A.; PI060064 to A.X.), Generalitat de Catalunya AGAUR (SGR 661 to J.A.), Generalitat Valenciana (AICO/2016/085 to J.P.), and predoctoral fellowships from the Ministerio de Ciencia e Innovación (to AG) and CONACYT (to R.L.).
Abbreviations used:
- AFM
atomic force microscopy
- α-SMA
alpha smooth muscle actin
- E
Young’s elastic modulus
- EMT
epithelial–mesenchymal transition
- FAK
focal adhesion kinase
- TGF-β1
transforming growth factor beta1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0026) on October 18, 2017.
REFERENCES
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with Image J. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Acerbi I, Luque T, Gimenez A, Puig M, Reguart N, Farre R, Navajas D, Alcaraz J. Integrin-specific mechanoresponses to compression and extension probed by cylindrical flat-ended AFM tips in lung cells. PLoS One. 2012;7:e32261. doi: 10.1371/journal.pone.0032261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz J, Buscemi L, Grabulosa M, Trepat X, Fabry B, Farre R, Navajas D. Microrheology of human lung epithelial cells measured by atomic force microscopy. Biophys J. 2003;84:2071–2079. doi: 10.1016/S0006-3495(03)75014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz J, Mori H, Ghajar CM, Brownfield D, Galgoczy R, Bissell MJ. Collective epithelial cell invasion overcomes mechanical barriers of collagenous extracellular matrix by a narrow tube-like geometry and MMP14-dependent local softening. Integr Biol (Camb) 2011;3:1153–1166. doi: 10.1039/c1ib00073j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcaraz J, Otero J, Jorba I, Navajas D. Bidirectional mechanobiology between cells and their local extracellular matrix probed by atomic force microscopy. Semin Cell Dev Biol. 2017 doi: 10.1016/j.semcdb.2017.07.020. doi: 10.1016/j.semcdb.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Arora PD, Narani N, McCulloch CA. The compliance of collagen gels regulates transforming growth factor-beta induction of alpha-smooth muscle actin in fibroblasts. Am J Pathol. 1999;154:871–882. doi: 10.1016/s0002-9440(10)65334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrini JL, Chaudhry S, Sarrazy V, Koehler A, Hinz B. The mechanical memory of lung myofibroblasts. Integr Biol. 2012;4:410–421. doi: 10.1039/c2ib00149g. [DOI] [PubMed] [Google Scholar]
- Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, Mikels A, Vaysberg M, Ghermazien H, Wai C, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:U1009–U1107. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important. Thorax. 2014;69:760–765. doi: 10.1136/thoraxjnl-2013-204608. [DOI] [PubMed] [Google Scholar]
- Bhaskaran M, Kolliputi N, Wang Y, Gou D, Chintagari NR, Liu L. Trans-differentiation of alveolar epithelial type II cells to type I cells involves autocrine signaling by transforming growth factor beta 1 through the Smad pathway. J Biol Chem. 2007;282:3968–3976. doi: 10.1074/jbc.M609060200. [DOI] [PubMed] [Google Scholar]
- Blackwell TS, Tager AM, Borok Z, Moore BB, Schwartz DA, Anstrom KJ, Bar-Joseph Z, Bitterman P, Blackburn MR, Bradford W, et al. Future directions in idiopathic pulmonary fibrosis research. Am J Respir Crit Care Med. 2014;189:214–222. doi: 10.1164/rccm.201306-1141WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco R, Iwakawa R, Tang M, Kohno T, Angulo B, Pio R, Montuenga LM, Minna JD, Yokota J, Sanchez-Cespedes M. A gene-alteration profile of human lung cancer cell lines. Hum Mutat. 2009;30:1199–1206. doi: 10.1002/humu.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AC, Fiore VF, Sulchek TA, Barker TH. Physical and chemical microenvironmental cues orthogonally control the degree and duration of fibrosis-associated epithelial-to-mesenchymal transitions. J Pathol. 2013;229:25–35. doi: 10.1002/path.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley ST, Medina C, Davies AM, Ehrhardt C. Cytoskeletal re-arrangement in TGF-beta 1-induced alveolar epithelial-mesenchymal transition studied by atomic force microscopy and high-content analysis. Nanomed Nanotechnol Biol Med. 2012;8:355–364. doi: 10.1016/j.nano.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nat Rev Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G-Q, Zheng A, Tang Q, White ES, Chou C-F, Gladson CL, Olman MA, Ding Q. Downregulation of FAK-related non-kinase mediates the migratory phenotype of human fibrotic lung fibroblasts. Exp Cell Res. 2010;316:1600–1609. doi: 10.1016/j.yexcr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom A, Galgoczy R, Almendros I, Xaubet A, Farre R, Alcaraz J. Oxygen diffusion and consumption in extracellular matrix gels: Implications for designing three-dimensional cultures. J Biomed Mater Res Part A. 2014;102:2776–2784. doi: 10.1002/jbm.a.34946. [DOI] [PubMed] [Google Scholar]
- Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, McMahon FB, Gleaves LA, Blackwell TS, Lawson WE. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299:L442–L452. doi: 10.1152/ajplung.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Cai G-Q, Hu M, Yang Y, Zheng A, Tang Q, Gladson CL, Hayasaka H, Wu H, You Z, et al. FAK-related nonkinase is a multifunctional negative regulator of pulmonary fibrosis. Am J Pathol. 2013;182:1572–1584. doi: 10.1016/j.ajpath.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara T, Venkatesan N, Tanaka R, Ludwig MS. Changes in extracellular matrix and tissue viscoelasticity in bleomycin-induced lung fibrosis: temporal aspects. Am J Respir Crit Care Med. 2000;162:1569–1576. doi: 10.1164/ajrccm.162.4.9912011. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forino M, Torregrossa R, Ceol M, Murer L, Della Vella M, Del Prete D, D’Angelo A, Anglani F. TGF beta 1 induces epithelial-mesenchymal transition, but not myofibroblast transdifferentiation of human kidney tubular epithelial cells in primary culture. Int J Exp Pathol. 2006;87:197–208. doi: 10.1111/j.1365-2613.2006.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabasa M, Ikemori R, Hilberg F, Reguart N, Alcaraz J. Nintedanib selectively inhibits the activation and tumor-promoting effects of fibroblasts from lung adenocarcinoma patients. Br J Cancer. 2017;117:1128–1136. doi: 10.1038/bjc.2017.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabasa M, Royo D, Molina-Molina M, Roca-Ferrer J, Pujols L, Picado C, Xaubet A, Pereda J. Lung myofibroblasts are characterized by down-regulated cyclooxygenase-2 and its main metabolite, prostaglandin E2. PLoS One. 2013;8:e65445. doi: 10.1371/journal.pone.0065445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgoczy R, Pastor I, Colom A, Gimenez A, Mas F, Alcaraz J. A spectrophotometer-based diffusivity assay reveals that diffusion hindrance of small molecules in extracellular matrix gels used in 3D cultures is dominated by viscous effects. Colloid Surf B Biointerfaces. 2014;120:200–207. doi: 10.1016/j.colsurfb.2014.05.017. [DOI] [PubMed] [Google Scholar]
- Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, Eliceiri KW, Weaver VM, Calderwood DA, Keely PJ. Filamin A-b1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20:3224–3238. doi: 10.1091/mbc.E08-12-1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech. 2014;7:193–203. doi: 10.1242/dmm.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez A, Jose Uriarte J, Vieyra J, Navajas D, Alcaraz J. Elastic properties of hydrogels and decellularized tissue sections used in mechanobiology studies probed by atomic force microscopy. Microsc Res Tech. 2017a;80:85–96. doi: 10.1002/jemt.22740. [DOI] [PubMed] [Google Scholar]
- Giménez A, Uriarte JJ, Vieyra J, Navajas D, Alcaraz J. Elastic properties of hydrogels and decellularized tissue sections used in mechanobiology studies probed by atomic force microscopy. Microsc Res Tech. 2017b;80:85–96. doi: 10.1002/jemt.22740. [DOI] [PubMed] [Google Scholar]
- Goldberg MT, Han Y-P, Yan C, Shaw MC, Garner WL. TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: An implication for abnormal wound healing. J Invest Dermatol. 2007;127:2645–2655. doi: 10.1038/sj.jid.5700890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande T, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, Grant Rowe R, Weiss SJ, Lopez-Novoa JM, Angela Nieto M. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–997. doi: 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- Hinz B. Mechanical aspects of lung fibrosis: a spotlight on the myofibroblast. Proc Am Thorac Soc. 2012;9:137–147. doi: 10.1513/pats.201202-017AW. [DOI] [PubMed] [Google Scholar]
- Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbian G. Recent developments in myofibroblast biology paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis-a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10:390–402. doi: 10.1038/nrrheum.2014.53. [DOI] [PubMed] [Google Scholar]
- Hostettler KE, Zhong J, Papakonstantinou E, Karakiulakis G, Tamm M, Seidel P, Sun Q, Mandal J, Lardinois D, Lambers C, Roth M. Anti-fibrotic effects of nintedanib in lung fibroblasts derived from patients with idiopathic pulmonary fibrosis. Respir Res. 2014;15:157. doi: 10.1186/s12931-014-0157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber DE. Mechanobiology and diseases of mechanotransduction. Ann Med. 2003;35:564–577. doi: 10.1080/07853890310016333. [DOI] [PubMed] [Google Scholar]
- Jolly MK, Tripathi SC, Jia D, Mooney SM, Celiktas M, Hanash SM, Mani SA, Pienta KJ, Ben-Jacob E, Levine H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 2016;7:27067–27084. doi: 10.18632/oncotarget.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, Sheppard D, Chapman HA. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–13185. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, Chapman HA. Epithelial cell alpha 3 beta 1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–224. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TE, Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi: 10.1016/S0140-6736(11)60052-4. [DOI] [PubMed] [Google Scholar]
- Lagares D, Busnadiego O, Garcia-Fernandez RA, Kapoor M, Liu S, Carter DE, Abraham D, Shi-Wen X, Carreira P, Fontaine BA, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagares D, Grasberger P, Probst C, Engler A, Tager AM. Therapeutic targeting of fibroblast durotaxis: a novel class of anti-fibrotic therapies for IPF. Am J Respir Crit Care Med. 2016;193 [Google Scholar]
- Lieber M, Smith B, Szakal A, Nelsonrees W, Todaro G. Continuous tumor-cell line from a human lung carcinoma with properties of type-ii alveolar epithelial cells. Int J Cancer. 1976;17:62–70. doi: 10.1002/ijc.2910170110. [DOI] [PubMed] [Google Scholar]
- Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, Tschumperlin DJ. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. 2010;190:693–706. doi: 10.1083/jcb.201004082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lugo R, Gabasa M, Andriani F, Puig F, Facchinetti F, Ramírez J, Gómez-Caro A, Pastorino U, Fuster G, Almendros I, et al. Heterotypic paracrine signaling drives fibroblast senescence and tumor progression of large cell carcinoma of the lung. Oncotarget. 2016;7:82324–82337. doi: 10.18632/oncotarget.10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of colocalization of objects in dual-color confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Melo E, Cardenes N, Garreta E, Luque T, Rojas M, Navajas D, Farre R. Inhomogeneity of local stiffness in the extracellular matrix scaffold of fibrotic mouse lungs. J Mech Behav Biomed Mater. 2014;37:186–195. doi: 10.1016/j.jmbbm.2014.05.019. [DOI] [PubMed] [Google Scholar]
- Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: In command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- Molina-Vila MA, Bertran-Alamillo J, Reguart N, Taron M, Castella E, Llatjos M, Costa C, Mayo C, Pradas A, Queralt C, et al. A sensitive method for detecting EGFR mutations in Non-small cell lung cancer samples with few tumor cells. J Thorac Oncol. 2008;3:1224–1235. doi: 10.1097/JTO.0b013e318189f579. [DOI] [PubMed] [Google Scholar]
- Moreno-Bueno G, Peinado H, Molina P, Olmeda D, Cubillo E, Santos V, Palacios J, Portillo F, Cano A. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat Protoc. 2009;4:1591–1613. doi: 10.1038/nprot.2009.152. [DOI] [PubMed] [Google Scholar]
- O’Connor JW, Gomez EW. Biomechanics of TGFbeta-induced epithelial-mesenchymal transition: implications for fibrosis and cancer. Clin Transl Med. 2014;3:23–23. doi: 10.1186/2001-1326-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Puig M, Lugo R, Gabasa M, Gimenez A, Velasquez A, Galgoczy R, Ramirez J, Gomez-Caro A, Busnadiego O, Rodriguez-Pascual F, et al. Matrix stiffening and beta(1) integrin drive subtype-specific fibroblast accumulation in lung cancer. Mol Cancer Res. 2015;13:161–173. doi: 10.1158/1541-7786.MCR-14-0155. [DOI] [PubMed] [Google Scholar]
- Roca-Cusachs P, Alcaraz J, Sunyer R, Samitier J, Farre R, Navajas D. Micropatterning of single endothelial cell shape reveals a tight coupling between nuclear volume in G1 and proliferation. Biophys J. 2008;94:4984–4995. doi: 10.1529/biophysj.107.116863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BLM. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D, Baronsky T, Pietuch A, Rother J, Oelkers M, Fichtner D, Wedlich D, Janshoff A. Tension monitoring during epithelial-to-mesenchymal transition links the switch of phenotype to expression of moesin and cadherins in NMuMG cells. PLoS One. 2013;8:e80068. doi: 10.1371/journal.pone.0080068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotton CJ, Chambers RC. Molecular targets in pulmonary fibrosis: the myofibroblast in focus. Chest. 2007;132:1311–1321. doi: 10.1378/chest.06-2568. [DOI] [PubMed] [Google Scholar]
- Shannon JM, Mason RJ, Jennings SD. Functional-differentiation of alveolar type-ii epithelial-cells invitro: effects of cell-shape, cell matrix interactions and cell cell-interactions. Biochim Biophys Acta. 1987;931:143–156. doi: 10.1016/0167-4889(87)90200-x. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–820. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Simonetti S, Angel Molina M, Queralt C, de Aguirre I, Mayo C, Bertran-Alamillo J, Javier Sanchez J, Luis Gonzalez-Larriba J, Jimenez U, Isla D, et al. Detection of EGFR mutations with mutation-specific antibodies in stage IV non-small-cell lung cancer. J Transl Med. 2010;8:135. doi: 10.1186/1479-5876-8-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Henke CA, Horowitz JC, Noble PW, Roman J, Sime PJ, Zhou Y, Wells RG, White ES, Tschumperlin DJ. Matrix biology of idiopathic pulmonary fibrosis: a workshop report of the National Heart, Lung, and Blood Institute. Am J Pathol. 2014;184:1643–1651. doi: 10.1016/j.ajpath.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor-beta 1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Thomson S, Buck E, Petti F, Griffin G, Brown E, Ramnarine N, Iwata KK, Gibson N, Haley JD. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005;65:9455–9462. doi: 10.1158/0008-5472.CAN-05-1058. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- van der Wekken AJ, Saber A, Hiltermann TJN, Kok K, van den Berg A, Groen HJM. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit Rev Oncol Hematol. 2016;100:107–116. doi: 10.1016/j.critrevonc.2016.01.024. [DOI] [PubMed] [Google Scholar]
- Vicens-Zygmunt V, Estany S, Colom A, Montes-Worboys A, Machahua C, Sanabria AJ, Llatjos R, Escobar I, Manresa F, Dorca J, et al. Fibroblast viability and phenotypic changes within glycated stiffened three-dimensional collagen matrices. Respir Res. 2015;16:15. doi: 10.1186/s12931-015-0237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizoso M, Puig M, Carmona FJ, Maqueda M, Velásquez A, Gómez A, Labernadie A, Lugo R, Gabasa M, Rigat de Brugarolas LG, et al. Aberrant DNA methylation in non small cell lung cancer associated fibroblasts. Carcinogenesis. 2015;36:1453–1463. doi: 10.1093/carcin/bgv146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki T, Wysolmerski RB, Elson EL. Mechanics of cell spreading: role of myosin II. J Cell Sci. 2003;116:1617–1625. doi: 10.1242/jcs.00340. [DOI] [PubMed] [Google Scholar]
- Wang G, Yang X, Jin Y, Deng Y, Luo X, Hu J, Wang J. TGF-β regulates the proliferation of lung adenocarcinoma cells by inhibiting PIK3R3 expression. Mol Carcinog. 2015;54:E162–E171. doi: 10.1002/mc.22243. [DOI] [PubMed] [Google Scholar]
- Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. 2006;3:377–382. doi: 10.1513/pats.200601-004TK. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor Nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349:209–220. doi: 10.1124/jpet.113.208223. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X, Xu J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an α-smooth muscle actin-Cre transgenic mouse. Respir Res. 2007;8:1–11. doi: 10.1186/1465-9921-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Hirai S, Tanaka Y, Sumi T, Miyajima M, Mishina T, Yamada G, Otsuka M, Hasegawa T, Kojima T, et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial–mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab Invest. 2017;97:232–242. doi: 10.1038/labinvest.2016.135. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–8698. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- Yu W, Fang X, Ewald A, Wong K, Hunt CA, Werb Z, Matthay MA, Mostov K. Formation of cysts by alveolar type II cells in three-dimensional culture reveals a novel mechanism for epithelial morphogenesis. Mol Biol Cell. 2007;18:1693–1700. doi: 10.1091/mbc.E06-11-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M, Bonner G, Maeshima Y, Colorado P, Muller GA, Strutz F, Kalluri R. Renal fibrosis - Collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am J Pathol. 2001;159:1313–1321. doi: 10.1016/S0002-9440(10)62518-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FJ, Nielsen LD, Lucas JJ, Mason RJ. Transforming growth factor-beta antagonizes alveolar type II cell proliferation induced by keratinocyte growth factor. Am J Respir Cell Mol Biol. 2004;31:679–686. doi: 10.1165/rcmb.2004-0182OC. [DOI] [PubMed] [Google Scholar]
- Zhang H-J, Wang H-Y, Zhang H-T, Su J-M, Zhu J, Wang H-B, Zhou W-Y, Zhang H, Zhao M-C, Zhang L, Chen X-F. Transforming growth factor-beta 1 promotes lung adenocarcinoma invasion and metastasis by epithelial-to-mesenchymal transition. Mol Cell Biochem. 2011;355:309–314. doi: 10.1007/s11010-011-0869-3. [DOI] [PubMed] [Google Scholar]
- Zhao L, Yee M, O’Reilly MA. Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-beta and BMP signaling. Am J Physiol Lung Cell Mol Physiol. 2013;305:L409–L418. doi: 10.1152/ajplung.00032.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X-K, Cheng Y, Cheng ML, Yu L, Mu M, Li H, Liu Y, Zhang B, Yao Y, Guo H, et al. Focal adhesion kinase regulates fibroblast migration via integrin beta-1 and plays a central role in fibrosis. Sci Rep. 2016;6:19276. doi: 10.1038/srep19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.