

Abstract
The bis(pyridine)silver (I) permanganate promoted hydroxylation of diketopiperazines has served as a pivotal transformation in the synthesis of complex epipolythiodiketopiperazine alkaloids. This late-stage C–H oxidation chemistry is strategically critical to access N-acyl iminium ion intermediates necessary for nucleophilic thiolation of advanced diketopiperazines en route to potent epipolythiodiketopiperazine anticancer compounds. In this study, we develop an informative mathematical model using hydantoin derivatives as a training set of substrates by relating the relative rates of oxidation to various calculated molecular descriptors. The model prioritizes Hammett values and percent buried volume as key contributing factors in the hydantoin series while correctly predicting the experimentally observed oxidation sites in various complex diketopiperazine case studies. Thus, a method is presented by which to use simplified training molecules and resulting correlations to explain and predict reaction behavior for more complex substrates.
INTRODUCTION
Epipolythiodiketopiperazine (ETP) alkaloids are a diverse and structurally complex class of natural products.1,2 As illustrated in Figure 1, the diketopiperazine substructure of these alkaloids is commonly adorned with a polysulfane motif that is known to be essential to their biological activity,3 including the potent anticancer activity of both natural and designed ETP derivatives.4,5 The combination of their fascinating molecular architecture and biological activity has prompted significant interest in chemical synthesis of ETPs.6,7 One of our laboratories has developed a general strategy for conversion of complex diketopiperazines to the corresponding epipolythiodiketopiperazines in the context of several synthetic campaigns.2,4,6 A critical step in our approach to stereo-, regio-, and congener-specific sulfidation of complex diketopiperazines is the C–H hydroxylation of the diketopiperazine heterocycle. In particular, our discovery of the bis(pyridine)silver (I) permanganate8 promoted oxidation of diketopiperazines has enabled the chemical synthesis of various epipolythiodiketopiperazines.4,6 We have found the corresponding tetra-n-butyl ammonium permanganate9 to be a milder oxidant, as illustrated in our synthesis of (+)-gliocladins B and C.6c While mechanistic details10 of the permanganate promoted C–H oxidation reaction in complex settings are of ongoing interest, we are likewise intent to experimentally identify substrate structural parameters that greatly impact the success of this late-stage hydroxylation reaction. One of our laboratories has developed an effective methodology for parameterization of complex chemical transformations to glean key insights that inform further development and application of the chemistry, as demonstrated in several case studies including fluorination, gold catalyzed cyclization, and rhodium-catalyzed C–H functionalization.11 Herein, we describe the development of an informative model using a library of simple hydantoins as a training substrate-set by relating the relative rates of oxidation to various calculated molecular descriptors. The resulting model highlights the impact of the Hammett parameter and percent buried volume on the oxidation outcome, and when applied to analysis of new case studies, the model can reliably predict and explain the experimentally observed oxidation of complex diketopiperazine substrates.
Figure 1.
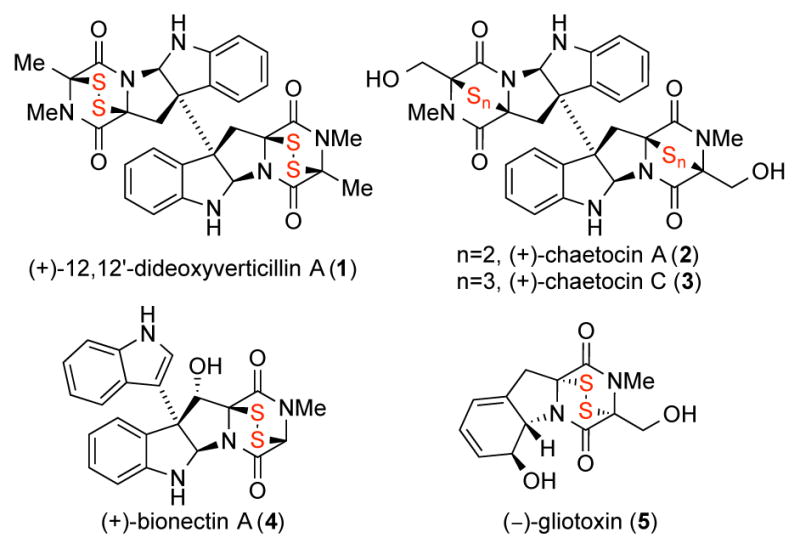
Representative epipolythiodiketopiperazine natural products.
RESULTS AND DISCUSSION
The sensitivity of the epipolythiodiketopiperazines’ polysulfane bridge to various reducing or oxidizing reaction conditions, and their propensity toward elimination and degradation requires precise timing for their introduction into complex molecular frameworks. These considerations are compounded in the context of dimeric epipolythiodiketopiperazines that require the introduction of challenging quaternary stereogenic centers.2 Informed by prior biosynthetic studies of sirodesmin by Howlett,12a and the cysteine feeding experiments by Kirby,12b and given the presence of various polysulfane congeners in distinct families of natural ETPs, we posited2a,6a that the introduction of the carbon–sulfur bonds in the biosynthesis of these alkaloids may involve a C–H hydroxylation followed by nucleophilic glutathione thiolation of N-acyl iminium ion intermediates (Scheme 1).2,6a–b
Scheme 1.
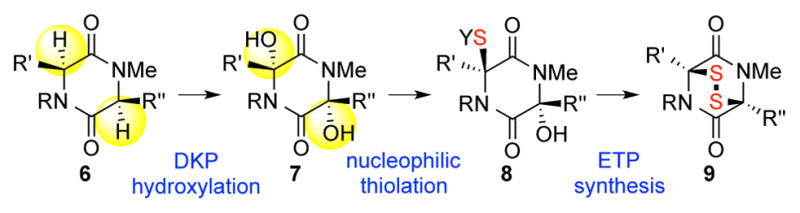
Key steps in the conversion of diketopiperazines (DKPs) to the corresponding epidithiodiketopiperazines (ETPs).
Importantly, this biogenetically inspired approach to the chemical synthesis of epipolythiodiketopiperazines led to the development of our permanganate promoted hydroxylation of diketopiperazines and laid the foundation for the synthesis of a number of natural and designed complex epipolythiodiketopiperazines.2a Additionally, consistent with this hypothesis, C–H hydroxylation of a phenylalanine-serine diketopiperazine followed by nucleophilic addition of glutathione has recently been experimentally observed in the biosynthesis of (−)-gliotoxin (5, Figure 1).13 While our late-stage permanganate promoted C–H hydroxylation of complex diketopiperazines has enabled strategic access to the corresponding N-acyl iminium ions as a prelude to our epipolythiodiketopiperazine syntheses,2a we have sought to better understand the critical substrate characteristics that govern the reaction outcome.
Application of this permanganate oxidation to complex diketopiperazines has proven successful in a variety of total synthetic efforts.6 As illustrated in Scheme 2A, the bis(pyridine)silver (I) permanganate promoted oxidation of dimeric diketopiperazine (+)-10a (4.80 equiv) led to the formation of the corresponding tetrahydroxylated dimer (+)-11a, with hydroxylation at C11 and C15, en route to the synthesis of (+)-12,12′-dideoxyverticillin A (1).6a Similarly, hydroxylation of dimeric diketopiperazine (+)-10b afforded the corresponding tetrahydroxylated dimer (+)-11b (Scheme 2A. 8.00 equiv). Notably, the desired hydroxylation proceeds even at the more electron-deficient C15-center next to the acetoxy group to give tetraol (+)-11b.14 However, oxidation of the C11-epimer of dimeric diketopiperazine (+)-10a (not shown) under identical conditions only afforded a diol product where C–H oxidation is limited to the C15-positions without oxidation at the C11-positions,6a highlighting the impact of the diketopiperazine stereochemistry on the reaction outcome.6a The hydroxylation of diketopiperazine (+)-12 using the tetra-n-butyl ammonium permanganate reagent provided diol (−)-13, which served as a key intermediate en route to (+)-gliocladins B and C (Scheme 2B, 3.79 equiv).6c Notably, bis(pyridine)silver (I) permanganate promoted hydroxylation of diketopiperazine (−)-14 resulted in the triketopiperazine alcohol (−)-15 en route to (+)-bionectin A (4, Scheme 2C, 8.00 equiv).6d While we have reasoned that the C11-stereochemistry of alcohol (−)-15 is a consequence of the C12 substituent, the double oxidation at C15 was surprising given the monohydroxylation of the structurally related diketopiperazine of (+)-12. Interestingly, bis(pyridine)silver (I) permanganate promoted hydroxylation of diketopiperazine (+)-16, a substrate with the same diketopiperazine stereochemistry as substrates (+)-10a–10b, led to alcohol (+)-17 (Scheme 2D, 3.00 equiv), along with recovery of 41% of the substrate (+)-16, without oxidation at C15-position, illustrating the strong impact of the N-formyl group. Furthermore, hydroxylation of diketopiperazine (+)-18 gave the triketopiperazine (+)-19 (Scheme 2E, 3.00 equiv) with double oxidation at the methylene, consistent with our observations in the oxidation of diketopiperazine (−)-14, without C–H oxidation adjacent to the acetylated diketopiperazine nitrogen, consistent with the lack of oxidation at C15 with diketopiperazine (+)-16. Given the nuanced reaction outcomes in the representative cases illustrated in Scheme 2, we envisioned a substrate based parameterization of our permanganate promoted diketopiperazine hydroxylation reaction could provide a detailed analysis of these reactivity trends and form the basis for more informed future applications of the chemistry.
Scheme 2.

Representative application of our permanganate–mediated diketopiperazine oxidation chemistry.
While we have gathered a wide range of experimental observations related to this hydroxylation chemistry (Scheme 2), the multiple oxidation outcomes in these complex examples make it difficult to separate individual factors that impact the reaction outcome. To avoid the complexity of studying multiple and non-independent oxidation events in these substrates, we proposed that analysis of a versatile model substrate class could offer informative insight into substrate structural parameters that influence this reaction. We identified hydantoins (Scheme 3) as a versatile class of substrates for our planned investigation as these substrates would allow simplification of the oxidation analysis to a single event, and provide excellent substrate variability needed for model development. Notably, these substrates offer control over both the steric and electronic environment of the single activated C–H bond that would be subject to hydroxylation. An array of hydantoins 21 was assembled on multi-gram scale from readily available amino acid derivatives 20 and phenyl isocyanate.15 Hydantoins 21 along with the corresponding N-substituted hydantoins 22 provided a diverse training set of substrates for our study.
Scheme 3.
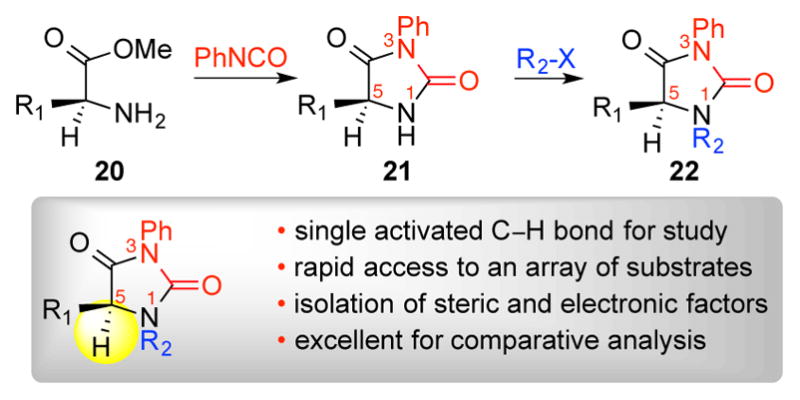
General preparation of hydantoins.
We focused on the use of bis(pyridine)silver(I) permanganate as the oxidant for hydantoin hydroxylation (Table 1) under typical conditions employed in the case studies illustrated in Scheme 2. The bis(pyridine)silver(I) permanganate oxidant was selected over the tetra-n-butylammonium permanganate reagent for this study due to the former oxidant’s broader utility in hydroxylation of complex diketopiperazines.2a Importantly, a standard set of hydroxylation reaction conditions were used for all hydantoins in this study, unless noted otherwise, in order to quantify the relative success of each individual case.16 We began our investigation with the oxidation of the simple alanine derived hydantoin 23a, which underwent permanganate promoted oxidation to provide alcohol 23b (Table 1, entry 1). As we increased the size of the N1-substituents to methyl and phenyl (24a and 25a, respectively), we observed a decrease in the reaction rate and the isolated yield of the corresponding alcohols 24b–25b (Table 1, entries 2–3). Increasing the size of the amino acid side chain, as in the valine derived hydantoin 26a (Table 1, entry 4), decreased the reaction rate relative to hydantoin 23a, but oxidation proceeded completely to alcohol 26b in a good yield under the standard conditions. Increasing the size of the N1 substituent in the presence of the isopropyl group at C5 caused a significant reduction in the reaction rate and yield (Table 1, entries 5–6). Hydroxylation of N-methylated hydantoin 27a afforded only 41% yield of alcohol 27b with a significant amount of starting material remaining, while the N-phenyl hydantoin 28a proved highly recalcitrant toward permanganate oxidation providing 13% yield of alcohol 28b and returning 71% of substrate 28a.
Table 1.
Bis(pyridine)silver (I) Permanganate promoted hydroxylation of hydantoins.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Yield (%) | Recovered Substrate (%) | krela |
| 1 | 23a | Me | H | 78 | 0 | 1 |
| 2 | 24a | Me | Me | 60 | 11 | 0.21 |
| 3 | 25a | Me | Ph | 54 | 18 | 0.14 |
| 4 | 26a | (CH3)2CH | H | 79 | 0 | 0.17 |
| 5 | 27a | (CH3)2CH | Me | 41 | 26 | 0.04 |
| 6 | 28a | (CH3)2CH | Ph | 13 | 71 | 0.12 |
| 7 | 29a | Ph | H | 72 | 0 | Xb |
| 8 | 30a | Ph | Me | 85 | 0 | Xb |
| 9 | 31a | Ph | Ph | 64 | 0 | 0.58 |
| 10 | 32a | CH2CH2CH2 | 16 | 70 | 0.12 | |
| 11 | 33a | (CH3)3C | H | 21 | 30 | 0.17 |
| 12 | 34a | (CH3)2CH2CH | H | 65 | 0 | 0.90 |
| 13 | 35a | (CH3)2CH2CH | Me | 62 | 10 | 0.32 |
| 14 | 36a | Cyclohexyl | H | 57 | 0 | 0.48 |
| 15c | 37a | Cyclohexyl | Me | 72 | 0 | 0.16 |
| 16 | 38a | Cyclohexyl | Ph | 30 | 49 | 0.98 |
| 17 | 39a | Me | (CH3)2CHCH2 | 64 | 8 | 0.26 |
| 18 | 40a | Me | CF3CH2 | 33 | 35 | Xb |
| 19 | 41a | Me | Bn | 30 | 5 | 0.10 |
| 20 | 42a | (CH3)2CH | CF3CH2 | 9 | 71 | 0.02 |
| 21 | 43a | (CH3)2CH | Bn | 7 | 70 | 0.03 |
| 22 | 44a | Me | Ac | 0 | 95 | 0 |
Conditions: substrate (1 equiv), py2AgMnO4 (2 equiv), CH2Cl2 (0.1 M), 23 °C, 1 h, 0.1 mmol scale. Yield of product and recovered starting material after chromatography.
Conditions for relative rate measurement: 23a (1 equiv), substrate (1 equiv), py2AgMnO4 (0.4 equiv), 1-methylnaphthalene (1 equiv), CH2Cl2 (0.1 M), 30 °C, 1 h, 0.1 mmol scale
Accurate relative rate not determined due to poor starting material solubility.
0.2 mmol scale, py2AgMnO4 (4 equiv).
We next sought to vary the C5–H bond environment. Use of phenyl glycine to form hydantoin 29a provided a substrate that has the benefit of a weakened C–H bond due to the adjacent π system. In spite of increased steric encumbrance, oxidation of hydantoin 29a (Table 1, entry 7) to alcohol 29b proceeded efficiently to give the product in 72% yield. Substitution of the N1 position of the phenyl glycine derived hydantoins did not significantly decrease the rate and efficiency of the hydroxylation reaction. Both hydantoins 30a and 31a were oxidized completely under standard conditions, providing products 30b and 31b, respectively (Table 1, entries 8–9). The complete oxidation of the phenyl glycine derived hydantoins is consistent with a weakened C–H bond due to optimal alignment of the π system of the phenyl group with the C–H orbitals.
The oxidation rate of bicyclic proline hydantoin 32a was surprisingly slow and alcohol 32b was isolated in 16% yield (Table 1, entry 10). The 5,5-bicycle likely forces the C–H bond slightly out of the optimal orientation with respect to the carbonyl leading to an increased bond strength and slower oxidation. Further increase of the size of the C5 residue to a tert-butyl group as in hydantoin 33a (Table 1, entry 11), significantly slowed the oxidation to alcohol 33b, even with no substitution at N1. Similar trends were observed for both the leucine derived hydantoins 34a–35a and the cyclohexyl glycine derived hydantoins 36a–38a (Table 1, entry 12–16).
We also prepared hydantoins with varied N1 steric hindrance using hydantoins 23a as starting material. We prepared N1-iso-butyl hydantoin 39a. Hydroxylation of hydantoin 39a proceeded efficiently to alcohol 39b (Table 1, entry 17), demonstrating that steric hindrance at N1, slightly removed from the site of reactivity, has a measurable but reduced impact on the yield of the product. In an effort towards further electronic variation at N1, introduction of a 2,2,2-trifluoroethyl group at N1 gave hydantoins 40a. Hydroxylation of hydantoin 40a proceeded to give alcohol 40b in 33% yield along with returning 35% of starting hydantoin 40a (Table 1, entry 18). Hydroxylation of N1-benzyl hydantoin 41a provided an internal competition between the C5–H and the benzylic CHs. Interestingly, the only isolable oxidation product for hydantoin 41a was alcohol 41b (Table 1, entries 19), albeit in diminished yields compared to the standard substrate 23a, and no benzylic oxidation products were obtained. To probe the interaction between C5 and N1 substituents, we prepared the corresponding valine derived hydantoins 42a and 43a. Hydroxylation of hydantoin 42a exhibited an even greater decrease in reaction rate, compared to hydantoin 40a, providing only 9% of the corresponding alcohol 42b with 71% recovered hydantoin (Table 1, entry 20). Similarly, hydantoin 43a showed greatly diminished reactivity, as compared to hydantoin 41a, providing alcohol 43b in a 7% yield with 70% of the starting hydantoin 43a recovered (Table 1 entry 21). Consistent with the reaction outcome of diketopiperazines (+)-16 and (+)-18 (Scheme 2D&E), exposure of acylated hydantoin 44a to the same reaction conditions resulted in no oxidation (Table 1, entry 22).
While the mechanism of permanganate oxidation of toluene to benzoic acid has been studied computationally,10 supporting a C–H abstraction–rebound pathway, there is need for additional informative experimental data to assist with a detailed analysis of this reaction in complex settings. We initiated our studies by preparing the C5–D hydantoin 24a-d1 and comparing the rate of oxidation to that of hydantoin 24a (Scheme 4A). Both competition reactions and independent initial rate measurements confirmed that the rate determining step in the reaction involves C–H bond cleavage. However, the disparity between the internal and external KIE values, although previously described,17 may be a consequence of the amount of oxidant used (super-stoichiometric for external and sub-stoichiometric for internal), choice of solvent, and the level of solubility of the oxidant.
Scheme 4.

Mechanistic investigations of permanganate mediated oxidation of hydantoins. Reaction conditions are as described in Table 1 with excess Py2AgMnO4 (2.00 equiv).
Throughout our synthetic studies, the permanganate mediated diketopiperazine dihydroxylation has been highly diastereoselective, often yielding a single diastereomer. In fact, the data are consistent with a stereospecific hydroxylation except in cases that prevent this outcome due to severe steric blocking leading to a competing reaction manifold or a post-oxidation ionization step.6d This observation has been both synthetically useful and mechanistically insightful, allowing us to support the hypothesis that the oxygen rebound to the transiently generated radical center is rapid enough to prevent inversion of the radical.6a The hydroxylation of enantiomerically enriched hydantoin 33a provided further support for a stereospecific and stereoretentive hydroxylation under typical conditions (2.0 equiv oxidant, 1 h).16 While the oxidation proceeded further in acetonitrile as compared to dichloromethane as solvent, in both solvents the product 33b was isolated without loss of enantiomeric enrichment (Scheme 4B).
We also carried out the hydroxylation reaction with a set of hydantoins that informed a relative rate analysis as a function of the N-aryl substituent wherein a more electron poor substrate was modestly faster (Scheme 4C). While C–H abstraction is the rate determining step based on KIE experiments, this trend is consistent with the electron withdrawing substituent slightly better facilitating the formation of the short lived radical intermediate, likely due to enhancement of the C4-carbonyl for a captodative stabilization of the radical.
We next focused on how the oxidation rate of a series of hydantoins could be used to approximate selectivity of more complex diketopiperazine oxidations. To accomplish this, competitive rate measurements were performed between a training set of hydantoins with differing R1 and R2 groups (Scheme 3) and hydantoin 23a (Table 1, Entry 1) with the goal of identifying the factors that contribute to the reaction success. A relative rate constant, krel, was obtained for each substrate, and this was converted to a transition state energy difference of ΔΔG‡ using the equation ΔΔG‡ = −RTln(krel).16 Several molecular descriptors, which have the potential to correlate with ΔΔG‡, were then calculated from structures of the substrates computationally optimized using the M06-2X functional and Def2TZVP basis set. These parameters include sterimol values of the R1 and R2 substituents, IR vibrations and intensities,18 13C and 1H NMR shifts, effective charges,19 Hammett20 and Taft21 parameters, various molecular angles, and several percent buried volume (%Vbur) measurements, a parameter traditionally used in organometallic chemistry,22 with spherical radii varying between 1.75 and 3.50 Å (Figure 2). After parameter collection, an optimized correlation between these parameters and ΔΔG‡ was achieved using linear regression fitting to quantitatively analyze the substituent effects on the oxidation rate.18, 19
Figure 2.
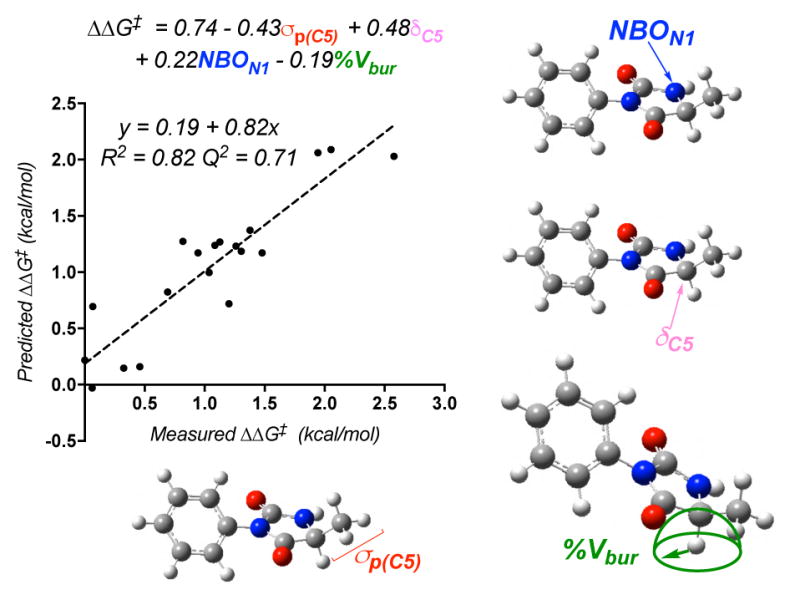
Model comparing predicted ΔΔG‡ to measured ΔΔG‡ for the hydantoin library utilizing the Taft parameter σp, a nitrogen NBO charge, a 13C NMR shift, and percent buried volume of the abstracted hydrogen.
Previous studies from one of our groups23 and others24 have identified numerous multi-dimensional correlations between molecular parameters and reaction outcomes. The computationally derived parameters in this study only revealed relatively complex models to describe the rate measurement, the simplest of which involved four unique terms. These parameters include the Hammett parameter σp of the carbon substituent (σp(C5)), the NBO charge of the nitrogen adjacent to the hydrogen to be abstracted (NBON1), the calculated 13C NMR shift of the carbon which is oxidized (δC5), and the %Vbur at a spherical radius of 2.0 Å of the abstracted hydrogen (Figure 2). An internal validation employing a leave-one-out analysis (Q2=0.71) suggests a relatively robust model. Considering the apparent complexity of the model likely resulting from the structural variance included in the training set, we selected to deconstruct the terms to facilitate understanding as to how selectivity is imparted by the oxidant.
Isolating the model’s parameters revealed notable trends among varying subsets of hydantoins. Variations in the N1 substituent are described by σp(C5) and the N1 NBO charge. In considering σp(C5), it is clear that hydantoins with the same C5 substituent cluster together. However, N1-phenyl substituents are unique with a much higher σp(C5) in each set (Figure 3), corresponding with a moderately low relative rate for these substrates. Because σp describes resonance and electronic effects, it is reasonable that phenyl would have a greater impact on the models than the remainder of the aliphatic substituents. When N1 substituents were compared, the N1–H substituted hydantoins were found to have a much lower N1 NBO charge than the remainder of the hydantoins, accompanied by their high rate of oxidation (Figure 4A). Generation of a model excluding the N1-H subset resulted in a reasonable model similar to that in Figure 2, with an R2 of 0.75 and a Q2 of 0.56 with the exclusion of the N1 NBO charge.16 Thus, this parameter mainly functions to normalize the N1–H subset.
Figure 3.
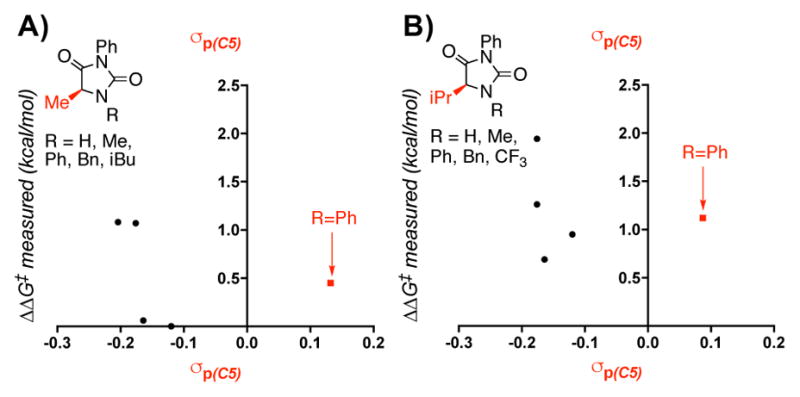
Descriptions of variations in the electronic contribution of the N1 substituent to the model.
Figure 4.
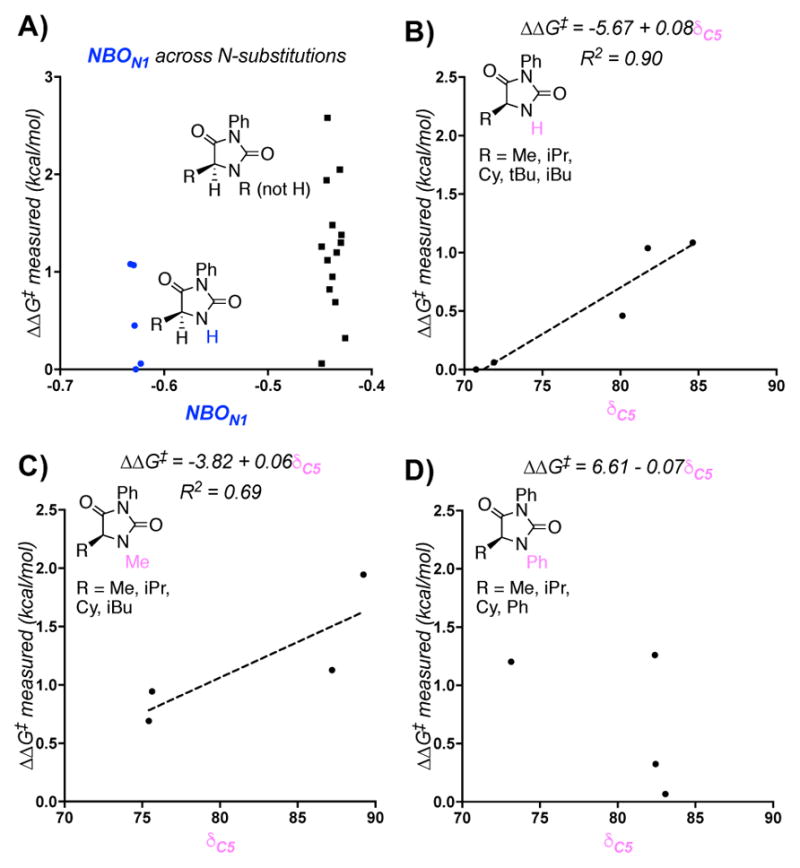
The NBO charge of N1 describes the N1-H subset (A) and the 13C NMR shift describes variation in the C5 substituent (B,C,D).
While σp(C5) and the N1 NBO charge shed light on effects of the N1 substituent, variations in C5 are described by examining the isotropic 13C NMR shift of C5. Holding the N1-substituent constant, several trends were apparent. For the N1–H and N1–Me subsets, as well as a subset describing a variety of other R groups, the energy barrier increases as a function of the 13C NMR chemical shift (Figure 4B and 4C). Greater electron density thus appears to stabilize radical formation, in agreement with the KIE studies that suggest rate limiting hydrogen atom abstraction. This trend is not observed in the set of hydantoins with N1-phenyl substitution (Figure 4D). In the case of these substrates, no clear trend is present. This suggests that the contributions of the N1-phenyl substituent have a larger impact on the nature of C5 than any substitution at C5. The %Vbur measurement did not yield clear patterns when compared to relative rates for hydantoin subsets with various N1 and C5 substituents. This likely occurs because this parameter accounts for integrated steric effects of both substituents.
Using the model developed above (Figure 2) and the mechanistic insight regarding influential substrate parameters governing the outcome of this hydroxylation reaction, we sought to evaluate the effectiveness of this model in the analysis of complex diketopiperazine substrate case studies illustrated in Scheme 2. We began by correlating the hydantoin and diketopiperazine structures (Figure 5), and identifying 14 distinct sites of oxidation (Scheme 2). The C2-symmetric dimeric diketopiperazines were modeled as simplified C3-tert-butyl variants and the Ca–H bonds of six diketopiperazine substrates were computationally analyzed, including three positions that do not undergo oxidation, to afford the same parameters that were extracted in our hydantoin analysis (Figure 5).16
Figure 5.
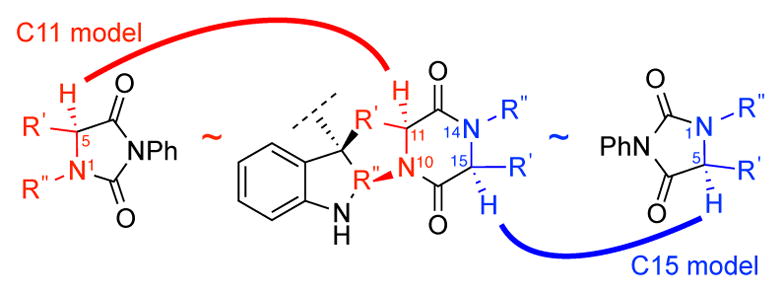
Structural correlation used to calculate parameters for hydantoins and the corresponding diketopiperazines.
Using the model generated from the hydantoin derivatives, the predicted ΔΔG‡ was calculated for each of the diketopiperazine substrates. This required that the parameters for hydantoin and diketopiperazine reactive sites be normalized together, but the intention of this analysis was to use the information gained from the hydantoin series to explain diketopiperazine trends and not to directly compare the diketopiperazine reactivity to the hydantoin reactivity. It therefore must be emphasized that the calculated ΔΔG‡ does not represent an actual energy barrier between the diketopiperazines and substrate 23a, but rather an abstract, but nevertheless descriptive, numerical output by which the model generated by the hydantoin derivatives can categorize the diketopiperazine derivatives. These calculations divided the diketopiperazine reactive sites into two subsets: one with a calculated energy barrier of >0.15 kcal/mol corresponding to oxidized diketopiperazine sites, and another with a calculated energy barrier of <0.15 kcal/mol corresponding with diketopiperazine sites that fail to undergo oxidation. For this analysis, we viewed a successful diketopiperazine oxidation site as one that provides the corresponding alcohol product, whereas a diketopiperazine site that does not yield the corresponding alcohol is viewed as a failed oxidation site. In this significant extrapolation of the simple model system, 13 out the 14 sites evaluated are correctly predicted and consistent with observed experimental data (Figure 6).
Figure 6.
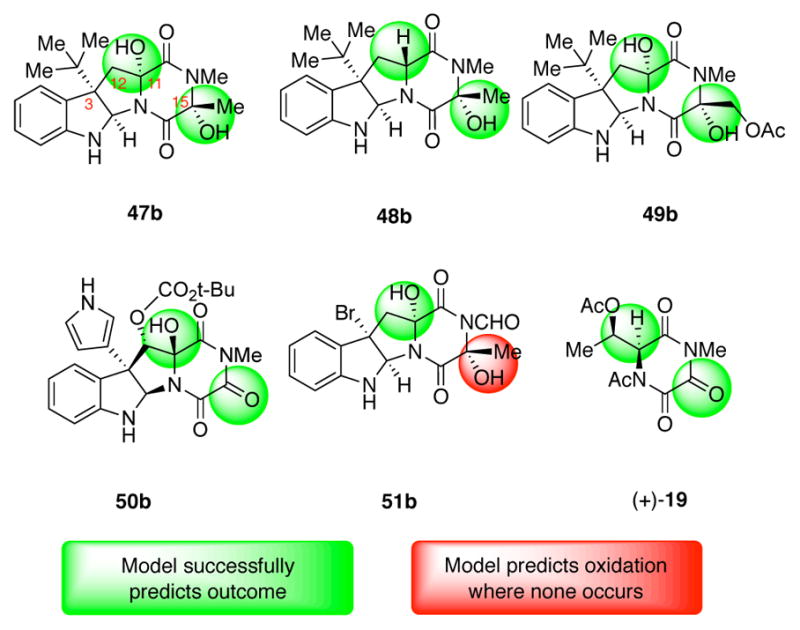
Relation of complex diketopiperazine oxidation outcomes predicted by model to experimental outcomes.
As described above, a C3-tert-butyl diketopiperazine was used to represent the dimeric diketopiperazine (+)-10a (Scheme 2A) and the model developed in this study accurately predicts the successful oxidation of both C11- and C15-positions to give diol 47b (Figure 6).25 Furthermore, when the corresponding C11-epimeric diketopiperazine was used as substrate for this analysis the model correctly predicts the mono oxidation at C15-position to give alcohol 48b consistent with prior experimental data.6a The model also predicted the formation of dihydroxylated C3-t-butyl diketopiperazine 49b, an outcome reminiscent of the oxidation of diketopiperazine (+)-10b to tetraol (+)-11b (Scheme 2A). Interestingly, application of the model to the C3-pyrrole diketopiperazine as a surrogate for diketopiperazine (−)-14 (Scheme 2C) led to the correct prediction of oxidation at both C11- and C15-positions to afford triketopiperazine 50b (Figure 6), corresponding to triketopiperazine (−)-15 (Scheme 2C).26 While the model successfully predicts hydroxylation at C11-position of diketopiperazine (+)-16 (Scheme 2D), it fails to predict the lack of reactivity at the C15-position and offers diol 51b (Figure 6) as the oxidation product instead of the alcohol (+)-17 (Scheme 2D). However, application of the model to the analysis of the simpler diketopiperazine (+)-18 (Scheme 2E) led to the correct prediction for the formation of triketopiperazine (+)-19 (Figure 6) as the oxidation product in agreement with our experimental findings. Importantly, the model not only predicts double oxidation at the methylene of diketopiperazine (+)-18, it also predicts the lack of oxidation at carbon adjacent to the acetylated nitrogen. Indeed, this model demonstrates high fidelity with our experimental results and holds great promise for further development and application to complex substrates as a predictive tool.
While the model correctly predicts the experimental outcome in the majority of cases, collecting multiple parameters and performing a linear regression analysis can be time-consuming. Thus, the model’s parameters were also examined individually to determine whether a single parameter might differentiate between sites that were oxidized and those that were not oxidized in several complex diketopiperazines. The %Vbur for a spherical radius of 2.00 Å was found to be higher than 75% for substrates that did not undergo oxidation, and lower than 75% for those which were oxidized. This single parameter approximation proved slightly less descriptive than the full model prediction, accurately predicting 12 of the 14 diketopiperazine potential sites of oxidation. The %Vbur parameter incorrectly predicts oxidation at the C15 site to yield 51b (Figure 6), the same site incorrectly predicted by the model. This parameter also incorrectly predicts that oxidation will not occur at one C11 position, which is oxidized, thus yielding a triketopiperazine derivative of 50b with no oxidation at the C11 site. Because %Vbur is expected to account jointly for steric effects of both N1 and C5 substituents while the remaining parameters focus on one of the two substituents, its predictive ability indicates that the combined steric effects of both substituents play the most significant role in predicting success of oxidation in the diketopiperazine substrates. These results also suggest that the %Vbur parameter, traditionally used to describe ligand-metal complexes, may be a significant measurement predicting the reactivity of organic substrates.
CONCLUSIONS
In summary, the hydroxylation of complex diketopiperazines promoted by bis(pyridine)silver (I) permanganate has been a critical transformation for the total synthesis of a variety of epipolythiodiketopiperazines. We have described the development of a mathematical model for this hydroxylation reaction using hydantoin derivatives by relating the relative rates of oxidation to various calculated molecular descriptors. The use of hydantoins as the training set of substrates was desirable as this allowed simplification of the oxidation analysis to a single event, and provided excellent substrate variability with control over both the steric and electronic environment of the activated C–H bond subject to hydroxylation. The model prioritizes Hammett values and %Vbur as key contributing factors in the hydantoin series. Importantly, the model may be applied to more complex substrates as illustrated in Figure 6 and it can correctly predict the hydroxylation outcome with a high level of agreement with experimental results. This will provide a roadmap to synthetic design and application of this late-stage oxidation reaction in complex synthesis and provides a strategic guide into mapping other site selective oxidation processes.
Supplementary Material
Acknowledgments
M.M. and B.M.N. acknowledge financial support by NIH-NIGMS (GM089732) and Amgen. B.M.N. thanks the National Science Foundation for a graduate fellowship. We thank the NSF under CCI Center for Selective C–H Functionalization (CHE-1205646). A.J.B. thanks the Center for Selective C–H Functionalization for the CCHF Summer Undergraduate Research Program for an undergraduate fellowship.
Footnotes
Notes
The authors declare no competing financial interests.
Experimental procedures, spectroscopic data, and copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For reviews on epipolythiodiketopiperazines, see: Jordan TW, Cordiner SJ. Trends Pharmacol Sci. 1987;8:144.Waring P, Eichner RD, Müllbacher A. Med Res Rev. 1988;8:499. doi: 10.1002/med.2610080404.Gardiner DM, Waring P, Howlett BJ. Microbiology. 2005;151:1021. doi: 10.1099/mic.0.27847-0.Patron NJ, Waller RF, Cozijnsen AJ, Straney DC, Gardiner DM, Nierman WC, Howlett BJ. BMC Evol Biol. 2007;7:174. doi: 10.1186/1471-2148-7-174.Huang R, Zhou X, Xu T, Yang X, Liu Y. Chem Biodiversity. 2010;7:2809. doi: 10.1002/cbdv.200900211.Iwasa E, Hamashima Y, Sodeoka M. Isr J Chem. 2011;51:420.
- 2.(a) Kim J, Movassaghi M. Acc Chem Res. 2015;48:1159. doi: 10.1021/ar500454v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim J, Movassaghi M. Chem Soc Rev. 2009;38:3035. doi: 10.1039/b819925f. [DOI] [PubMed] [Google Scholar]
- 3.(a) Rightsel WA, Schneider HG, Sloan BJ, Graf PR, Miller FA, Bartz QR, Ehrlich J, Dixon GJ. Nature. 1964;204:1333. doi: 10.1038/2041333b0. [DOI] [PubMed] [Google Scholar]; (b) Müllbacher A, Waring P, Tiwari-Palni U, Eichner RD. Mol Immunol. 1986;23:231. doi: 10.1016/0161-5890(86)90047-7. [DOI] [PubMed] [Google Scholar]; (c) Soledade M, Pedras C, Sèguin-Swartz G, Abrams SR. Phytochemistry. 1990;29:777. [Google Scholar]; (d) Waring P, Beaver J. Gen Pharmacol. 1996;27:1311. doi: 10.1016/s0306-3623(96)00083-3. [DOI] [PubMed] [Google Scholar]; (e) Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM. Cancer Cell. 2004;6:33. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]; (f) Vigushin DM, Mirsaidi N, Brooke G, Sun C, Pace P, Inman L, Moody CJ, Coombes RC. Med Oncol. 2004;21:21. doi: 10.1385/MO:21:1:21. [DOI] [PubMed] [Google Scholar]; (g) Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Nat Chem Biol. 2005;1:143. doi: 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]; (h) Yanagihara M, Sasaki-Takahashi N, Sugahara T, Yamamoto S, Shinomi M, Yamashita I, Hayashida M, Yamanoha B, Numata A, Yamori T, Andoh T. Cancer Sci. 2005;96:816. doi: 10.1111/j.1349-7006.2005.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Dong JY, He HP, Shen YM, Zhang KQ. J Nat Prod. 2005;68:1510. doi: 10.1021/np0502241. [DOI] [PubMed] [Google Scholar]; (j) Zheng CJ, Kim CJ, Bae KS, Kim YH, Kim WJ. J Nat Prod. 2006;69:1816. doi: 10.1021/np060348t. [DOI] [PubMed] [Google Scholar]; (k) Isham CR, Tibodeau JD, Jin W, Timm MM, Bible KC. Blood. 2007;109:2579. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Cook KM, Hilton ST, Mecinovic J, Motherwell WB, Figg WD, Schofield CJ. J Biol Chem. 2009;39:26831. doi: 10.1074/jbc.M109.009498. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Lakshmikuttyamma A, Scott SA, DeCoteau JF, Geyer CR. Oncogene. 2010;29:576. doi: 10.1038/onc.2009.361. [DOI] [PubMed] [Google Scholar]; (n) Jiang CS, Guo YW. Mini-Rev Med Chem. 2011;11:728. doi: 10.2174/138955711796355276. [DOI] [PubMed] [Google Scholar]; (o) Coleman JJ, Ghosh S, Okoli I, Mylonakis E. PLOS ONE. 2011;6:e25321. doi: 10.1371/journal.pone.0025321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyer N, Morrison KC, Kim J, Hergenrother PJ, Movassaghi M. Chem Sci. 2013;4:1646. doi: 10.1039/C3SC50174D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Nicolaou KC, Lu M, Totokotsopoulos S, Heretsch P, Giguère D, Sun YP, Sarlah D, Nguyen TH, Wolf IC, Dmee DF, Day CW, Bopp S, Winzeler EA. J Am Chem Soc. 2012;134:17320. doi: 10.1021/ja308429f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeLorbe JE, Horne D, Jove R, Mennen SM, Nam S, Zhang FL, Overman LE. J Am Chem Soc. 2013;135:4117. doi: 10.1021/ja400315y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Baumann M, Dieskau AP, Loertscher BM, Walton MC, Nam S, Xie J, Horne D, Overman LE. Chem Sci. 2015;6:4451. doi: 10.1039/c5sc01536g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238. doi: 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim J, Movassaghi M. J Am Chem Soc. 2010;132:14376. doi: 10.1021/ja106869s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Boyer N, Movassaghi M. Chem Sci. 2012;3:1798. doi: 10.1039/C2SC20270K. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Coste A, Kim J, Adams TC, Movassaghi M. Chem Sci. 2013;4:3191. doi: 10.1039/C3SC51150B. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Adams TC, Payette JN, Cheah JH, Movassaghi M. Org Lett. 2015;17:4268. doi: 10.1021/acs.orglett.5b02059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For selected epidithiodiketopiperazine total syntheses, see: Trown PW. Biochem Biophys Res Commun. 1968;33:402. doi: 10.1016/0006-291x(68)90585-8.Hino T, Sato T. Tetrahedron Lett. 1971;12:3127.Öhler E, Poisel H, Tataruch F, Schmidt U. Chem Ber. 1972;105:635. doi: 10.1002/cber.19721050229.Öhler E, Tataruch F, Schmidt U. Chem Ber. 1973;106:165. doi: 10.1002/cber.19731060120.Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6490. doi: 10.1021/ja00800a078.Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6492. doi: 10.1021/ja00800a078.Kishi Y, Nakatsuka S, Fukuyama T, Havel M. J Am Chem Soc. 1973;95:6493. doi: 10.1021/ja00800a079.Yoshimura J, Nakamura H, Matsunari K, Sugiyama Y. Chem Lett. 1974:559.Ottenheijm HCJ, Herscheid JDM, Kerkhoff GPC, Spande TF. J Org Chem. 1976;41:3433. doi: 10.1021/jo00883a024.Fukuyama T, Kishi Y. J Am Chem Soc. 1976;98:6723. doi: 10.1021/ja00437a063.Herscheid JDM, Nivard RJF, Tijhuis MW, Ottenheijm HCJ. J Org Chem. 1980;45:1885.Williams RM, Rastetter WH. J Org Chem. 1980;45:2625.Srinivasan A, Kolar AJ, Olsen RK. J Heterocycl Chem. 1981;18:1545.Fukuyama T, Nakatsuka S, Kishi Y. Tetrahedron. 1981;37:2045.Shimazaki N, Shima I, Hemmi K, Tsurumi Y, Hashimoto M. Chem Pharm Bull. 1987;35:3527. doi: 10.1248/cpb.35.3527.Miknis GF, Williams RM. J Am Chem Soc. 1993;115:536.Wu Z, Williams LJ, Danishefsky SJ. Angew Chem, Int Ed. 2000;39:3866. doi: 10.1002/1521-3773(20001103)39:21<3866::AID-ANIE3866>3.0.CO;2-E.Aliev AE, Hilton ST, Motherwell WB, Selwood DL. Tetrahedron Lett. 2006;47:2387.Overman LE, Sato T. Org Lett. 2007;9:5267. doi: 10.1021/ol702518t.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J Am Chem Soc. 2010;132:4078. doi: 10.1021/ja101280p.DeLorbe JE, Jabri SY, Mennen SM, Overman LE, Zhang FL. J Am Chem Soc. 2011;133:6549. doi: 10.1021/ja201789v.Nicolaou KC, Totokotsopoulos S, Giguère D, Sun YP, Sarlah D. J Am Chem Soc. 2011;133:8150. doi: 10.1021/ja2032635.Codelli JA, Puchlopek ALA, Reisman SE. J Am Chem Soc. 2012;134:1930. doi: 10.1021/ja209354e.Jabri SY, Overman LE. J Org Chem. 2013;78:8766. doi: 10.1021/jo4015479.DeLorbe JE, Horne D, Jove R, Mennen SM, Nam S, Zhang FL, Overman LE. J Am Chem Soc. 2013;135:4117. doi: 10.1021/ja400315y.Takeuchi R, Shimokawa J, Fukuyama T. Chem Sci. 2014;5:2003.Kurogi T, Okaya S, Fujiwara H, Okano K, Tokuyama H. Angew Chem Int Ed. 2016;55:283. doi: 10.1002/anie.201507830.Wang H, Regan CJ, Codelli JA, Romanato P, Puchlopek-Dermenci ALA, Reisman SE. Org Lett. 2017;19:1698. doi: 10.1021/acs.orglett.7b00418.
- 8.Firouzabadi H, Vessal B, Naderi M. Tetrahedron Lett. 1982;23:1847. [Google Scholar]
- 9.Sala T, Sargent MV. J Chem Soc Chem Commun. 1978:253. [Google Scholar]
- 10.(a) Cullis CF, Ladbury JW. J Chem Soc. 1955:555. [Google Scholar]; (b) Gardner KA, Mayer JM. Science. 1995;269:1849. doi: 10.1126/science.7569922. [DOI] [PubMed] [Google Scholar]; (c) Strassner T, Houk KN. J Am Chem Soc. 2000;122:7821. [Google Scholar]
- 11.(a) Miller JJ, Sigman MS. Angew Chem Int Ed. 2008;47:771. doi: 10.1002/anie.200704257. [DOI] [PubMed] [Google Scholar]; (b) Harper KC, Sigman MS. Science. 2011;333:1875. doi: 10.1126/science.1206997. [DOI] [PubMed] [Google Scholar]; (c) Harper KC, Sigman MS. Proc Natl Acad Sci U S A. 2011;108:2179. doi: 10.1073/pnas.1013331108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Harper KC, Bess EN, Sigman MS. Nat Chem. 2012;4:366. doi: 10.1038/nchem.1297. [DOI] [PubMed] [Google Scholar]; (e) Harper KC, Vilardi SC, Sigman MS. J Am Chem Soc. 2013;135:2482. doi: 10.1021/ja4001807. [DOI] [PubMed] [Google Scholar]; (f) Bess EN, Bischoff AJ, Sigman MS. Proc Natl Acad Sci U S A. 2014;111:14698. doi: 10.1073/pnas.1409522111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Bess EN, DeLuca RJ, Tindall DJ, Oderinde MS, Roizen JL, Du Bois J, Sigman MS. J Am Chem Soc. 2014;136:5783. doi: 10.1021/ja5015508. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Milo A, Bess EN, Sigman MS. Nature. 2014;507:210. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]; (i) Bess EN, Guptill DM, Davies HML, Sigman MS. Chem Sci. 2015;6:3057. doi: 10.1039/c5sc00357a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Milo A, Neel AJ, Toste FD, Sigman MS. Science. 2015;347:737. doi: 10.1126/science.1261043. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Neel AJ, Milo A, Sigman MS, Toste FD. J Am Chem Soc. 2016;138:3863. doi: 10.1021/jacs.6b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Yamamoto E, Hilton MJ, Orlandi M, Saini V, Toste FD, Sigman MS. J Am Chem Soc. 2016;138:15877. doi: 10.1021/jacs.6b11367. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Niemeyer ZL, Milo A, Hickey DP, Sigman MS. Nature Chem. 2016;8:610. doi: 10.1038/nchem.2501. [DOI] [PubMed] [Google Scholar]; (n) Sigman MS, Harper KC, Bess EN, Milo A. Acc Chem Res. 2016;49:1292. doi: 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]; (o) Christian AH, Niemeyer ZL, Sigman MS, Toste FD. ACS Catal. 2017;7:3973. doi: 10.1021/acscatal.7b00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Fox EM, Howlett BJ. Mycol Res. 2008;112:162. doi: 10.1016/j.mycres.2007.08.017. [DOI] [PubMed] [Google Scholar]; (b) Kirby GW, Robins DJ. In: The Biosynthesis of Mycotoxins: A Study in Secondary Metabolism. Steyn PS, editor. Academic Press; New York: 1980. p. 301. [Google Scholar]
- 13.(a) Scharf DH, Remme N, Habel A, Chankhamjon P, Scherlach K, Heinekamp T, Hortschansky P, Brakhage AA, Hertweck C. J Am Chem Soc. 2011;133:12322. doi: 10.1021/ja201311d. [DOI] [PubMed] [Google Scholar]; (b) Scharf DH, Habel A, Heinekamp T, Brakhage AA, Hertweck C. J Am Chem Soc. 2014;136:11674. doi: 10.1021/ja5033106. [DOI] [PubMed] [Google Scholar]
- 14.For discussion on impact of electron withdrawing groups on C–H oxidation, see: Ishihara Y, Baran PS. Synlett. 2010;12:1733.Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960. doi: 10.1002/anie.201201666.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. Chem Soc Rev. 2016;45:546. doi: 10.1039/c5cs00628g.Engle KM, Yu JQ. J Org Chem. 2013;78:8927. doi: 10.1021/jo400159y.Neufeldt SR, Jimènez-Osès G, Huckins JR, Thiel OR, Houk KN. J Am Chem Soc. 2015;137:9843. doi: 10.1021/jacs.5b03535.Gormisky PE, White MC. J Am Chem Soc. 2013;135:14052. doi: 10.1021/ja407388y.Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936. doi: 10.1021/ar300014f.Fan Z, Ni J, Zhang A. J Am Chem Soc. 2016;138:8470. doi: 10.1021/jacs.6b03402.Liao K, Negretti S, Musaev DG, Bacsa J, Davies HML. Nature. 2016;533:230. doi: 10.1038/nature17651.Gensch T, Hopkinson MN, Glorius F, Wencel-Delord J. Chem Soc Rev. 2016;45:2900. doi: 10.1039/c6cs00075d.
- 15.(a) Ware E. Chem Rev. 1950;46:403. doi: 10.1021/cr60145a001. [DOI] [PubMed] [Google Scholar]; (b) Konnert L, Colacino E. Chem Rev. 2017 doi: 10.1021/acs.chemrev.7b00067. [DOI] [PubMed] [Google Scholar]
- 16.See the Supporting Information for details.
- 17.(a) Ye X, Liu G, Popp BV, Stahl SS. J Org Chem. 2011;76:1031. doi: 10.1021/jo102338a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Simmons EM, Hartwig JF. Angew Chem Int Ed. 2012;51:3066. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 18.Milo A, Bess EN, Sigman MS. Nature. 2014;507:210–214. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- 19.Santiago CB, Milo A, Sigman MS. J Am Chem Soc. 2016;138:13424. doi: 10.1021/jacs.6b08799. [DOI] [PubMed] [Google Scholar]
- 20.(a) Hammett LP. Chem Rev. 1935;17:125. [Google Scholar]; (b) Pratt DA, DiLabio GA, Mulder P, Ingold KU. Acc Chem Res. 2004;37:334. doi: 10.1021/ar010010k. [DOI] [PubMed] [Google Scholar]
- 21.(a) Taft RW. J Am Chem Soc. 1952;74:2729. [Google Scholar]; (b) Taft RW. J Am Chem Soc. 1952;74:3120. [Google Scholar]; (c) Taft RW. J Am Chem Soc. 1953;75:4538. [Google Scholar]; (d) Hansch C, Leo A, Hoekman D. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology. American Chemical Society; Washington DC: 1995. [Google Scholar]
- 22.(a) Clavier H, Nolan SP. Chem Commun. 2010;46:841. doi: 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]; (b) Falivene L, Credendino R, Poater A, Petta A, Serra L, Oliva R, Scarano V, Cavallo L. Organometallics. 2016;35(13):2286. [Google Scholar]
- 23.(a) Harper KC, Sigman MS. Science. 2011;333(6051):1875. doi: 10.1126/science.1206997. [DOI] [PubMed] [Google Scholar]; (b) Harper KC, Bess EN, Sigman MS. Nat Chem. 2012;4:366. doi: 10.1038/nchem.1297. [DOI] [PubMed] [Google Scholar]; (c) Milo A, Neel AJ, Toste FD, Sigman MS. Science. 2015;347:737. doi: 10.1126/science.1261043. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mougel V, Santiago CB, Zhizhko PA, Bess EN, Varga J, Frater G, Sigman MS, Coperet C. J Am Chem Soc. 2015;137:6699. doi: 10.1021/jacs.5b03344. [DOI] [PubMed] [Google Scholar]; (e) Neel AJ, Milo A, Sigman MS, Toste FD. J Am Chem Soc. 2016;138:3863. doi: 10.1021/jacs.6b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Niemeyer ZL, Milo A, Hickey DP, Sigman MS. Nat Chem. 2016;8:610. doi: 10.1038/nchem.2501. [DOI] [PubMed] [Google Scholar]; (g) Santiago CB, Milo A, Sigman MS. J Am Chem Soc. 2016;138:13424. doi: 10.1021/jacs.6b08799. [DOI] [PubMed] [Google Scholar]; (h) Sigman MS, Harper KC, Bess EN, Milo A. Acc Chem Res. 2016;49:1292. doi: 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]; (i) Orlandi M, Coelho JAS, Hilton MJ, Toste FD, Sigman MS. J Am Chem Soc. 2017;139:6803. doi: 10.1021/jacs.7b02311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Piou T, Romanov-Michailidis F, Romanova-Michaelides M, Jackson KE, Semakul N, Taggart TD, Newell BS, Rithner CD, Paton RS, Rovis T. J Am Chem Soc. 2017;139:1296–1310. doi: 10.1021/jacs.6b11670. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu K, Doyle AG. Nat Chem. 2017;9:779. doi: 10.1038/nchem.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.The model determines the potential for oxidation at each position and does not provide stereochemical information. For illustrative purposes, the alcohol stereochemistry of the predicted products is depicted consistent with prior experimental observations, see reference 2a.
- 26.For substrates (−)-14 and (+)-18, each diketopiperazine–methylene C–H bond was considered independently, and parameters were collected from the starting material. Additionally, parameters were collected for partially oxidized hemiaminal intermediates in which the C15 position was oxidized cis or trans with respect to the C11 methine. These structures were used to probe reactivity of the methylene C–H bonds and did not display a significant difference in modeling or prediction.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.