

Abstract
Purpose of review
Post-translational modifications (PTMs), specifically serine phosphorylation, are essential for determination and tuning up an activity of many proteins, including those that are involved in the control of gene transcription. Transcription factors PPARγ2 and RUNX2 are essential for mesenchymal stem cell (MSC) commitment to either adipocyte or osteoblast lineage. This review is summarizing current knowledge how serine phosphorylation PTMs regulate activities of both transcription factors and MSCs lineage commitment.
Recent finding
Both PPARγ2 and RUNX2 transcriptional activities are regulated by similar PTMs, however with an opposite outcome. The same p38 MAPK mediates serine phosphorylation that leads to activation of RUNX2 and inactivation of PPARγ2. The process of protein phosphorylation is balanced with a process of protein dephosphorylation. Protein phosphatase 5 simultaneously dephosphorylates both proteins, which results in activation of PPARγ2 and inactivation of RUNX2.
Summary
This review provides a summary of the “yinyang” fine-tuned mechanism by which p38 MAPK and PP5 regulate MSCs lineage commitment.
Keywords: osteoblasts, adipocytes, p38 MAPK, PP5, RUNX2, PPARγ2, bone, rosiglitazone
Introduction
It has been accepted that marrow mesenchymal stem cells (MSCs) lineage commitment occurs by a stochastic mechanism in which factors specific to one lineage dominate and suppress the activity of factors of another lineage [1]. The lineage commitment can be triggered by environmental cues consisting of BMP and Wnt signaling for osteoblast and fatty acids and hormonal stimulation for adipocytes resulting in transcriptional upregulation of two key proteins: the runt-related transcription factor 2/core-binding factor-α1 (RUNX2/CBFA1; also known as AML3 and PEBP2αA) and the peroxisome proliferator-activated receptor gamma2 (PPARγ2 or NR1C3) [2]. Reciprocal regulation of expression of these two transcription factors includes the activity of Wnt signaling which upregulates RUNX2 and suppresses PPARγ2, and suppressive effect of PPARγ2 on Wnt signaling activity and RUNX2 expression [3, 4, 2]. An additional mechanism which operates at the level of activity of both proteins includes TAZ (transcriptional coactivator with PDZ-binding motif) which co-activates RUNX2-dependent gene transcription while repressing PPARγ-dependent gene transcription [5].
Activation of PPARγ nuclear receptor consists of binding specific ligand and formation of heterodimer with another nuclear receptor, retinoid X receptor alpha (RXRα). The antidiabetic drugs thiazolidinediones (TZDs) act as PPARγ full agonists which commit MSCs to adipocyte and irreversibly suppress osteoblast lineage [6]. The induction of adipocyte and suppression of osteoblast lineage by TZDs includes immediate upregulation of PPARγ2-dependent gene transcription and immediate downregulation of RUNX2-dependent gene transcription, both events occurring before changes in the expression of these factors are observed [7]. Such rapid response is consistent with fine-tuned regulation of protein activities at the level of their post-translational modifications (PTMs).
PPARγ and RUNX2 activities are regulated at the level of serine phosphorylation
Protein phosphorylation is a common PTM for as many as 30% of all proteins and encompasses tens of thousands of distinct phosphorylation sites [8]. The phosphorylation process may be specific to particular serine, threonine, or tyrosine residues in the target protein [9]. Phosphorylation introduces a charged and hydrophilic group in the side chain of amino acids, possibly changing a protein's structure by altering interactions with nearby amino acids. Some phosphorylation sites appear to have evolved as conditional ‘on’switches, allowing these proteins, such as RUNX2, to adopt an active conformation only in response to a specific signal [10]. While other phosphorylation sites have evolved as conditional ‘off’ switches blocking the activity these proteins, such as PPARγ.
PPARγ2 activities are linked to the status of serine phosphorylation by members of the ERK and MAP kinase family (p38 MAPK and JNK) and CDK5 [11, 12]. PPARγ2 can be phosphorylated by p38 MAPK at serine 112 (S112), which severely decreases pro-adipocytic transcriptional activity of the receptor [13, 14]. Dephosphorylation of S112 is an immediate response to activation of PPARγ2 with full agonist rosiglitazone and switching on a pro-adipocytic program [15]. In contrast, the inverse PPARγ agonist SR10171, which increases levels of S112 phosphorylation, inhibits proadipocytic activity and renders animals resistant to high fat diet induced obesity [16]. PPARγ2 can also be phosphorylated at serine 273 (S273), and this phosphorylation event occurs shortly after the onset of high fat diet feeding and increases with progressive obesity [17]. Both, phosphorylation of S112 and S273, correlate with dysregulation and decreased expression of PPARγ target genes including adiponectin. Insulin sensitization provided by full and partial PPARγ agonists correlates with their ability to block phosphorylation of PPARγ2 at S273, leading to consideration of this PTM as sensitizing to insulin [17]. The surrounding of S273 amino acids in the PPARγ2 protein form a consensus site favored by CDK5 kinase [18]. A unique feature of CDK5 is that it is activated by system of p35/p25 kinases instead of cyclins [19]. On the organismal level, CDK5 is activated by pro-inflammatory cytokines and circulating free fatty acids which levels increases in obesity [20]. We have recently showed that the phosphorylation status of S273 is also involved in a regulation of osteoclastogenesis. Thus, dephosphorylation of S273, an event that sensitizes to insulin, increases osteoclast differentiation from hematopoietic precursor and increases support for osteoclastogenesis by increasing RANKL expression in cells of mesenchymal lineage [16]. Perhaps it sounds provocative at this point, but the regulation of bone resorption and insulin sensitivity by the same PTM suggests interdependence of these two processes; perhaps as a part of a mechanism leading to the release from the bone matrix of the bioactive and insulin sensitizing form of osteocalcin [21].
RUNX2 is also a phosphorylation target for ERK/MAPK pathway. RUNX2 has several serine residues that are identified as sites for phosphorylation and correlate with either a positive or a negative regulation of RUNX2 activity measured as target gene expression. For instance, phosphorylation at S104 and S451 negatively regulates RUNX2 activity [22, 23]. These PTMs cause RUNX2 ubiquitination and eventual proteasomal degradation. In addition, phosphorylation of RUNX2 at the S369, S373, and S377 by glycogen synthase kinase 3 beta (GSK-3β) is also associated with the loss of activity of this transcription factor [24]. YES-associated protein interacts with RUNX2 resulting in tyrosine phosphorylation and suppression of RUNX2 transcriptional activity [25]. However, MAPK-dependent phosphorylation of S301 and S319 sites is indispensable for activation of RUNX2 and osteoblast differentiation [26]. Besides activation of RUNX2 through phosphorylation of S301 and S319 [27], p38 MAPK can also phosphorylate S28, S31, S244, and S472, which probably act as permissive sites for RUNX2 association with the transcriptional co-activator CREB-binding protein and increase in transcriptional activity [28].
p38 MAPK regulation of adipocyte and osteoblast differentiation
The p38 MAPK family is composed of four proteins: p38α, p38β, p38γ, and p38δ encoded by Mapk14, Mapk11, Mapk12, and Mapk13 genes, respectively. Their coding genes have a distinct tissue distribution and they appear differentially expressed, with p38a the most highly expressed isoform in osteoblasts [29]. The p38 pathway has been implicated in controlling differentiation of mesenchymalcells including marrow MSCs. Thep38 MAPK signaling pathway is critical for skeleton development, maintenance of bone homeostasis, and osteoblast differentiation [30]. On the other hand, the p38 MAPK inhibits adipogenesis on multiple levels including inhibition of upstream regulators of PPARγ activity such as C/EBPα [31], C/EBPα [32], and NFATc4 [33], as well as PPARγ2 through phosphorylation at S112 [34].
Downregulation of p38 activity correlates positively with adipocyte differentiation. Activation of JNK or suppression of p38 MAPK is required for differentiation of 3T3-L1 cells to adipocytes [35]. p38 MAPK activity seems to decrease during 3T3-L1 adipocyte differentiation, and a decrease in p38 MAPK activity correlates with PPARv increased transcriptional activity and adipocyte gene markers expression [35]. Furthermore, pharmacological inhibition or genetic disruption of p38 MAPK has also been shown to increase PPARγ2 transcriptional activity and expression of adiponectin and leptin in vitro [34]. In contrast, rescue of p38 MAPK in mouse embryonic fibroblast p38 MAPK knockout cells reduced PPARγ2 activity to the basal level of wild-type cells [34].
Conversely, the p38 MAPK plays a pivotal role in different steps of osteoblast differentiation. The differentiation of primary human MSCs or murine C2C12 cell line toward osteoblasts requires p38 MAPK activity [36, 37], while its pharmacologic inhibition with SB203580 impairs osteoblast differentiation of MC3T3 cells and expression of phenotype-specific markers including alkaline phosphatase, osteocalcin, and collagen [38, 39]. In vivo deletion of p38 MAPK or its upstream activator TAK1 hampers osteoblast and osteocytes terminal differentiation and function [27, 30, 40]. In fact multiple models of p38 conditional knockout show the overlapping phenotype with models carrying RUNX2 functional deficiency, including animal phenocopy of human cleidocranial dysplasia syndrome that is related to the mutation in Runx2 gene locus. Mutations in genes affecting the outcome of the p38 MAPK pathway can cause developmental bone disorders such as chondrodysplasia, cleidocranial dysplasia or faciogenital dysplasia. In addition, the activity of this signaling pathway is altered in the context of osteoporosis, inflammatory osteolysis, obesity and osteopetrosis. As seen in specific knockout animal models, deleting some key upstream targets of the p38 MAPK pathway, TAK1, MLK3, MKK3/6, NBR1 affects different stages of osteoblast differentiation [30, 40].
Role of protein phosphatase 5 (PP5) in regulation of RUNX2 and PPARγ2 activities
Although a critical role of MAPK activities in the regulation of MSCs differentiation toward osteoblast and away from adipocytes is well documented, until recently there has been essentially no information about the mechanisms which regulate the opposite, PPARγ and RUNX2 dephosphorylation. It is a logical possibility that the fine balance between phosphorylation and dephosphorylation of both proteins constitutes a mechanism responsible for the maintenance of MSCs undifferentiated phenotype and their responsiveness to differentiating factors. For example, activation of PPARγ with full agonist rosiglitazone leads to rapid up-regulation of adipocytic gene expression which possess PPRE sequences in their promoter region, but at the same time there is a PPRE-independent rapid downregulation of osteoblast-specific genes [7]. This suggests that the mechanism suppressing osteoblast gene expression is independent from PPARγ direct transcriptional activity. It has been reported that MAPK activity is decreased as a result of prolonged treatment with rosiglitazone and it correlates with decreased RUNX2 phosphorylation and activity [41]. It has been also shown that the expression of Runx2 gene is decreased in U33/γ2 cells after 72 h of rosiglitazone treatment, much later than the RUNX2-dependent osteoblast gene markers which expression is decreased rapidly 2 h after treatment [7]. This suggests a synchronized and reciprocal mechanism which in the yin yang manner regulates rapid switch in MSCs differentiation toward osteoblasts or adipocytes.
Protein phosphatase 5 (PP5) is a member of the phosphoprotein phosphatase family with specificity for serine and threonine residues [42-44]. However, PP5 is unique in that it contains three consecutive tetratricopeptide repeat (TPR) motifs [45, 46]. The TPR motifs contain binding sites for long-chain, polyunsaturated fatty acids [47, 48] as well as binding sites for HSP90 protein chaperon [49]. Unlike other members of the phosphoprotein phosphatase family in solution PP5 has little catalytic activity, because the TPR domains fold over the catalytic site blocking substrate access [47]. This state of auto-inhibition is reversed when PP5 binds to free fatty acids or HSP90 via its TPR domains, which triggers a conformational change and allows substrate access to the PP5 catalytic site [50].
PP5 is a multi-tasking mediator of cellular responses to environmental and endogenous stimuli. To date, PP5 has been identified as a key effector for inactivation of three major MAPK signal components, Rac GTPase, Raf, and ASK1 [51]. PP5 also plays a role in cell cycle progression in several ways. First, treatment of cells with PP5 antisense RNA leads to hyperphosphorylation of p53 and subsequent cell cycle arrest in the G1 phase [52, 53, 46] Second, PP5 also binds to two proteins, CDC16 and CDC27, which are members of the anaphase-promoting complex (APC) which is required for anaphase initiation and the exit from mitosis [54, 53, 46]. Finally, PP5 plays an important role in DNA-damage repair and cell cycle arrest by attenuating the activities of a checkpoint kinase, ATM (ataxia telangiectasia mutated) [55]. Additionally, PP5 interacts with transcription factors including estrogen receptor [56], glucocorticoid receptor [57], and PPARγ [13].
Upon rosiglitazone treatment PP5 dephosphorylates PPARγ at S112 and promotes pro-adipocytic transcriptional activity in mouse embryonic fibroblasts [13]. Most recently, it has also been shown that in response to rosiglitazone in marrow MSCs PP5 forms complexes with both PPARγ and RUNX2, which results in dephosphorylation of S112 in PPARγ and activation of adipocytic, and dephosphorylation of S319 in RUNX2 and inhibition of osteoblastic activities [15]. In both cases, HSP90 has been found to be present in the complex suggesting that PP5 activation and interaction with PPARγ2 and RUNX2 are mediated through TPR domains. These findings reconcile a concept that PPARγ2 and RUNX2 transcriptional activities are regulated simultaneously albeit with opposite effect on MSCs differentiation. Accordingly, dephosphorylation of PPARγ at S112 and RUNX2 at S319 by PP5 induces adipocytic and suppresses osteoblastic differentiation, whereas phosphorylation of PPARγ at S112 and RUNX2 at S319 by p38 MAPK suppresses adipocytic and induces osteoblastic differentiation. Figure 1 summarizes this relationship.
Figure 1.
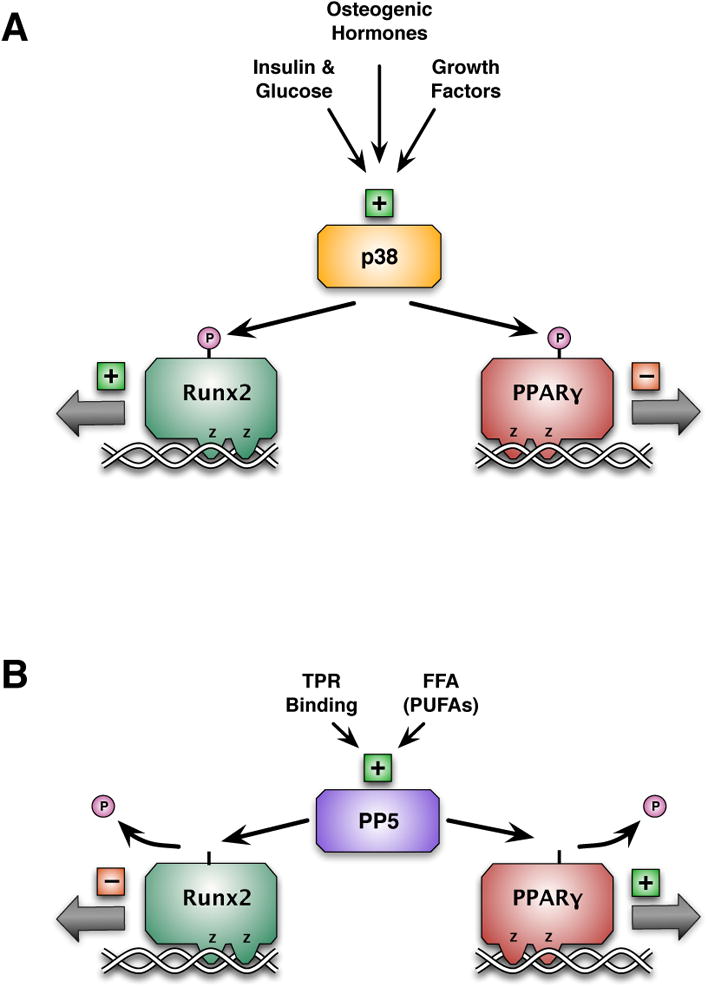
Regulation of RUNX2 and PPARγ at the level of serine phosphorylation. A. p38 MAPK is activated by growth factors, osteogenic hormones, insulin and glucose. Activation of p38 MAPK results in the phosphorylation of both RUNX2 and PPARγ. Phosphorylation of RUNX2 increases its activity, whereas phosphorylation of PPARγ decreases its activity. This result in MSCs allocation shifted to osteoblastic lineage. B. PP5 is activated by free fatty acids and protein interaction through TPR motifs. Activation of PP5 results in dephosphorylation of both RUNX2 and PPARγ.Dephosphorylation of RUNX2 decreases its activity, whereas dephosphorylation of PPARγ increases its activity. This result in MSCs allocation shifted to adipocytic lineage.
PP5 conveys the pro-adipocytic and anti-osteoblastic effects of rosiglitazone
An ultimate support for PP5 role in reciprocal regulation of adipocyte and osteoblast differentiation has been provided by animal model of PP5 deficiency and its skeletal response to pharmacologic treatment with rosiglitazone. Thus, mice with global deficiency in PP5 are characterized with increased bone mass and decreased volume of marrow adipose tissue (MAT). Ex vivo, MSCs deficient in PP5 have an increased propensity to differentiate to osteoblasts and a compromised differentiation to adipocytes. This correlates with increased phosphorylation of S112 in PPARγ2 and S319 in RUNX2 in MSCs [15].
As shown extensively in animals and humans, pharmacologic treatment with TZDs including rosiglitazone leads to bone loss and increased skeletal fragility [58-62]. In mice, rosiglitazone-induced bone loss results from unbalanced bone remodeling with decreased bone formation and increased bone resorption, and is associated with a massive accumulation of MAT [59]. Consistently with PP5 reciprocal regulation of PPARγ2 and RUNX2 activities, animals deficient in PP5 are resistant to rosiglitazone-induced bone loss. Thus, feeding mice rosiglitazone-supplemented diet which induced up to 50% of trabecular bone loss in WT animals, did not affect bone mass in PP5 deficient mice and did not compromise osteoblast activity. Moreover, lack of PP5 protected from accumulation of MAT [15].
Interestingly, PP5 deficiency protected the skeleton entirely from the negative effect of rosiglitazone. This is surprising from two points. First, that PP5 is an exclusive phosphatase conveying rosiglitazone effect on PPARγ2 and RUNX2 activities. Indeed, a series of in vitro tests consistently showed that PP5 is sufficient to convey an entire effect of rosiglitazone on PPARγ2 and RUNX2 activities and MSCs differentiation toward adipocytes and osteoblasts [15]. Second, that PP5 regulates osteoclast differentiation and function known to be stimulated in the PPARγ dependent manner with rosiglitazone. This additional activity of PP5 requires more studies to characterize it in the tissue-specific context. Most importantly, the role of PP5 in regulation of energy metabolism through PPARγ and bone metabolism through PPARγ2/RUNX2 warrants more specific studies to determine the role of this protein in the cross talk between bone and energy metabolism.
Conclusions
The close relationship between energy and bone metabolism includes variety cues and outcomes which provide for mechanistic responses. The inter-relationship between p38 MAPK and PP5 signaling is one of the paradigms of yin yang regulation of MSCs differentiation and regulation of bone and energy metabolism. Corroboration of these mechanisms may identify pharmacologic targets for simultaneous treatment of bone and energy metabolism diseases.
Acknowledgments
This work was supported by the American Diabetes Association (ADA) grant #1-17-PDF-067 to LAS and grants from NIH DK105825 and ADA 7-13-BS-089 to BLC.
Footnotes
Compliance with Ethical Standards: Conflict of Interest: Lance A. Stechschulte and BeataLecka-Czernik each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent: This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Madras N, Gibbs AL, Zhou Y, Zandstra PW, Aubin JE. Modeling stem cell development by retrospective analysis of gene expression profiles in single progenitor-derived colonies. Stem Cells. 2002;20(3):230–40. doi: 10.1634/stemcells.20-3-230. [DOI] [PubMed] [Google Scholar]
- 2•.Bianco P, Robey PG. Skeletal stem cells. Development. 2015;142(6):1023–7. doi: 10.1242/dev.102210. This ia a comprehensive review of skeletal stem cell biology and potential therapeutic uses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem. 2009;106(2):232–46. doi: 10.1002/jcb.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rahman S, Czernik PJ, Lu Y, Lecka-Czernik B. beta-Catenin Directly Sequesters Adipocytic and Insulin Sensitizing Activities but Not Osteoblastic Activity of PPARgamma2 in Marrow Mesenchymal Stem Cells. PLoS One. 2012;7(12):e51746. doi: 10.1371/journal.pone.0051746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309(5737):1074–8. doi: 10.1126/science.1110955. [DOI] [PubMed] [Google Scholar]
- 6.Lecka-Czernik B, Gubrij I, Moerman EJ, Kajkenova O, Lipschitz DA, Manolagas SC, et al. Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2. J Cell Biochem. 1999;74(3):357–71. [PubMed] [Google Scholar]
- 7•.Shockley KR, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 Regulates a Molecular Signature of Marrow Mesenchymal Stem Cells. PPAR Res. 2007;2007:81219. doi: 10.1155/2007/81219. This analysis demonstrates that MSCs “stemness” is under control of PPARγ2 protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen P. The regulation of protein function by multisite phosphorylation--a 25 year update. Trends Biochem Sci. 2000;25(12):596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 9.Cohen P. Protein phosphorylation and hormone action. Proceedings of the Royal Society of London Series B, Biological sciences. 1988;234(1275):115–44. doi: 10.1098/rspb.1988.0040. [DOI] [PubMed] [Google Scholar]
- 10.Pearlman SM, Serber Z, Ferrell JE., Jr A mechanism for the evolution of phosphorylation sites. Cell. 2011;147(4):934–46. doi: 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem. 1997;272(8):5128–32. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 12.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274(5295):2100–3. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 13.Hinds TD, Jr, Stechschulte LA, Cash HA, Whisler D, Banerjee A, Yong W, et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma) J Biol Chem. 2011;286(50):42911–22. doi: 10.1074/jbc.M111.311662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosooka T, Noguchi T, Kotani K, Nakamura T, Sakaue H, Inoue H, et al. Dok1 mediates high-fat diet-induced adipocyte hypertrophy and obesity through modulation of PPAR-gamma phosphorylation. Nat Med. 2008;14(2):188–93. doi: 10.1038/nm1706. [DOI] [PubMed] [Google Scholar]
- 15••.Stechschulte LA, Ge C, Hinds TD, Jr, Sanchez ER, Franceschi RT, Lecka-Czernik B. Protein Phosphatase PP5 Controls Bone Mass and the Negative Effects of Rosiglitazone on Bone through Reciprocal Regulation of PPARgamma (Peroxisome Proliferator-activated Receptor gamma) and RUNX2 (Runt-related Transcription Factor 2) J Biol Chem. 2016;291(47):24475–86. doi: 10.1074/jbc.M116.752493. This study demonstrates that PP5 reciprocally regulates activities of PPARγ and RUNX2 and MSCs lineage commitment, and that PP5 deficiency protects bone entirely from the rosiglitazone-induced bone loss. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16••.Stechschulte LA, Czernik PJ, Rotter ZC, Tausif FN, Corzo CA, Marciano DP, et al. PPARG Post-translational Modifications Regulate Bone Formation and Bone Resorption. EBioMedicine. 2016;10:174–84. doi: 10.1016/j.ebiom.2016.06.040. This study demonstrates for the first time that the same PPARγ PTM (S273) which regulates insulin sensitivity also regulates osteoclast differentiation independently from S112 which regulates adipocyte differentiation from MSCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi JH, Banks AS, Kamenecka TM, Busby SA, Chalmers MJ, Kumar N, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477(7365):477–81. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2(10):749–59. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 19.Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275(22):17166–72. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 20.Utreras E, Futatsugi A, Rudrabhatla P, Keller J, Iadarola MJ, Pant HC, et al. Tumor necrosis factor-alpha regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J Biol Chem. 2009;284(4):2275–84. doi: 10.1074/jbc.M805052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferron M, Wei J, Yoshizawa T, Del Fattore A, Depinho RA, Teti A, et al. Insulin Signaling in Osteoblasts Integrates Bone Remodeling and Energy Metabolism. Cell. 2010;142(2):296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wee HJ, Huang G, Shigesada K, Ito Y. Serine phosphorylation of RUNX2 with novel potential functions as negative regulatory mechanisms. EMBO Rep. 2002;3(10):967–74. doi: 10.1093/embo-reports/kvf193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001;20(4):723–33. doi: 10.1093/emboj/20.4.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kugimiya F, Kawaguchi H, Ohba S, Kawamura N, Hirata M, Chikuda H, et al. GSK-3beta controls osteogenesis through regulating Runx2 activity. PLoS One. 2007;2(9):e837. doi: 10.1371/journal.pone.0000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaidi SK, Sullivan AJ, Medina R, Ito Y, van Wijnen AJ, Stein JL, et al. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. Embo J. 2004;23(4):790–9. doi: 10.1038/sj.emboj.7600073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. 2009;284(47):32533–43. doi: 10.1074/jbc.M109.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120(7):2457–73. doi: 10.1172/JCI42285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou W, Greenblatt MB, Shim JH, Kant S, Zhai B, Lotinun S, et al. MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J Clin Invest. 2011;121(11):4383–92. doi: 10.1172/JCI59041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429(3):403–17. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez-Carballo E, Gamez B, Ventura F. p38 MAPK Signaling in Osteoblast Differentiation. Front Cell Dev Biol. 2016;4:40. doi: 10.3389/fcell.2016.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. 1996;272(5266):1347–9. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 32.Yang TT, Xiong Q, Enslen H, Davis RJ, Chow CW. Phosphorylation of NFATc4 by p38 mitogen-activated protein kinases. Mol Cell Biol. 2002;22(11):3892–904. doi: 10.1128/MCB.22.11.3892-3904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan J, Gan L, Chen D, Sun C. Adiponectin impairs chicken preadipocytes differentiation through p38 MAPK/ATF-2 and TOR/p70 S6 kinase pathways. PLoS One. 2013;8(10):e77716. doi: 10.1371/journal.pone.0077716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aouadi M, Laurent K, Prot M, Le Marchand-Brustel Y, Binetruy B, Bost F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes. 2006;55(2):281–9. doi: 10.2337/diabetes.55.02.06.db05-0963. [DOI] [PubMed] [Google Scholar]
- 35.Feng M, Tian L, Gan L, Liu Z, Sun C. Mark4 promotes adipogenesis and triggers apoptosis in 3T3-L1 adipocytes by activating JNK1 and inhibiting p38MAPK pathways. Biol Cell. 2014;106(9):294–307. doi: 10.1111/boc.201400004. [DOI] [PubMed] [Google Scholar]
- 36.Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, Pittenger MF. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem. 2000;275(13):9645–52. doi: 10.1074/jbc.275.13.9645. [DOI] [PubMed] [Google Scholar]
- 37.Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S, et al. Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone. 2001;28(5):491–8. doi: 10.1016/s8756-3282(01)00415-x. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki A, Guicheux J, Palmer G, Miura Y, Oiso Y, Bonjour JP, et al. Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone. 2002;30(1):91–8. doi: 10.1016/s8756-3282(01)00660-3. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki A, Palmer G, Bonjour JP, Caverzasio J. Regulation of alkaline phosphatase activity by p38 MAP kinase in response to activation of Gi protein-coupled receptors by epinephrine in osteoblast-like cells. Endocrinology. 1999;140(7):3177–82. doi: 10.1210/endo.140.7.6857. [DOI] [PubMed] [Google Scholar]
- 40.Thouverey C, Caverzasio J. Focus on the p38 MAPK signaling pathway in bone development and maintenance. Bonekey Rep. 2015;4:711. doi: 10.1038/bonekey.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41••.Ge C, Cawthorn WP, Li Y, Zhao G, MacDougald OA, Franceschi RT. Reciprocal Control of Osteogenic and Adipogenic Differentiation by ERK/MAP Kinase Phosphorylation of Runx2 and PPARgamma Transcription Factors. J Cell Physiol. 2016;231(3):587–96. doi: 10.1002/jcp.25102. This study demonstrates for the first time that PPARγ and RUNX2 activities are regulated by the same MAPKs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chinkers M. Targeting of a distinctive protein-serine phosphatase to the protein kinase-like domain of the atrial natriuretic peptide receptor. ProcNatlAcadSci U S A. 1994;91(23):11075–9. doi: 10.1073/pnas.91.23.11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen MX, McPartlin AE, Brown L, Chen YH, Barker HM, Cohen PT. A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 1994;13(18):4278–90. doi: 10.1002/j.1460-2075.1994.tb06748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker W, Kentrup H, Klumpp S, Schultz JE, Joost HG. Molecular cloning of a protein serine/threonine phosphatase containing a putative regulatory tetratricopeptide repeat domain. J Biol Chem. 1994;269(36):22586–92. [PubMed] [Google Scholar]
- 45.Chen MX, Cohen PT. Activation of protein phosphatase 5 by limited proteolysis or the binding of polyunsaturated fatty acids to the TPR domain. FEBS Lett. 1997;400(1):136–40. doi: 10.1016/s0014-5793(96)01427-5. [DOI] [PubMed] [Google Scholar]
- 46.Hinds TD, Jr, Sanchez ER. Protein phosphatase 5. Int J Biochem Cell Biol. 2008;40(11):2358–62. doi: 10.1016/j.biocel.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang H, Sayner SL, Gross KL, Russell LC, Chinkers M. Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry. 2001;40(35):10485–90. doi: 10.1021/bi010999i. [DOI] [PubMed] [Google Scholar]
- 48.Ramsey AJ, Chinkers M. Identification of potential physiological activators of protein phosphatase 5. Biochemistry. 2002;41(17):5625–32. doi: 10.1021/bi016090h. [DOI] [PubMed] [Google Scholar]
- 49.Russell LC, Whitt SR, Chen MS, Chinkers M. Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J Biol Chem. 1999;274(29):20060–3. doi: 10.1074/jbc.274.29.20060. [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Roe SM, Cliff MJ, Williams MA, Ladbury JE, Cohen PT, et al. Molecular basis for TPR domain-mediated regulation of protein phosphatase 5. EMBO J. 2005;24(1):1–10. doi: 10.1038/sj.emboj.7600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. MolCell. 2005;17(2):215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 52.Zuo Z, Dean NM, Honkanen RE. Serine/threonine protein phosphatase type 5 acts upstream of p53 to regulate the induction of p21(WAF1/Cip1) and mediate growth arrest. J Biol Chem. 1998;273(20):12250–8. doi: 10.1074/jbc.273.20.12250. [DOI] [PubMed] [Google Scholar]
- 53.Chinkers M. Protein phosphatase 5 in signal transduction. Trends EndocrinolMetab. 2001;12(1):28–32. doi: 10.1016/s1043-2760(00)00335-0. [DOI] [PubMed] [Google Scholar]
- 54.Ollendorff V, Donoghue DJ. The serine/threonine phosphatase PP5 interacts with CDC16 and CDC27, two tetratricopeptide repeat-containing subunits of the anaphase-promoting complex. J Biol Chem. 1997;272(51):32011–8. doi: 10.1074/jbc.272.51.32011. [DOI] [PubMed] [Google Scholar]
- 55.Yong W, Bao S, Chen H, Li D, Sanchez ER, Shou W. Mice lacking protein phosphatase 5 are defective in ataxia telangiectasia mutated (ATM)-mediated cell cycle arrest. J Biol Chem. 2007;282(20):14690–4. doi: 10.1074/jbc.C700019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ikeda K, Ogawa S, Tsukui T, Horie-Inoue K, Ouchi Y, Kato S, et al. Protein phosphatase 5 is a negative regulator of estrogen receptor-mediated transcription. MolEndocrinol. 2004;18(5):1131–43. doi: 10.1210/me.2003-0308. [DOI] [PubMed] [Google Scholar]
- 57.Chen MS, Silverstein AM, Pratt WB, Chinkers M. The tetratricopeptide repeat domain of protein phosphatase 5 mediates binding to glucocorticoid receptor heterocomplexes and acts as a dominant negative mutant. J Biol Chem. 1996;271(50):32315–20. doi: 10.1074/jbc.271.50.32315. [DOI] [PubMed] [Google Scholar]
- 58.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B. Bone is a target for the antidiabetic compound rosiglitazone. Endocrinology. 2004;145(1):401–6. doi: 10.1210/en.2003-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B. Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone. Endocrinology. 2007;148(6):2669–80. doi: 10.1210/en.2006-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwartz AV. TZDs and Bone: A Review of the Recent Clinical Evidence. PPAR Res. 2008;2008:297893. doi: 10.1155/2008/297893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zinman B, Haffner SM, Herman WH, Holman RR, Lachin JM, Kravitz BG, et al. Effect of rosiglitazone, metformin, and glyburide on bone biomarkers in patients with type 2 diabetes. The Journal of clinical endocrinology and metabolism. 2010;95(1):134–42. doi: 10.1210/jc.2009-0572. [DOI] [PubMed] [Google Scholar]
- 62•.Schwartz AV, Chen H, Ambrosius WT, Sood A, Josse RG, Bonds DE, et al. Effects of TZD Use and Discontinuation on Fracture Rates in ACCORD Bone Study. The Journal of clinical endocrinology and metabolism. 2015;100(11):4059–66. doi: 10.1210/jc.2015-1215. This review summarizes the effect of anti-diabetic thiazolidinediones on bone loss and fracture risk in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]