

Abstract
It is well established that genetic variability has a major impact on susceptibility to common diseases, responses to drugs and toxicants, and influences disease-related outcomes. The appreciation that epigenetic marks also vary across the population is growing with more data becoming available from studies in humans and model organisms. In addition, the links between genetic variability, toxicity outcomes and epigenetics are being actively explored. Recent studies demonstrate that gene-by-environment interactions involve both chromatin states and transcriptional regulation, and that epigenetics provides important mechanistic clues to connect expression-related quantitative trait loci (QTL) and disease outcomes. However, studies of Gene×Environment×Epigenetics further extend the complexity of the experimental designs and create a challenge for selecting the most informative epigenetic readouts that can be feasibly performed to interrogate multiple individuals, exposures, tissue types and toxicity phenotypes. We propose that among the many possible epigenetic experimental methodologies, assessment of chromatin accessibility coupled with total RNA levels provides a cost-effective and comprehensive option to sufficiently characterize the complexity of epigenetic and regulatory activity in the context of understanding the inter-individual variability in responses to toxicants.
Genetic variability
Estimation of the degree of inter-individual variability in the population is a required step in assessing the human health hazard posed by environmental chemicals. Indeed, the National Academies report Science and Decisions [1] called for the need to better “account for differences among humans in cancer susceptibility other than from possible early-life susceptibility.” Recent advances in the ability to conduct genome-wide association studies (GWAS) that identify quantitative trait loci (QTL) have enabled identification of genetic variants associated with important diseases [2]. It is clear that genetic variation influences the response of an individual to drugs and chemicals [3]. The blossoming field of personalized medicine now brings GWAS-enabled understanding of basic biology into clinical practice to determine how the knowledge of genetic variation can make therapies safer and more effective by tailoring selection and dosing of drugs for an individual patient [4].
GWAS that characterize effects of environmental toxicants on humans are usually based on epidemiological data, not controlled exposures [5]. This makes it a challenge to interpret findings from human cohorts exposed in the occupational or environmental settings. In addition, collection of tissues (with the exception of blood) from a wide variety of anatomical sites or developmental stages is not possible in humans that have been exposed to environmental toxicants. These limitations can be alleviated, at least partially, by the use of appropriate genetically-diverse laboratory animal-based model systems [6].
The mouse is a popular in vivo model for which genetic resources with publicly available genetic maps across dozens of strains are now available [7]. Mouse populations, such as the Collaborative Cross [8], provide an excellent testing system for evaluation of complexities in toxicokinetics and toxicodynamics [6, 9-11]. In the past decade, it has been demonstrated convincingly that genetic diversity in the mouse can be used to identify sensitive sub-populations using a mouse model of the human population approach [12-25]. Most of the genetic variability among mouse strains has been focused on SNPs; however, variation in structure of DNA regions affecting DNA sequence length and/or orientation that includes deletions, insertions, copy-number gains, inversions, and transposable elements, may also underpin susceptibility traits [26]. In addition, while inbred mouse strains are considered isogenic, intra-strain differences and their influence on experimental outcomes have been identified [27, 28].
While advances in sequencing technologies, statistical genetics analysis methods and clinical trial designs have shown promise for the discovery of variants associated with drug response, interpretation of both human and mouse GWAS through identifying causal variants is a challenge, and the translation of the findings to the clinic and/or regulatory actions is slow. On the one hand, it remains difficult to interpret the outcomes of GWAS and validate genes underlying QTLs with certainty, due in part to not knowing which organs, tissues, and/or cell types any particular QTL is having a significant functional effect. On the other hand, the GWAS-driven attempts to disentangle treatment responders from non-responders via genetic predictors in pharmacogenetics studies have not been uniformly successful [29].
Linkages between genetic, transcriptional, and epigenetic variability
Comprehensive maps of human and mouse regulatory DNA were recently published by the ENCODE (Encyclopedia of DNA Elements) Consortium [30], mouse ENCODE [31], and the Roadmap Epigenomics Project [32]. These studies comprehensively characterized the location and relationships between chromatin accessibility, histone modifications, chromatin looping, transcription, DNA methylation and the occupancy of sequence-specific factors. The wide spectrum of different cultured cell lines and tissues that were assayed have identified over a million common and cell-type specific gene regulatory elements. Genome-wide chromatin accessibility analyses, originally performed by DNase-seq [33] and more recently by ATAC-seq [34], have become invaluable approaches for mapping the genomic location of transcriptionally-active chromatin. While consortia such as ENCODE and Roadmap have identified large numbers of putative regulatory elements, little is known about how these elements are affected by variation in genetics, sex, or exposure to individual or complex combinations of environmental stimuli.
Studies in a large and genetically heterogeneous collection of human lymphoblast cell lines (LCLs) [32] and tissues [35] have identified heritable variation in gene expression across humans. These expression quantitative trait loci (eQTL) studies have been complemented by a more limited number of chromatin studies that have identified QTLs that impact DNaseI sensitivity (dsQTL; [36]), histone modification chromatin (cQTLs; [37]), DNA methylation [38], and transcription factors binding sites [37, 39]. These studies demonstrate the versatility and complexity of gene regulation, whereby modulation of gene expression is executed by different elements forming intricate networks that include changes in chromatin activity. In addition, these studies show how genetic variants identified in GWAS can be linked through a regulatory network to the associated gene. For example, it was shown that both locally and distally acting genetic variants exhibit strong influence on expression and chromatin [37, 40]. It was also found that two-thirds of local eQTLs were also local dsQTLs or cQTLs [36], which means that the variation in chromatin is associated with variation in the expression levels of nearby genes. At the same time, a total of 15% of proximal histone QTLs were associated with changes in chromatin states at distal genomic regions with which they interact physically [41]. These data show that specific genetic variants modulating regulatory element activity may concordantly affect local and distal chromatin modifications and gene expression.
While population variability in DNA- and chromatin-related epigenetic marks is well recognized, it has been shown that variability in miRNA expression in the population may be negligible as compared to the genetically-determined variability in mRNA expression [20, 42]. Specifically, few eQTLs were observed for miRNAs in various tissues in population studies in mice [13, 43, 44]. The stability of miRNA expression in a genetically diverse population suggests that miRNAs may be a much more reliable population-wide biomarker of the effects of chemicals on epigenetic mechanisms of toxicity, as compared to changes in DNA methylation, chromatin and/or histone modifications. Indeed, chemical-induced disruptions in miRNA expression, a phenomenon established for a large number of toxicants, is recognized as an important toxicity mechanism [45]. Post-transcriptional regulation of mRNA levels by miRNAs is not a true epigenetic process. For the remainder of this review, though, we include miRNAs when discussing the epigenome for the sake of simplicity as their primary function is to regulate gene expression.
Environmental agents cause toxicity through epigenetic mechanisms
Epigenetic reprogramming has been proposed as an integral part of the “genome instability” enabling characteristic of cancer cells [46] and it is well established that chemical carcinogens may affect the cellular epigenetic state [47]. Changes in DNA methylation, histone/chromatin remodeling, and altered expression of miRNAs represent the most frequently reported toxicant-induced alterations of the epigenome [48]. Because of the potential impact of these epigenotoxic effects on gene expression patterns and, consequently, on the toxicity phenotypes, epigenetic changes have been proposed as biomarkers of carcinogen exposure and effect [49, 50].
One of the first examples of the linkages between environment, epigenetics and phenotypes were studies of in utero exposure to environmental agents that can also disrupt the epigenome. The agouti mouse model was used to demonstrate that environmental factors may affect the fetal epigenome [51]. Using this mouse model, maternal exposure to the endocrine disruptor BPA causes loss of DNA methylation at key loci, resulting in a shift in coat color of offspring [52]. Normal methylation patterns can then be restored with maternal dietary supplementation using methyl donors like folic acid. Evidence shows along with DNA methylation, variable histone modifications affect the inter-individual epigenetic variation of this metastable epiallele [53].
Another prominent example of how environmental toxicants may have epigenetic effects are studies on the mechanism of carcinogenesis for metals [54]. Arsenic, a ubiquitous environmental contaminant, disrupts the normal epigenome transforming the epigenetic landscape to reflect that of a cancer cell [55]. Exposure to metals like arsenic causes significant epigenetic modifications such as changes in global histone methylation levels [56]. After exposure to arsenic compounds, human lung carcinoma A549 cells showed an increase in global levels of H3K4me3 and H3K9me2 [57]. Similarly, human peripheral blood nuclear cells extracted from subjects exposed to high levels of arsenic in water had an increase in H3K9me2 levels [58].
Environmental contaminants can also alter gene expression by epigenetically reprogramming tissues. Neonatal BPA exposure increases H3K4me3 levels in promoters of genes associated with prostate cancer through activation of histone methyltransferase MLL1 [59]. Although there was no difference in basal expression of levels of BPA reprogrammed genes, once challenged with hormone treatment, there is enhanced gene-specific transcription. It is thought the change in levels of H3K4me3 primes these genes for an enhanced response. Additionally, evidence suggests that BPA exposure during prostate development could epigenetically reprogram the expression of Scgb2a1 in the adult prostate [60].
Epigenetic changes may be a consequence of DNA damage [61], or may be part of the non-genotoxic mechanisms of carcinogenesis [62]. The interplay between chemical-induced DNA damage response and transcription, DNA replication, and repair has only recently been linked to chromatin dynamics, especially to histone modifications and post-repair chromatin restoration at the sites of DNA damage [63]. For example, a local response to DNA double-stranded breaks gives rise to chromatin condensation which spreads at least over several Kb from the damage sites and can induce epigenetic silencing of the nearby genes [64]. In addition, it was shown that levels of the heterochromatin-associated histone modification H3K9me3 accounted for more than 40% of mutation rate variation, providing striking evidence that mutation rates in cancer genomes are related closely to chromatin organization [65]. Besides that, DNA repair can cause local chromatin state transitions eventually resulting in prolonged inactivation of transcription via not yet fully established gene silencing mechanisms. Modulation of the epigenetic status of damaged genes potentially expands the field of DNA damage into the sphere of regulation of gene expression [61]. While the interest in the role of epigenome in toxicity mechanisms is growing, the genotoxicity of chemicals has been more thoroughly studied and characterized, as evidenced by a systematic review of published studies of genotoxic carcinogens that investigated epigenetic endpoints [66].
DNA methylation is another key epigenetic mechanism, regulating both gene expression and chromatin stability. DNA methylation studies have been recently combined with RNA-seq and ChIP-seq to identify the role of the changes in the methylome in disease pathogenesis [67]. DNA methylation and genetic polymorphisms have important concomitant regulatory effects on transcription factor-driven gene expression [68]. Aberrant DNA methylation patterns due to exposure to environmental chemicals are also well-characterized. Exposure to benzene, metals, and traffic pollution are all examples of toxicants that can have an effect on DNA methylation [69-72].
Environmental effects on the epigenome in the context of genetic variability
There is now overwhelming evidence that connects genetic variability and epigenetic marks and that chemical exposures can exert toxicity through epigenetic mechanisms; yet less is known about how the effects on the epigenome may vary in the population. Intriguing novel insights into linkages between genotoxic and epigenetic mechanisms of carcinogenesis, and the role of genetic variability among individuals have been provided by studies of a classical genotoxic carcinogen 1,3-butadiene. It is a genotoxic chemical with DNA damaging effects that vary among genetically distinct individuals. Butadiene is a major industrial chemical used in the production of synthetic rubbers and polymers. It is also a ubiquitous environmental contaminant that is found in cigarette smoke and automobile exhaust. IARC has classified butadiene as a known human carcinogen [73]. It is well established that the mechanism of carcinogenicity is due to butadiene's reactive metabolites. These epoxides interact directly with DNA and form mutagenic DNA adducts. Butadiene also elicits an epigenetic response, causing significant loss of global DNA methylation as well as a decrease in H3K9, H3K27, and H3K20 trimethylation in C57BL/6J mouse liver [74].
Using a mouse population-based model it was shown that inter-individual (e.g., inter-strain) differences exist in both genotoxic and epigenotoxic effects of 1,3-butadiene exposure and that the chromatin remodeling response is at least one mechanism for the inter-strain differences in 1,3-butadiene-induced DNA damage [14]. Specifically, it was shown that 1,3-butadiene alters bulk chromatin histone mark levels resulting in strain-specific abundances of these marks. In particular, CAST/EiJ and C57BL/6J mice, two genetically distinct strains, exhibited basal and treatment-induced differences in overall levels of these histone marks.
Opinion: Studies of Gene×Environment×Epigenome in genetically diverse populations can provide a mechanistic explanation for expression and QTL effects
Studies of Gene×Environment×Epigenome seek to uncover not only relationships between environment exposures and gene expression levels, but also to determine the epigenetic gene regulatory mechanisms altered by these exposures that contribute to the observed expression changes (Figure 1). These studies further extend the complexity of the experimental designs and require choosing the most informative epigenetic readouts to interrogate multiple individuals, exposures, tissue types and toxicity phenotypes (Figure 2). One possibility would be to perform ChIP-seq for many different histone marks or transcription factors, but this would be costly since multiple experiments would be required for each sample. It may also be impractical as these assays typically require non-trivial amounts of sample material limiting the number of experiments that could be performed, thus requiring an uninformed selection of assays to be made. RNA-seq protocols are able to comprehensively quantify multiple types of RNA transcripts, including protein-coding mRNAs, mircoRNAs (miRNAs), long non-coding RNAs (lncRNAs), enchancer RNAs (eRNAs), and circular RNAs, (circRNAs), and chemical modifications of these molecules [75, 76]. These data are able to characterize variation in transcription generation and RNA processing, including alternative transcript initiation and splicing, providing detailed information about transcriptional outputs that affect cellular function. Therefore, a more cost-effective and unbiased approach would be to assess chromatin accessibility coupled with various RNA levels to comprehensively characterize epigenetic and transcriptional activity in the context of inter-individual variability in responses to toxicants.
Figure 1.
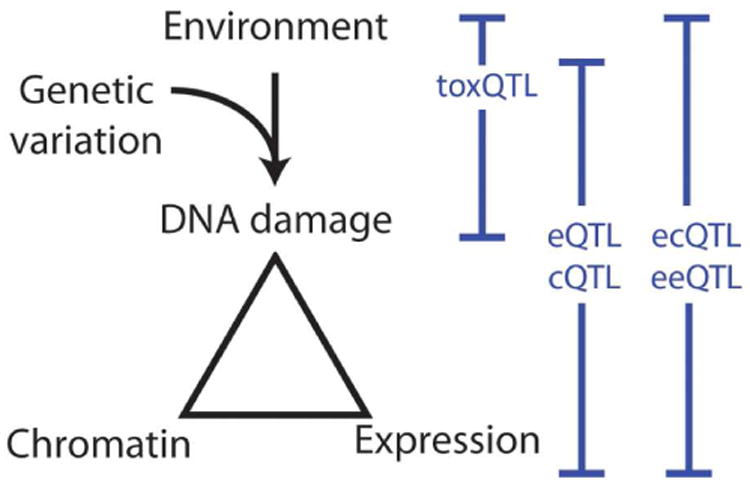
A combination of toxicity phenotyping, transcriptomics, chromatin state analyses, and genetic variability in a population-based model allow exploration of Gene×Environment×Epigenome interactions. Abbreviations: eQTL, expression quantitative trait loci; cQTL, chromatin QTL; ecQTLs, environmental chromatin QTL; toxQTL, toxicity QTL; eeQTL, environmental expression QTL.
Figure 2.
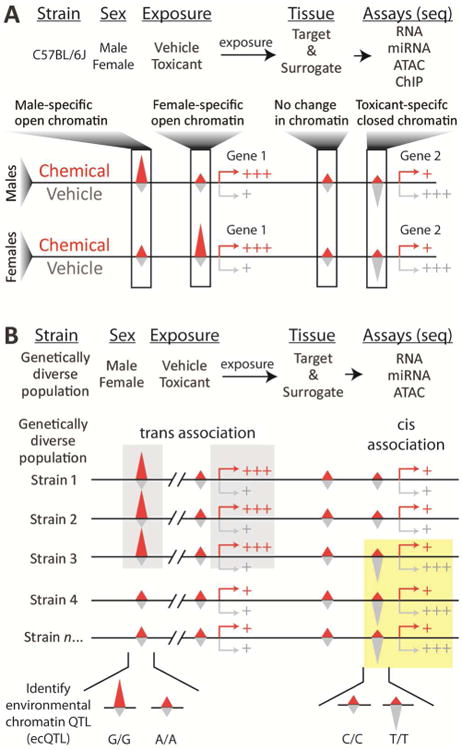
Hypothetical study designs to evaluate toxicant-induced effects on the epigenome in (A) single genetic background, or (B) population-wide experimental animal model.
The ideal way to identify gene regulatory elements would be a single empirical method that exploits a feature held in common between all functional classes of regulatory elements [77]. Identification of “accessible chromatin” through techniques like DNase-seq or ATAC-seq are currently the closest to this, because nucleosome depletion at active regulatory regions is a conserved feature of eukaryotic chromatin [78, 79]. The sequence-specific binding of transcription factors and the recruitment of active histone marks at promoters, enhancers, and insulators is typically associated with nucleosome depletion in eukaryotic cells [80, 81]. DNase-seq and ATAC-seq reveal these active regulatory elements by preferential digestion of or transposon insertion into nucleosome-depleted regions [34, 80, 82]. Until recently, DNase-seq was one of the few methods for assessing chromatin accessibility [83-86], and was used in the analysis of >100 human cell types and tissues [85, 87].
While DNase-seq and ATAC-seq are both designed to measure accessible chromatin, DNase-seq technique is a derivative of the DNaseI hypersensitivity assay first described nearly 40 years ago [88]. Therefore, there is a massive amount of literature identifying and characterizing DNaseI hypersensitive sites, which supports the value of this method [77]. ATAC-seq was devised more recently, and has been shown to identify largely similar, but not identical, regions to that of DNase-seq [89]. The ATAC-seq protocol is less technically challenging and faster which facilitated many more labs to adopt the ATAC-seq over DNase-seq. It is not surprising that there are some differences between ATAC-seq and DNAse-seq, and while some of this may be due to different sequence biases of the Tn5 transposase or the DNase enzyme, it is not yet clear what other factors may contribute to the differences [90, 91]. An additional advantage of ATAC-seq is the ability to characterize much smaller amounts of cells or tissue [89], which is essential for experiments where there is no renewable resources, such as biobanked tissues. While a modified version of DNase-seq exists for small samples [92], this protocol has not been as widely adopted as ATAC-seq or single cell ATAC-seq [93-95]. However, generation of ATAC-seq libraries is more expensive due to the cost of Tn5 transposase and the need for requiring deeper sequencing (up to 50% of ATAC-seq reads come from the mitochondrial genome). Analysis of DNase-seq and ATAC-seq data is fairly similar.
We posit that data from strand-specific RNA-seq and ATAC-seq represents a cost-effective approach to identify and resolve regions of active or repressed enhancers and promoters. A common challenge among all regulatory element assays is determining the target gene(s) being regulated. Chromatin conformation capture assays provide the best evidence for interactions between regulatory elements and target genes. While these assays are cost-prohibitive to run on all samples, data has been generated from an increasing number of diverse cell-types and tissues through individual labs and within the ENCODE consortium that are being compiled and visualized in browsers [96]. In addition, software such as TargetFinder [97] can predict gene targets in specific samples using accessible chromatin data. In conjunction with RNA-seq data, alterations in chromatin accessibility correlated with expression changes in target genes either due to differences in genetic background or in response to environmental stimuli provide evidence of regulatory element function. ATAC-seq also does not immediately reveal what factors are bound in accessible chromatin regions. The DNA sequence binding preferences for an increasing number of factors is being defined [98], which can be used to predict which factors are present. Transcription factor footprinting using accessible chromatin data [99] provide additional evidence for sites where factors are bound. Admittedly, ChIP-seq data for particular factors and histone modifications provide more direct evidence of regulatory element function. Analysis of chromatin accessibility data can reveal which additional assays may provide the most relevant additional information.
Concentrating on just these two assays makes feasible the generation of data from sufficient samples of genetically diverse backgrounds to further link gene expression and epigenetic changes to genetic variability. Along with environmental phenotype data of interest, the stage is set for a full Gene×Environment×Epigenome analysis. The optimal study design would include not only post-environmental exposure data, but also matched pre-exposure data from control subjects. Susceptibility to damaging effects of toxicants may be due not only to genetic variability driving differential responses, but also may depend on baseline transcriptional and epigenetic states, also influenced by genetic background, that may be better primed in certain individuals to defend against injury [100].
Highlights.
Genetic variability is a major driver for susceptibility to disease
Recent studies identified epigenetic variability factors linked to genetic variants
Environmental agents may cause toxicity through epigenetic mechanisms
Environmental effects on the epigenome in the context of genetic variability is a budding research area
Studies of Gene×Environment×Epigenome can provide important mechanistic clues in toxicology
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.National Research Council. Science and Decisions: Advancing Risk Assessment. National Academies Press; Washington, DC: 2009. [PubMed] [Google Scholar]
- 2.Hirschhorn JN. Genomewide association studies--illuminating biologic pathways. N Engl J Med. 2009;360(17):1699–1701. doi: 10.1056/NEJMp0808934. [DOI] [PubMed] [Google Scholar]
- 3.Wheeler HE, Maitland ML, Dolan ME, Cox NJ, Ratain MJ. Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet. 2013;14(1):23–34. doi: 10.1038/nrg3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashley EA. Towards precision medicine. Nat Rev Genet. 2016;17(9):507–22. doi: 10.1038/nrg.2016.86. [DOI] [PubMed] [Google Scholar]
- 5.Simonds NI, Ghazarian AA, Pimentel CB, Schully SD, Ellison GL, Gillanders EM, Mechanic LE. Review of the Gene-Environment Interaction Literature in Cancer: What Do We Know? Genet Epidemiol. 2016;40(5):356–65. doi: 10.1002/gepi.21967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rusyn I, Gatti DM, Wiltshire T, Kleeberger SR, Threadgill DW. Toxicogenetics: population-based testing of drug and chemical safety in mouse models. Pharmacogenomics. 2010;11(8):1127–36. doi: 10.2217/pgs.10.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448(7157):1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 8.Threadgill DW, Churchill GA. Ten years of the Collaborative Cross. Genetics. 2012;190(2):291–4. doi: 10.1534/genetics.111.138032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeise L, Bois FY, Chiu WA, Hattis D, Rusyn I, Guyton KZ. Addressing human variability in next-generation human health risk assessments of environmental chemicals. Environmental health perspectives. 2013;121(1):23–31. doi: 10.1289/ehp.1205687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cichocki JA, Furuya S, Venkatratnam A, McDonald TJ, Knap AH, Wade T, Sweet S, Chiu WA, Threadgill DW, Rusyn I. Characterization of Variability in Toxicokinetics and Toxicodynamics of Tetrachloroethylene Using the Collaborative Cross Mouse Population. Environmental health perspectives. 2017;125(5):057006. doi: 10.1289/EHP788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venkatratnam A, Furuya S, Kosyk O, Gold A, Bodnar W, Konganti K, Threadgill DW, Gillespie KM, Aylor DL, Wright FA, Chiu WA, Rusyn I. Collaborative Cross mouse population enables refinements to characterization of the variability in toxicokinetics of trichloroethylene and provides genetic evidence for the role of PPAR pathway in its oxidative metabolism. Toxicological sciences : an official journal of the Society of Toxicology. 2017 doi: 10.1093/toxsci/kfx065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuchiya M, Ji C, Kosyk O, Shymonyak S, Melnyk S, Kono H, Tryndyak V, Muskhelishvili L, Pogribny IP, Kaplowitz N, Rusyn I. Interstrain differences in liver injury and one-carbon metabolism in alcohol-fed mice. Hepatology. 2012;56(1):130–9. doi: 10.1002/hep.25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatti DM, Lu L, Williams RW, Sun W, Wright FA, Threadgill DW, Rusyn I. MicroRNA expression in the livers of inbred mice. Mutat Res. 2011;714(1-2):126–33. doi: 10.1016/j.mrfmmm.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koturbash I, Scherhag A, Sorrentino J, Sexton K, Bodnar W, Swenberg JA, Beland FA, Pardo-Manuel Devillena F, Rusyn I, Pogribny IP. Epigenetic mechanisms of mouse interstrain variability in genotoxicity of the environmental toxicant 1,3-butadiene. Toxicological sciences : an official journal of the Society of Toxicology. 2011;122(2):448–56. doi: 10.1093/toxsci/kfr133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradford BU, Lock EF, Kosyk O, Kim S, Uehara T, Harbourt D, DeSimone M, Threadgill DW, Tryndyak V, Pogribny IP, Bleyle L, Koop DR, Rusyn I. Interstrain differences in the liver effects of trichloroethylene in a multistrain panel of inbred mice. Toxicological sciences : an official journal of the Society of Toxicology. 2011;120(1):206–17. doi: 10.1093/toxsci/kfq362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gatti DM, Zhao N, Chesler EJ, Bradford BU, Shabalin AA, Yordanova R, Lu L, Rusyn I. Sex-specific gene expression in the BXD mouse liver. Physiological genomics. 2010;42(3):456–68. doi: 10.1152/physiolgenomics.00110.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatti DM, Harrill AH, Wright FA, Threadgill DW, Rusyn I. Replication and narrowing of gene expression quantitative trait loci using inbred mice. Mammalian genome : official journal of the International Mammalian Genome Society. 2009;20(7):437–46. doi: 10.1007/s00335-009-9199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pogribny IP, Tryndyak VP, Bagnyukova TV, Melnyk S, Montgomery B, Ross SA, Latendresse JR, Rusyn I, Beland FA. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. Journal of hepatology. 2009;51(1):176–86. doi: 10.1016/j.jhep.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrill AH, Watkins PB, Su S, Ross PK, Harbourt DE, Stylianou IM, Boorman GA, Russo MW, Sackler RS, Harris SC, Smith PC, Tennant R, Bogue M, Paigen K, Harris C, Contractor T, Wiltshire T, Rusyn I, Threadgill DW. Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome research. 2009;19(9):1507–15. doi: 10.1101/gr.090241.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gatti D, Maki A, Chesler EJ, Kirova R, Kosyk O, Lu L, Manly KF, Williams RW, Perkins A, Langston MA, Threadgill DW, Rusyn I. Genome-level analysis of genetic regulation of liver gene expression networks. Hepatology. 2007;46(2):548–57. doi: 10.1002/hep.21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu WA, Campbell JL, Clewell HJ, Zhou YH, Wright FA, Guyton KZ, Rusyn I. Physiologically-Based Pharmacokinetic (PBPK) Modeling of Inter-strain Variability in Trichloroethylene Metabolism in the Mouse. Environmental health perspectives. 2014;122(5):456–463. doi: 10.1289/ehp.1307623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aylor DL, Valdar W, Foulds-Mathes W, Buus RJ, Verdugo RA, Baric RS, Ferris MT, Frelinger JA, Heise M, Frieman MB, Gralinski LE, Bell TA, Didion JD, Hua K, Nehrenberg DL, Powell CL, Steigerwalt J, Xie Y, Kelada SN, Collins FS, Yang IV, Schwartz DA, Branstetter LA, Chesler EJ, Miller DR, Spence J, Liu EY, Mc Millan L, Sarkar A, Wang J, Wang W, Zhang Q, Broman KW, Korstanje R, Durrant C, Mott R, Iraqi FA, Pomp D, Threadgill D, Pardo-Manuel dV, Churchill GA. Genetic analysis of complex traits in the emerging collaborative cross. Genome research. 2011;21(8):1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillippi J, Xie Y, Miller DR, Bell TA, Zhang Z, Lenarcic AB, Aylor DL, Krovi SH, Threadgill DW, de Villena FP, Wang W, Valdar W, Frelinger JA. Using the emerging Collaborative Cross to probe the immune system. Genes Immun. 2014;15(1):38–46. doi: 10.1038/gene.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.French JE, Gatti DM, Morgan DL, Kissling GE, Shockley KR, Knudsen GA, Shepard KG, Price HC, King D, Witt KL, Pedersen LC, Munger SC, Svenson KL, Churchill GA. Diversity Outbred Mice Identify Population-Based Exposure Thresholds and Genetic Factors that Influence Benzene-Induced Genotoxicity. Environmental health perspectives. 2015;123(3):237–45. doi: 10.1289/ehp.1408202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrill AH, Desmet KD, Wolf KK, Bridges AS, Eaddy JS, Kurtz CL, Hall JE, Paine MF, Tidwell RR, Watkins PB. A mouse diversity panel approach reveals the potential for clinical kidney injury due to DB289 not predicted by classical rodent models. Toxicological sciences : an official journal of the Society of Toxicology. 2012;130(2):416–26. doi: 10.1093/toxsci/kfs238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keane TM, Wong K, Adams DJ, Flint J, Reymond A, Yalcin B. Identification of structural variation in mouse genomes. Front Genet. 2014;5:192. doi: 10.3389/fgene.2014.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watkins-Chow DE, Pavan WJ. Genomic copy number and expression variation within the C57BL/6J inbred mouse strain. Genome research. 2008;18(1):60–6. doi: 10.1101/gr.6927808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahajan VS, Demissie E, Mattoo H, Viswanadham V, Varki A, Morris R, Pillai S. Striking Immune Phenotypes in Gene-Targeted Mice Are Driven by a Copy-Number Variant Originating from a Commercially Available C57BL/6 Strain. Cell Rep. 2016;15(9):1901–9. doi: 10.1016/j.celrep.2016.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardon LR, Harris T. Precision medicine, genomics and drug discovery. Hum Mol Genet. 2016;25(R2):R166–R172. doi: 10.1093/hmg/ddw246. [DOI] [PubMed] [Google Scholar]
- 30.Encode Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS biology. 2011;9(4):e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, Shen Y, Pervouchine DD, Djebali S, Thurman RE, Kaul R, Rynes E, Kirilusha A, Marinov GK, Williams BA, Trout D, Amrhein H, Fisher-Aylor K, Antoshechkin I, DeSalvo G, See LH, Fastuca M, Drenkow J, Zaleski C, Dobin A, Prieto P, Lagarde J, Bussotti G, Tanzer A, Denas O, Li K, Bender MA, Zhang M, Byron R, Groudine MT, McCleary D, Pham L, Ye Z, Kuan S, Edsall L, Wu YC, Rasmussen MD, Bansal MS, Kellis M, Keller CA, Morrissey CS, Mishra T, Jain D, Dogan N, Harris RS, Cayting P, Kawli T, Boyle AP, Euskirchen G, Kundaje A, Lin S, Lin Y, Jansen C, Malladi VS, Cline MS, Erickson DT, Kirkup VM, Learned K, Sloan CA, Rosenbloom KR, Lacerda de Sousa B, Beal K, Pignatelli M, Flicek P, Lian J, Kahveci T, Lee D, Kent WJ, Ramalho Santos M, Herrero J, Notredame C, Johnson A, Vong S, Lee K, Bates D, Neri F, Diegel M, Canfield T, Sabo PJ, Wilken MS, Reh TA, Giste E, Shafer A, Kutyavin T, Haugen E, Dunn D, Reynolds AP, Neph S, Humbert R, Hansen RS, De Bruijn M, Selleri L, Rudensky A, Josefowicz S, Samstein R, Eichler EE, Orkin SH, Levasseur D, Papayannopoulou T, Chang KH, Skoultchi A, Gosh S, Disteche C, Treuting P, Wang Y, Weiss MJ, Blobel GA, Cao X, Zhong S, Wang T, Good PJ, Lowdon RF, Adams LB, Zhou XQ, Pazin MJ, Feingold EA, Wold B, Taylor J, Mortazavi A, Weissman SM, Stamatoyannopoulos JA, Snyder MP, Guigo R, Gingeras TR, Gilbert DM, Hardison RC, Beer MA, Ren B, Mouse EC. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515(7527):355–64. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roadmap Epigenomics Consortium. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahony S, Pugh BF. Protein-DNA binding in high-resolution. Crit Rev Biochem Mol Biol. 2015;50(4):269–83. doi: 10.3109/10409238.2015.1051505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21 29–1-9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45(6):580–5. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, Stephens M, Gilad Y, Pritchard JK. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482(7385):390–4. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waszak SM, Delaneau O, Gschwind AR, Kilpinen H, Raghav SK, Witwicki RM, Orioli A, Wiederkehr M, Panousis NI, Yurovsky A, Romano-Palumbo L, Planchon A, Bielser D, Padioleau I, Udin G, Thurnheer S, Hacker D, Hernandez N, Reymond A, Deplancke B, Dermitzakis ET. Population Variation and Genetic Control of Modular Chromatin Architecture in Humans. Cell. 2015;162(5):1039–50. doi: 10.1016/j.cell.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 38.Heyn H. Quantitative Trait Loci Identify Functional Noncoding Variation in Cancer. PLoS Genet. 2016;12(3):e1005826. doi: 10.1371/journal.pgen.1005826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McVicker G, van de Geijn B, Degner JF, Cain CE, Banovich NE, Raj A, Lewellen N, Myrthil M, Gilad Y, Pritchard JK. Identification of genetic variants that affect histone modifications in human cells. Science. 2013;342(6159):747–9. doi: 10.1126/science.1242429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grubert F, Zaugg JB, Kasowski M, Ursu O, Spacek DV, Martin AR, Greenside P, Srivas R, Phanstiel DH, Pekowska A, Heidari N, Euskirchen G, Huber W, Pritchard JK, Bustamante CD, Steinmetz LM, Kundaje A, Snyder M. Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell. 2015;162(5):1051–65. doi: 10.1016/j.cell.2015.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koch L. Genomics: Adding another dimension to gene regulation. Nat Rev Genet. 2015;16(10):563. doi: 10.1038/nrg4007. [DOI] [PubMed] [Google Scholar]
- 42.Bystrykh L, Weersing E, Dontje B, Sutton S, Pletcher MT, Wiltshire T, Su AI, Vellenga E, Wang J, Manly KF, Lu L, Chesler EJ, Alberts R, Jansen RC, Williams RW, Cooke MP, de Haan G. Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nature genetics. 2005;37(3):225–232. doi: 10.1038/ng1497. [DOI] [PubMed] [Google Scholar]
- 43.Zhao E, Keller MP, Rabaglia ME, Oler AT, Stapleton DS, Schueler KL, Neto EC, Moon JY, Wang P, Wang IM, Lum PY, Ivanovska I, Cleary M, Greenawalt D, Tsang J, Choi YJ, Kleinhanz R, Shang J, Zhou YP, Howard AD, Zhang BB, Kendziorski C, Thornberry NA, Yandell BS, Schadt EE, Attie AD. Obesity and genetics regulate microRNAs in islets, liver, and adipose of diabetic mice. Mammalian Genome. 2009;20(8):476–485. doi: 10.1007/s00335-009-9217-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parsons MJ, Grimm C, Paya-Cano JL, Fernandes C, Liu L, Philip VM, Chesler EJ, Nietfeld W, Lehrach H, Schalkwyk LC. Genetic variation in hippocampal microRNA expression differences in C57BL/6 J X DBA/2 J (BXD) recombinant inbred mouse strains. BMC Genomics. 2012;13:476. doi: 10.1186/1471-2164-13-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marrone AK, Beland FA, Pogribny IP. The role for microRNAs in drug toxicity and in safety assessment. Expert Opin Drug Metab Toxicol. 2015;11(4):601–11. doi: 10.1517/17425255.2015.1021687. [DOI] [PubMed] [Google Scholar]
- 46.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Smith MT, Guyton KZ, Gibbons CF, Fritz JM, Portier CJ, Rusyn I, DeMarini DM, Caldwell JC, Kavlock RJ, Lambert PF, Hecht SS, Bucher JR, Stewart BW, Baan RA, Cogliano VJ, Straif K. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environmental health perspectives. 2016;124(6):713–21. doi: 10.1289/ehp.1509912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marlowe J, Teo SS, Chibout SD, Pognan F, Moggs J. Mapping the epigenome--impact for toxicology. EXS. 2009;99:259–288. doi: 10.1007/978-3-7643-8336-7_10. [DOI] [PubMed] [Google Scholar]
- 49.Pogribny IP, Rusyn I. Environmental toxicants, epigenetics, and cancer. Advances in experimental medicine and biology. 2013;754:215–32. doi: 10.1007/978-1-4419-9967-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LeBaron MJ, Rasoulpour RJ, Klapacz J, Ellis-Hutchings RG, Hollnagel HM, Gollapudi BB. Epigenetics and chemical safety assessment. Mutat Res. 2010;705(2):83–95. doi: 10.1016/j.mrrev.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 51.Dolinoy DC. The agouti mouse model: an epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr Rev. 2008;66(1):S7–11. doi: 10.1111/j.1753-4887.2008.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dolinoy DC, Huang D, Jirtle RL. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A. 2007;104(32):13056–61. doi: 10.1073/pnas.0703739104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dolinoy DC, Weinhouse C, Jones TR, Rozek LS, Jirtle RL. Variable histone modifications at the A(vy) metastable epiallele. Epigenetics. 2010;5(7):637–44. doi: 10.4161/epi.5.7.12892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arita A, Costa M. Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics. 2009;1(3):222–8. doi: 10.1039/b903049b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brocato J, Costa M. Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit Rev Toxicol. 2013;43(6):493–514. doi: 10.3109/10408444.2013.794769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brocato J, Costa M. 10th NTES Conference: Nickel and Arsenic Compounds Alter the Epigenome of Peripheral Blood Mononuclear Cells. J Trace Elem Med Biol. 2015;31:209–13. doi: 10.1016/j.jtemb.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou X, Li Q, Arita A, Sun H, Costa M. Effects of nickel, chromate, and arsenite on histone 3 lysine methylation. Toxicol Appl Pharmacol. 2009;236(1):78–84. doi: 10.1016/j.taap.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chervona Y, Hall MN, Arita A, Wu F, Sun H, Tseng HC, Ali E, Uddin MN, Liu X, Zoroddu MA, Gamble MV, Costa M. Associations between arsenic exposure and global posttranslational histone modifications among adults in Bangladesh. Cancer Epidemiol Biomarkers Prev. 2012;21(12):2252–60. doi: 10.1158/1055-9965.EPI-12-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Q, Trevino LS, Wong RL, Medvedovic M, Chen J, Ho SM, Shen J, Foulds CE, Coarfa C, O'Malley BW, Shilatifard A, Walker CL. Reprogramming of the Epigenome by MLL1 Links Early-Life Environmental Exposures to Prostate Cancer Risk. Mol Endocrinol. 2016;30(8):856–71. doi: 10.1210/me.2015-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong RL, Wang Q, Trevino LS, Bosland MC, Chen J, Medvedovic M, Prins GS, Kannan K, Ho SM, Walker CL. Identification of secretaglobin Scgb2a1 as a target for developmental reprogramming by BPA in the rat prostate. Epigenetics. 2015;10(2):127–34. doi: 10.1080/15592294.2015.1009768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khobta A, Epe B. Interactions between DNA damage, repair, and transcription. Mutat Res. 2012;736(1-2):5–14. doi: 10.1016/j.mrfmmm.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 62.Pogribny IP, Rusyn I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett. 2014;342(2):223–30. doi: 10.1016/j.canlet.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu Q, Wani AA. Histone modifications: crucial elements for damage response and chromatin restoration. Journal of cellular physiology. 2010;223(2):283–8. doi: 10.1002/jcp.22060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141(6):970–81. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schuster-Bockler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488(7412):504–7. doi: 10.1038/nature11273. [DOI] [PubMed] [Google Scholar]
- 66.Chappell G, Pogribny IP, Guyton KZ, Rusyn I. Epigenetic alterations induced by genotoxic occupational and environmental human chemical carcinogens: A systematic literature review. Mutat Res Rev Mutat Res. 2016;768:27–45. doi: 10.1016/j.mrrev.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, Absher DM, Wold BJ, Myers RM. Dynamic DNA methylation across diverse human cell lines and tissues. Genome research. 2013;23(3):555–67. doi: 10.1101/gr.147942.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P, Vermaat M, van Iterson M, van Dijk F, van Galen M, Bot J, Slieker RC, Jhamai PM, Verbiest M, Suchiman HE, Verkerk M, van der Breggen R, van Rooij J, Lakenberg N, Arindrarto W, Kielbasa SM, Jonkers I, van 't Hof P, Nooren I, Beekman M, Deelen J, van Heemst D, Zhernakova A, Tigchelaar EF, Swertz MA, Hofman A, Uitterlinden AG, Pool R, van Dongen J, Hottenga JJ, Stehouwer CD, van der Kallen CJ, Schalkwijk CG, van den Berg LH, van Zwet EW, Mei H, Li Y, Lemire M, Hudson TJ, Consortium B, Slagboom PE, Wijmenga C, Veldink JH, van Greevenbroek MM, van Duijn CM, Boomsma DI, Isaacs A, Jansen R, van Meurs JB, Hoen PAt, Franke L, Heijmans BT. Disease variants alter transcription factor levels and methylation of their binding sites. Nature genetics. 2017;49(1):131–138. doi: 10.1038/ng.3721. [DOI] [PubMed] [Google Scholar]
- 69.Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67(3):876–80. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 70.Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, Zanobetti A, Sparrow D, Vokonas PS, Schwartz J. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–8. doi: 10.1164/rccm.200807-1097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reichard JF, Schnekenburger M, Puga A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem Biophys Res Commun. 2007;352(1):188–92. doi: 10.1016/j.bbrc.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takiguchi M, Achanzar WE, Qu W, Li G, Waalkes MP. Effects of cadmium on DNA-(Cytosine-5) methyltransferase activity and DNA methylation status during cadmium-induced cellular transformation. Exp Cell Res. 2003;286(2):355–65. doi: 10.1016/s0014-4827(03)00062-4. [DOI] [PubMed] [Google Scholar]
- 73.IARC. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 97. 1,3-butadiene, ethylene oxide and vinyl halides (vinyl fluoride, vinyl chloride and vinyl bromide) IARC Monogr Eval Carcinog Risks Hum. 2008;97:3–471. [PMC free article] [PubMed] [Google Scholar]
- 74.Koturbash I, Scherhag A, Sorrentino J, Sexton K, Bodnar W, Tryndyak V, Latendresse JR, Swenberg JA, Beland FA, Pogribny IP, Rusyn I. Epigenetic alterations in liver of C57BL/6J mice after short-term inhalational exposure to 1,3-butadiene. Environmental health perspectives. 2011;119(5):635–40. doi: 10.1289/ehp.1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bangru S, Kalsotra A. Advances in analyzing RNA diversity in eukaryotic transcriptomes: peering through the Omics lens. F1000Res. 2016;5:2668. doi: 10.12688/f1000research.9511.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–97. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 78.Costlow NA, Simon JA, Lis JT. A hypersensitive site in hsp70 chromatin requires adjacent not internal DNA sequence. Nature. 1985;313(5998):147–9. doi: 10.1038/313147a0. [DOI] [PubMed] [Google Scholar]
- 79.Levy A, Noll M. Chromatin fine structure of active and repressed genes. Nature. 1981;289(5794):198–203. doi: 10.1038/289198a0. [DOI] [PubMed] [Google Scholar]
- 80.Cockerill PN. Structure and function of active chromatin and DNase I hypersensitive sites. FEBS J. 2011;278(13):2182–210. [Google Scholar]
- 81.Hogan GJ, Lee CK, Lieb JD. Cell cycle-specified fluctuation of nucleosome occupancy at gene promoters. PLoS Genet. 2006;2(9):e158. doi: 10.1371/journal.pgen.0020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132(2):311–22. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McDaniell R, Lee BK, Song L, Liu Z, Boyle AP, Erdos MR, Scott LJ, Morken MA, Kucera KS, Battenhouse A, Keefe D, Collins FS, Willard HF, Lieb JD, Furey TS, Crawford GE, Iyer VR, Birney E. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010;328(5975):235–9. doi: 10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee BK, Bhinge AA, Battenhouse A, McDaniell RM, Liu Z, Song L, Ni Y, Birney E, Lieb JD, Furey TS, Crawford GE, Iyer VR. Cell-type specific and combinatorial usage of diverse transcription factors revealed by genome-wide binding studies in multiple human cells. Genome research. 2012;22(1):9–24. doi: 10.1101/gr.127597.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, Liu Z, London D, McDaniell RM, Shibata Y, Showers KA, Simon JM, Vales T, Wang T, Winter D, Zhang Z, Clarke ND, Birney E, Iyer VR, Crawford GE, Lieb JD, Furey TS. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome research. 2011;21(10):1757–67. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Birney E, Lieb JD, Furey TS, Crawford GE, Iyer VR. Allele-specific and heritable chromatin signatures in humans. Hum Mol Genet. 2010;19(R2):R204–9. doi: 10.1093/hmg/ddq404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu C, Bingham PM, Livak KJ, Holmgren R, Elgin SC. The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell. 1979;16(4):797–806. doi: 10.1016/0092-8674(79)90095-3. [DOI] [PubMed] [Google Scholar]
- 89.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, Liu XS, Brown M. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 2014;11(1):73–78. doi: 10.1038/nmeth.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ason B, Reznikoff WS. DNA sequence bias during Tn5 transposition. J Mol Biol. 2004;335(5):1213–25. doi: 10.1016/j.jmb.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 92.Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, Ni B, Sklar J, Przytycka TM, Childs R, Levens D, Zhao K. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015;528(7580):142–6. doi: 10.1038/nature15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348(6237):910–4. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Christiansen L, Amini S, Zhang F, Ronaghi M, Gunderson KL, Steemers FJ. Contiguity-Preserving Transposition Sequencing (CPT-Seq) for Genome-Wide Haplotyping, Assembly, and Single-Cell ATAC-Seq. Methods Mol Biol. 2017;1551:207–221. doi: 10.1007/978-1-4939-6750-6_12. [DOI] [PubMed] [Google Scholar]
- 95.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y, Zhang B, Zhang L, An L, Xu J, Li D, Choudhary M, Li Y, Hu M, Hardison R, Wang T, Yue F. The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. BioRxiv. 2017 doi: 10.1186/s13059-018-1519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nature genetics. 2016;48(5):488–96. doi: 10.1038/ng.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong X, Virgil D, Birney E, Hung JH, Weng Z. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41(Database issue):D171–6. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vierstra J, Stamatoyannopoulos JA. Genomic footprinting. Nat Methods. 2016;13(3):213–21. doi: 10.1038/nmeth.3768. [DOI] [PubMed] [Google Scholar]
- 100.Chappell Grace, Israel Jennifer W, Simon Jeremy M, Pott Sebastian, Safi Alexias, Eklund Karl, Sexton Kenneth G, Bodnar Wanda, Lieb Jason D, Crawford Gregory E, Rusyn Ivan, Furey Terrence S. Variation in DNA-damage responses to an inhalational carcinogen (1,3-butadiene) in relation to strain-specific differences in chromatin accessibility and gene transcription profiles in C57BL/6J and CAST/EiJ mice. Environ Health Perspect. 2017 doi: 10.1289/EHP1937. [DOI] [PMC free article] [PubMed] [Google Scholar]