

Abstract
Normal bone mineralization requires phosphate oversaturation in bone matrix vesicles, as well as normal regulation of phosphate metabolism via the interplay among bone, intestine, and kidney. In turn, derangement of phosphate metabolism greatly affects bone function and structure. The type III sodium-dependent phosphate transporters, PiT-1 and PiT-2, are believed to be important in tissue phosphate metabolism and physiological bone formation, but their requirement and molecular roles in bone remain poorly investigated. In order to decipher the role of PiT-2 in bone, we examined normal bone development, growth, and mineralization in global PiT-2 homozygous knockout mice. PiT-2 deficiency resulted in reduced vertebral column, femur, and tibia length as well as mandibular dimensions. Micro-computed tomography analysis revealed that bone mineral density in the mandible, femur, and tibia were decreased, indicating that maintenance of bone function and structure is impaired in both craniofacial and long bones of PiT-2 deficient mice. Both cortical and trabecular thickness and mineral density were reduced in PiT-2 homozygous knockout mice compared with wild-type mice. These results suggest that PiT-2 is involved in normal bone development and growth and plays roles in cortical and trabecular bone metabolism feasibly by regulating local phosphate transport and mineralization processes in the bone. Further studies that evaluate bone cell-specific loss of PiT-2 are now warranted and may yield insight into complex mechanisms of bone development and growth, leading to identification of new therapeutic options for patients with bone diseases.
Keywords: PiT-2, phosphate, phosphate transporters, bone mineral density, bone length
1. Introduction
Bone is a rigid and hard organ that forms the vertebrate skeleton. Bone is mainly composed of hydroxyapatite, a form of calcium-phosphate crystal, other minerals, and bone matrix proteins such as collagen and non-collagen fibrils [1]. Bone formation and its ongoing remodeling are critical for skeletal health. Both processes are mainly governed by osteoblasts, osteoclasts, and osteocytes under the coordination of a variety of minerals including inorganic phosphate (Pi), morphogens, hormones, and cytokines [2–4].
Experimental studies have shown that Pi promotes proliferation and differentiation of pre-osteoblasts and osteoblasts, and the mineralization of bone matrix as well as inhibition of osteoclastic activity [5–7]. Notably, for normal bone mineralization, Pi oversaturation in the bone matrix vesicles is critical [8]. Pi metabolism is tightly regulated by the balance among intestinal Pi uptake, renal Pi resorption, and the exchange between intracellular space and bone, which is controlled by various hormones [9]. Therefore, derangement of Pi metabolism in the bone is believed to greatly affect bone function and structure. When Pi is depleted in the body, bone mineral density (BMD) decreases and the bone quality degrades, ultimately leading to bone deformity and fragility fracture [10, 11].
There is a growing interest in the type III sodium-dependent Pi transporter family, which harbors just two members: Slc20a1/PiT-1 and Slc20a2/PiT-2. These two transporters are expressed ubiquitously in all organs at low levels, and thus were erroneously thought to play a minor role in maintaining homeostatic levels of intracellular Pi [12, 13]. However, recent studies have shown that PiT-1 is involved in matrix vesicle formation and bone mineralization [14]. Bourgine et al recently reported that hypomorphic mice with just 15% of normal expression of PiT-1 developed slightly shorter femurs than controls, but failed to display bone mineralization defects in vivo, possibly due to an observed upregulation of PiT-2 in this line, indicating a potentially important role of PiT-2 in the bone regulation [15]. PiT-1 and PiT-2 have also been suggested to regulate maternal-fetal Pi transport during development [16, 17]. More recently, PiT-1 loss driven by the col2a1 promoter on a Phospho1 null background inhibited bone formation in mice, leading to defects in skeletal mineralization as well as biogenesis and function of collagen derived matrix vesicles [18]. Previous studies have shown that PiT-2 is expressed in bone cells such as osteoblasts, chondrocytes in the growth plate, and osteoclasts [19–23]. However, little is known about the role of PiT-2 in skeletogenesis.
The aim of the present study was to test the hypothesis that PiT-2 mediates normal bone development, growth, and mineralization. In the study, we performed bone micro-computed tomography (micro CT) scans and assessed bone length and BMD in global PiT-2 homozygous knockout (PiT-2 KO) mice, which recapitulate many features seen in PiT-2 deficiency in humans [24]. We also calculated bone morphometric parameters. Herein, we report for the first time that BMD and dimensions of the craniofacial and long bones were indeed significantly reduced in PiT-2 deficient mice.
2. Materials and methods
2.1. Animal Work
Protocols were in compliance with the NIH Guidelines for the Care and Use of Laboratory Animals and have been approved by the Institutional Animal Care and Use Committee at the University of Washington. All mice were maintained in a specific pathogen-free environment and housed in a climate-controlled 12 hours day/night cycle and provided food and water ad libitum. Mice were fed a standard chow containing 0.98% Pi.
C57BL/6NTac-Slc20a2<tm1a (EUCOMM)Wtsi>/Ieg (Slc20a2 +/−) mice were purchased from the European Mouse Mutant Archive (EMMA) and used to generate PiT-2 KO mice and their wildtype (WT) littermates [24]. Ten-week-old male and female mice were euthanized by carbon dioxide asphyxiation, frozen, and analyzed, post-thaw, by micro CT as detailed below.
2.2. Micro CT
Micro CT scans were performed at the Small ANimal Tomographic Analysis (SANTA) facility located at the Seattle Children’s Research Institute using a SkyScan 1076 instrument (Skyscan, Kontich, Belgium). Full body scans were performed at an isotropic resolution of 34.42 μm (60 kV, 170 μA, 0.5 mm Aluminum filter, 120 ms exposure, rotation step of 0.6°, 180° scan, and 3 frame averaging). Raw data were reconstructed using the software NRecon V1.6.9 (SkyScan) and 3D rendered images of each dataset were generated using the Drishti V2 Volume Exploration software (http://sf.anu.edu.au/Vizlab/drishti).
2.3. Analysis of bone length and BMD
Length of vertebral column, femur, and tibia was measured using 3D rendered images. To compare mandibular morphology, mandibular height and length to the angular process and condylar process were also determined. BMD was measured at the mandibular ramus and distal femur using a standardized volume of interest (VOI) sphere created using micro CT scan software (Skyscan) following calibration with commercially supplied calcium hydroxyapatite phantoms of known density: 0.25 (min) and 0.75 (max) g/cm3 (Skyscan). Measurements were performed on each side (hemi-mandibles and femurs) and averaged for each mouse.
2.4. Quantitative assessment of static bone morphological parameters by micro CT
Morphological analysis of mouse femurs and determination of static bone parameters were performed as described previously [25]. Two regions were quantitatively analyzed in mice: the cortical bone region from 2.0 to 2.5 mm above the growth plate at the distal metaphysis; and the trabecular bone region from 0.1 to 1.1 mm above the growth plate at the distal metaphysis. From the reconstructed scan data, we calculated the following static bone parameters: bone volume/total volume; bone surface/total volume; trabecular number; trabecular thickness; trabecular separation; cortical bone area; total bone area; cortical bone area/total bone area; and cortical thickness. For each parameter, micro CT-derived standard bone morphometry nomenclature, symbols, and units were used [26].
2.5. Statistical Analyses
Data are expressed as mean ± S.E.M. Differences among groups were analyzed by un-paired t-test. Statistical significance was assessed at P<0.05. All statistical analyses were conducted by JMP version 13.2 (SAS Institute, NA, USA).
3. RESULTS
3.1. The impact of PiT-2 deficiency on body weight and bone length
Homozygous PiT-2 KO and WT mice were identified by genotyping using DNA extracted from tail biopsy specimens (Fig. 1A). Gross body size of female PiT-2 KO mice appeared to be smaller than WT mice (Fig. 1B) and this was supported by body weight measurements (Fig. 1C). Bone length measured on the 3D rendered micro CT data confirmed that PiT-2 deficiency resulted in shorter length of the vertebral column, femur, and tibia than WT mice (Fig. 1D–G). In addition, measurements of mandibular height (measurements 2, 3 and 4 in Fig. 2A) and length to the angular process (measurement 5 in Fig. 2A) were 8–10% smaller than controls (Fig. 2B and C). Of note, the length to the condylar process (measurement 1 in Fig. 2A) was only 5% smaller (Fig. 2C), consistent with the relative elongation of the condylar process that is evident when scaling and overlaying PiT-2 KO and WT hemi-mandibles (Fig. 2B), indicating an impact of PiT-2 loss on the primary growth center of the mandible.
Fig. 1. Reduced size, weight, and bone length in global Pit-2 homozygous knockout mice.
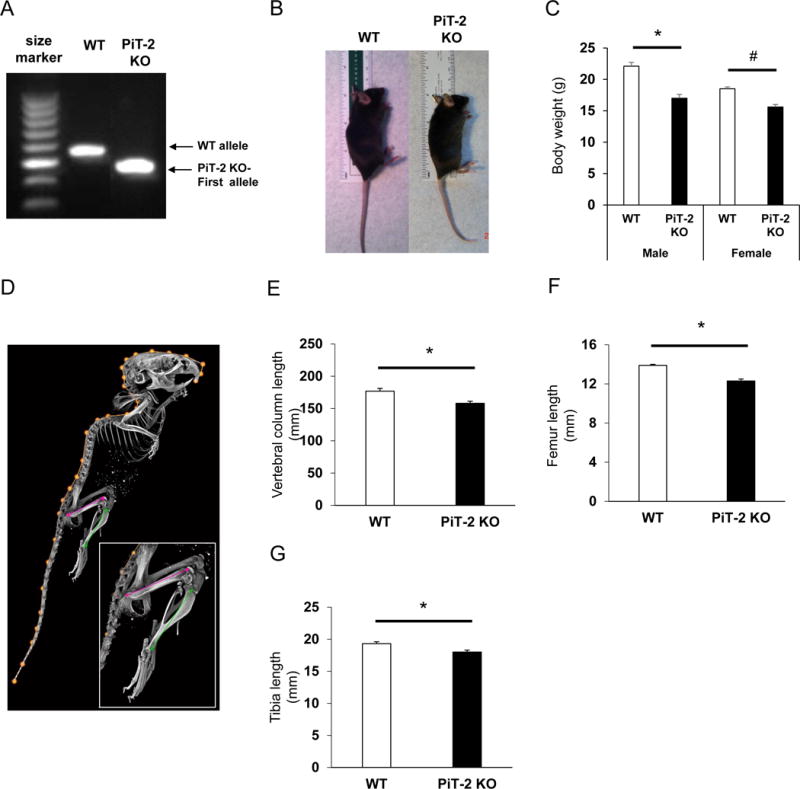
(A) Genomic DNA determined by RT-PCR in WT and PiT-2 KO mice. (B) Gross appearance of female mice. (C) Body weight of male and female mice. (D) Representative rendered micro CT scan showing the various bone length measurements taken. Length of (E) vertebral column, (F) femur, and (G) tibia of male mice. Data are presented as mean ± S.E.M (n=5). A two-tailed P-value < 0.05 was considered statistically significant. *P<0.05 versus WT group. #P<0.05 versus female WT group. Micro CT, micro-computed tomography; PiT-2 KO, PiT-2 homozygous knockout; RT-PCR, reverse transcriptase-polymerase chain reaction; WT, wild-type.
Fig. 2. Global PiT-2 homozygous knockout mice exhibit reduced mandibular size but relatively elongated condylar processes.
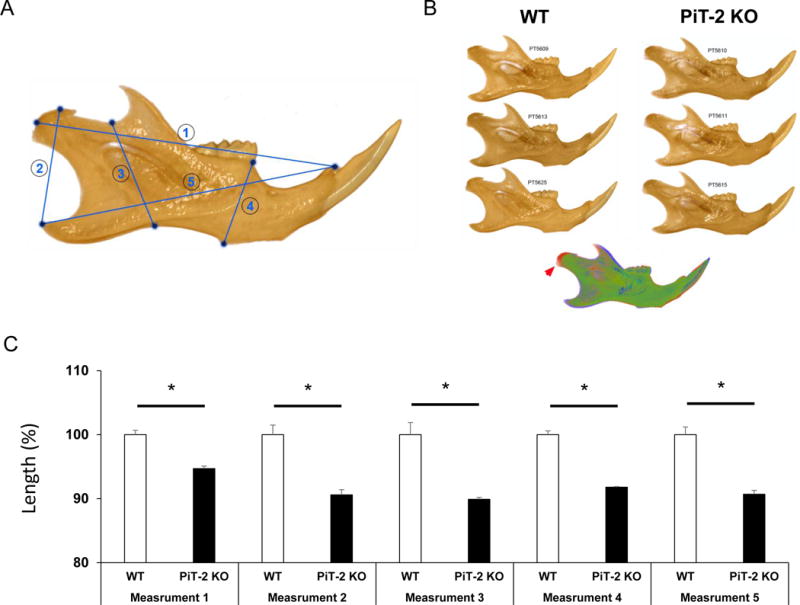
(A) Representative image of a 3D rendered mandible showing the five linear measurements collected from mandibles. (B) Scaled lateral views of representative mandibles of wildtype and PiT-2 KO male mice. The overlay of representative WT (green) and PiT-2 KO (red) mandibles after scaling shows the relative condylar process extension in PiT-2 KO mice (red arrowhead). (C) Comparison of five mandibular distances (denoted in (A)) between WT and PiT-2 KO mice. Data are presented as mean ± S.E.M (n= 3). Differences between groups were analyzed by an un-paired t-test. A two-tailed P-value < 0.05 was considered statistically significant. *P<0.05 versus WT group. PiT-2 KO, PiT-2 homozygous knockout; WT, wildtype.
3.2. PiT-2 deficiency decreased BMD in systemic bone
All scans, reconstructions, and 3D rendering were performed using the same settings for all animals, allowing for direct visual comparison between specimens. This visual analysis identified several bone abnormalities. Most notable was an apparent decrease in BMD in the maxilla, mandible, occipital bones, ribs, scapula, sternum, and vertebrae (as shown in the lateral upper body views in Fig. 3A) as well as in enamel mineral density in incisors and molars in PiT-2 KO mice. Subsequent quantification of mandible BMD revealed a significant decrease in both male and female PiT-2 deficient mice compared with WT mice, as shown in Fig. 3B.
Fig. 3. Global PiT-2 homozygous knockout mice exhibit reduced bone and tissue mineral density.
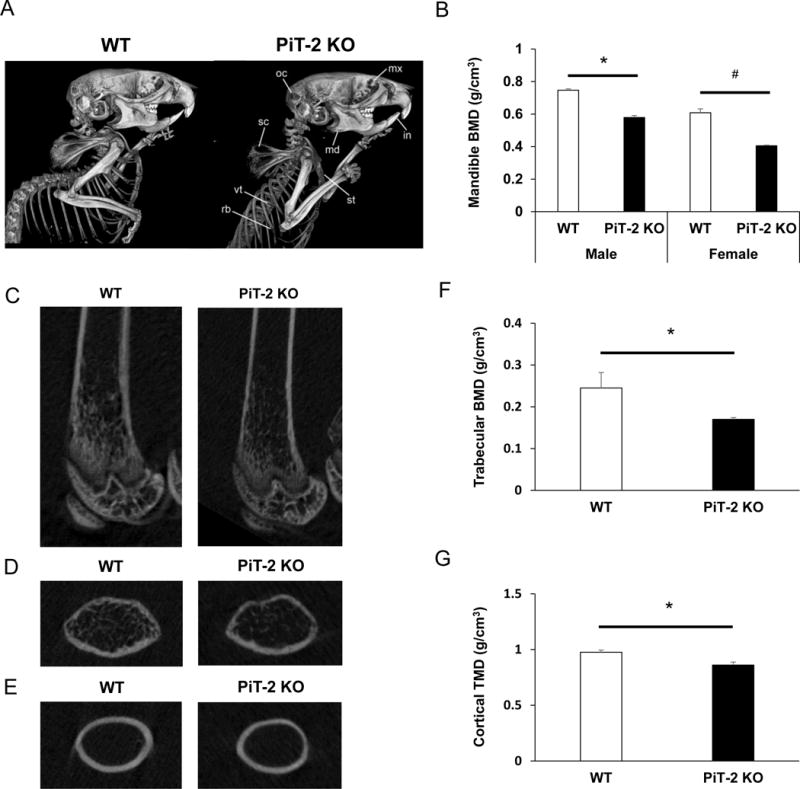
(A) Representative lateral views of the upper body of age-matched male mice scanned by micro CT. Several bone abnormalities were observed, including a suspected decreased in BMD in the maxillary (mx), mandible (md), incisors (in), occipital bones (oc), ribs (rb), scapula (sc), sternum (st), and vertebrae (vt). (B) BMD in the mandible of male and female mice. (C) Longitudinal images of femurs. (D) Cross-sectional images of femurs in trabecular bone area. (E) Cross-sectional images of femurs in cortical bone area. (F) Trabecular BMD of femurs. (G) Cortical BMD of femurs. Data are expressed as mean ± S.E.M (n= 3 or 5). Differences between groups were analyzed by un-paired t-test. A two-tailed P-value < 0.05 was considered statistically significant. *P<0.05 versus male WT group. #P<0.05 versus female WT group. BMD, bone mineral density; micro CT, micro-computed tomography; PiT-2 KO, PiT-2 homozygous knockout; WT, wildtype.
3.3. Global PiT-2 deficiency decreased both trabecular and cortical bone volume
To determine the impact of PiT-2 deficiency on long bones, we measured BMD of WT and PiT-2 KO mouse femurs by micro CT. Fig. 3C–E shows representative femur scans in longitudinal and cross-sectional views. When trabecular and cortical bone regions were analyzed separately, both trabecular and cortical BMD were significantly decreased in PiT-2 KO mice as compared with age-matched WT control mice (Fig. 3F and G). The decrease in trabecular BMD was greater than that in cortical BMD (31% versus 12%).
When static bone parameters were further analyzed, bone volume/total volume, bone surface/total volume, trabecular number, and trabecular thickness were significantly decreased in PiT-2 KO mice compared with WT mice (Fig. 4A–D). Although trabecular separation tended to be higher in PiT-2 KO mice, no significant difference was observed between the genotypes (Fig. 4E). As for cortical bone parameters, total area, bone area, bone area/total area, and cortical thickness were significantly decreased in PiT-2 KO mice compared with age-matched WT mice (Fig. 4F–I).
Fig. 4. Loss of PiT-2 impacts static bone morphometric parameters of femurs in male mice.
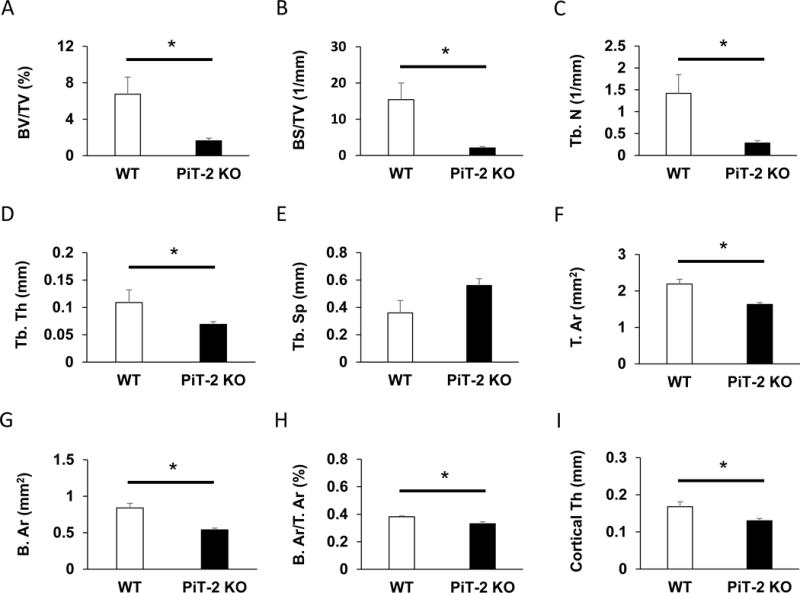
(A) BS/TV. (B) BV/TV. (C) Tb. Th. (D) Tb. N. (E) Tb. Sp. (F) T. Ar. (G) B. Ar. (H) B. Ar/T. Ar. (l) Cortical Th. Data are expressed as mean ± S.E.M. (n=3 or 5) and compared by un-paired t-test. A two-tailed P<0.05 was considered statically significant. *P<0.05 versus WT group. Ar, area; B, bone; BMD, bone mineral density; BS, bone surface; BV, bone volume; micro CT, micro-computed tomography; N, number; PiT-2 KO, PiT-2 homozygous knockout; Sp, separation; T, total; Tb, trabecular; Th, thickness; TV, total volume; WT, wild-type.
4. Discussion
The data presented herein report the first evaluation of the requirement for PiT-2 in normal bone growth and development in vivo. PiT-2 deficiency decreased BMD in both craniofacial and long bones and was associated with decreased length of the vertebral column, femur, and tibia. Reduced mandibular size with a differential impact on the condylar process was also identified. The lesser impact on the condyle is likely related to its being the major endochondral growth center of the mandible, and suggests that reduced mineralization may favor extended condylar cartilage growth. Quantitative analyses of micro CT data showed that static bone morphometric parameters in the trabecular and cortical bone were greatly decreased in PiT-2 KO mouse femurs. Our data therefore demonstrate that PiT-2 is required for physiological bone development and growth in both craniofacial and long bones, and PiT-2 loss affects both cortical and trabecular bone metabolism in mice.
In the clinical setting, impaired bone mineralization is often observed when Pi homeostasis is significantly disturbed such as in osteomalacia/rickets [10]. Osteomalacia/rickets is generally caused by vitamin D deficiency or Pi depletion (dietary or renal depletion) or hereditary disorders related to Pi, calcium, and vitamin D metabolism. The resultant hypophosphatemia leads to insufficient Pi delivery to the bone mineralization front, followed by increased osteoid volume and decreased mineral content [10, 27]. Notably, previous studies have shown that serum Pi and calcium levels were not altered by PiT-2 deficiency [24, 28]. Although the detailed mechanisms are unclear, our results suggest that PiT-2 deficiency causes bone loss independent of serum mineral parameters and failure to deliver Pi to the mineralizing front might be a possible cause for the decreased BMD in PiT-2 deficient mice.
In the present study, BMD was decreased in both craniofacial (mandible) and long bones (femur and tibia) in PiT-2 KO mice. Our results suggest that PiT-2 deficiency affects maintenance of both craniofacial and long bones. Given the robust expression of PiT-2 in osteoblasts [19–21], loss of PiT-2 in this cell type could explain the observed phenotypes. An additional cause for the decreased BMD may be increased osteoclast activity in the bone. Pi has been shown to inhibit osteoclastic activity [7] and bone volume is determined by the balance between osteoclasts and osteoblasts. Previous studies have shown that PiT-2 is also expressed in osteoclasts [22]. Thus, PiT-2 deficiency could also alter osteoclastic activity, thereby leading to decreased bone mineral content. Further work aimed at identifying the different bone cell types that are directly affected by PiT-2 deficiency will lead to a better understanding of PiT-2 roles in normal bone mineralization. In addition, histological analysis of neonates and juvenile mice will also likely be necessary to determine whether PiT-2 deficiency affects membranous and endochondral ossification, both of which are initiated in the early stage of bone development [1–3, 29, 30].
In our study, both cortical and trabecular BMD as well as other trabecular and cortical bone parameters were decreased in global PiT-2 deficient mice. Because bone volume and quality in both trabecular and cortical bone greatly affect bone strength [31], these data suggest that PiT-2 may be a therapeutic target for patients with bone disorders.
PiT-2 heterozygous knockout mice recapitulate some of the clinical characteristics of patients with idiopathic basal ganglion calcification (IBGC) including neurovascular calcification [24]. To our knowledge, there have been no reports on the development of low BMD in patients with IBGC. Thus, PiT-2 haploinsufficiency may be insufficient to reduce BMD. In human, PiT-1 or other transporters may compensate for the loss of PiT-2 in the bone. It may be mechanistically informative to examine the physiological role of PiT-2 in the bone development among patients with IBGC in the future.
In conclusion, PiT-2 deficiency decreased bone volume in systemic bone including craniofacial and long bones, shortened bone length, and affected mandibular morphology in mice. Our study is the first to show that PiT-2 is essential for normal bone development, growth, and mineralization, including both endochondral and membranous ossification. Studies, such as bone cell type-specific deletion of PiT-2 and more detailed histomorphometric analyses, may shed further insight into the molecular mechanisms by which PiT-2 regulates normal skeletogenesis.
Research highlights.
PiT-2 deficiency decreased mineral density in craniofacial and long bones in mice.
Bone length was reduced in long bones in PiT-2 deficient mice.
Trabecular and cortical bone mineral density were decreased in PiT-2 deficient mice.
Static bone morphometric parameters were altered in PiT-2 deficient mice.
PiT-2 deficiency altered craniofacial bone morphology.
Acknowledgments
SY is supported by the grant from the Japanese Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad (JSPS 20150701), the Uehara Memorial Foundation 2017, and the International Research Fund for Subsidy of Kyushu University School of Medicine Alumni 2015. MCW was supported by NHLBI Training Grant T32HL007828 and is currently supported by NICHD Pathway to Independence Grant K99HD090198. The work was further supported by grants from the National Institutes of Health (HL62329, HL081785, and HL114611) and the Department of Defense (OR120074) to CMG, and from the Laurel Foundation Endowment for Craniofacial Research (to TCC). The content of this project is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare that they have no conflict of interest.
References
- 1.Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol. 2008;(Suppl 3):S131–139. doi: 10.2215/CJN.04151206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teti A. Bone development: overview of bone cells and signaling. Curr Osteoporos Rep. 2011;9:264–273. doi: 10.1007/s11914-011-0078-8. [DOI] [PubMed] [Google Scholar]
- 3.Berendsen AD, Olsen BR. Bone development. Bone. 2015;80:14–18. doi: 10.1016/j.bone.2015.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 5.Kanatani M, Sugimoto T, Kano J, et al. IGF-I mediates the stimulatory effect of high phosphate concentration on osteoblastic cell proliferation. J Cell Physiol. 2002;190:306–312. doi: 10.1002/jcp.10067. [DOI] [PubMed] [Google Scholar]
- 6.Conrads KA, Yi M, Simpson KA, et al. A combined proteome and microarray investigation of inorganic phosphate-induced pre-osteoblast cells. Mol Cell Proteomics. 2005;4:1284–1296. doi: 10.1074/mcp.M500082-MCP200. [DOI] [PubMed] [Google Scholar]
- 7.Arioka M, Takahashi-Yanaga F, Tatsumoto N, et al. Inorganic phosphate-induced impairment of osteoclast cell-cell fusion by the inhibition of AP-1-mediated DC- STAMP expression. Biochem Biophys Res Commun. 2017;493:9–13. doi: 10.1016/j.bbrc.2017.09.096. [DOI] [PubMed] [Google Scholar]
- 8.Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–226. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- 9.Prié D, Ureña Torres P, Friedlander G. Latest findings in phosphate homeostasis. Kidney Int. 2009;75:882–889. doi: 10.1038/ki.2008.643. [DOI] [PubMed] [Google Scholar]
- 10.Elder CJ, Bishop NJ. Rickets. Lancet. 2014;383:1665–1676. doi: 10.1016/S0140-6736(13)61650-5. [DOI] [PubMed] [Google Scholar]
- 11.Feng JQ, Clinkenbeard EL, Yuan B, et al. Osteocyte regulation of phosphate homeostasis and bone mineralization underlies the pathophysiology of the heritable disorders of rickets and osteomalacia. Bone. 2013;54:213–221. doi: 10.1016/j.bone.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2008;23:22–44. doi: 10.2133/dmpk.23.22. [DOI] [PubMed] [Google Scholar]
- 13.Forster IC, Hernando N, Biber J, et al. Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med. 2013;34:386–395. doi: 10.1016/j.mam.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Yoshiko Y, Candeliere GA, Maeda N, et al. Osteoblast autonomous Pi regulation via Pit1 plays a role in bone mineralization. Mol Cell Biol. 2007;27:4465–4474. doi: 10.1128/MCB.00104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourgine A, Pilet P, Diouani S, et al. Mice with hypomorphic expression of the sodium-phosphate cotransporter PiT1/Slc20a1 have an unexpected normal bone mineralization. PLoS One. 2013;8:e65979. doi: 10.1371/journal.pone.0065979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallingford MC, Giachelli CM. Loss of PiT-1 results in abnormal endocytosis in the yolk sac visceral endoderm. Mech Dev. 2014;133:189–202. doi: 10.1016/j.mod.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallingford MC, Gammill HS, Giachelli CM. Slc20a2 deficiency results in fetal growth restriction and placental calcification associated with thickened basement membranes and novel CD13 and lamininα1 expressing cells. Reprod Biol. 2016;16:13–26. doi: 10.1016/j.repbio.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yadav MC, Bottini M, Cory E, et al. Skeletal Mineralization Deficits and Impaired Biogenesis and Function of Chondrocyte-Derived Matrix Vesicles in Phospho1(−/−) and Phospho1/Pi t1 Double-Knockout Mice. J Bone Miner Res. 2016;31:1275–1286. doi: 10.1002/jbmr.2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nielsen LB, Pedersen FS, Pedersen L. Expression of type III sodium-dependent phosphate transporters/retroviral receptors mRNAs during osteoblast differentiation. Bone. 2001;28:160–166. doi: 10.1016/s8756-3282(00)00418-x. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki A, Palmer G, Bonjour JP, et al. Stimulation of sodium-dependent inorganic phosphate transport by activation of Gi/o-protein-coupled receptors by epinephrine in MC3T3-E1 osteoblast-like cells. Bone. 2001;28:589–594. doi: 10.1016/s8756-3282(01)00459-8. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki A, Ghayor C, Guicheux J, et al. Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res. 2006;21:674–683. doi: 10.1359/jbmr.020603. [DOI] [PubMed] [Google Scholar]
- 22.Albano G, Moor S, Dolder S, et al. Sodium-dependent phosphate transporters in osteoclast differentiation and function. PLoS One. 2015;10:e0125104. doi: 10.1371/journal.pone.0125104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solomon DH, Wilkins RJ, Meredith D, et al. Characterisation of inorganic phosphate transport in bovine articular chondrocytes. Cell Physiol Biochem. 2007;20:99–108. doi: 10.1159/000104158. [DOI] [PubMed] [Google Scholar]
- 24.Wallingford MC, Chia J, Leaf EM, et al. SLC20A2 deficiency in mice leads to elevated phosphate levels in cerbrospinal fluid and glymphatic pathway-associated arteriolar calcification, and recapitulates human idiopathic basal ganglia calcification. Brain Pathol. 2017;27:64–76. doi: 10.1111/bpa.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bouxsein ML, Boyd SK, Christiansen BA, et al. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468–1486. doi: 10.1002/jbmr.141. [DOI] [PubMed] [Google Scholar]
- 26.Tatsumoto N, Arioka M, Yamada S, et al. Inhibition of GSK-3β increases trabecular bone volume but not cortical bone volume in adenine-induced uremic mice with severe hyperparathyroidism. Physiol Rep. 2016;4:e13010. doi: 10.14814/phy2.13010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elidrissy AT. The Return of Congenital Rickets, Are We Missing Occult Cases? Calcif Tissue Int. 2016;99:227–236. doi: 10.1007/s00223-016-0146-2. [DOI] [PubMed] [Google Scholar]
- 28.Jensen N, Autzen JK, Pedersen L. Slc20a2 is critical for maintaining a physiologic inorganic phosphate level in cerebrospinal fluid. Neurogenetics. 2016;17:125–130. doi: 10.1007/s10048-015-0469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackie EJ, Tatarczuch L, Mirams M. The skeleton: a multi-functional complex organ: the growth plate chondrocyte and endochondral ossification. J Endocrinol. 2011;211:109–121. doi: 10.1530/JOE-11-0048. [DOI] [PubMed] [Google Scholar]
- 30.Percival CJ, Richtsmeier JT. Angiogenesis and intramembranous osteogenesis. Dev Dyn. 2013;242:909–922. doi: 10.1002/dvdy.23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285:785–795. [Google Scholar]