

ABSTRACT
CD11b+Gr1+ myeloid-derived suppressor cells (MDSCs) suppress activation/proliferation of cytotoxic T cells, thereby hindering cancer immunotherapy. MDSCs are increased after adjuvant therapy with toll-like receptor (TLR) 2 ligands, such as Pam2CSK4, in tumor-bearing mice. However, it remains unknown if the activation of TLR2 in MDSCs affects their function and the therapeutic efficacy of TLR2 ligand. Here, we show that TLR2 signaling in CD11b+Ly6G−Ly6Chigh monocytic MDSCs (M-MDSCs), but not CD11b+Ly6G+Ly6Clow granulocytic MDSCs (G-MDSCs), enhances their immunosuppressive activity, thereby limiting anti-tumor T cell responses induced by TLR2-activated dendritic cells (DCs). iNOS induction was critical for Pam2CSK4-enhanced T cell suppression by M-MDSCs. iNOS was expressed in M-MDSC-derived macrophages, but not undifferentiated M-MDSCs, in cocultures with CD8+ T cells, CD11c+ DCs, antigen peptide and Pam2CSK4. Pam2CSK4 increased the differentiation frequency of M-MDSCs to macrophages, and iNOS expression required interferon-γ (IFN-γ) production by CD8+ T cells that had been transiently stimulated by M-MDSC-derived macrophages in an antigen/TLR2-dependent manner. Although Pam2CSK4 triggered DC maturation and tumor regression via induction of tumor antigen-specific cytotoxic T lymphocyte (CTL) responses in tumor-bearing mice, Pam2CSK4 plus antigen increased the frequency of iNOS+ macrophages in the tumor. Treatment with iNOS inhibitor enhanced the therapeutic efficacy of Pam2CSK4. Hence, the results suggest that TLR2 ligand and T cell-derived IFN-γ enhance M-MDSC-mediated immunosuppression, which may negatively regulate anti-tumor CTL response.
KEYWORDS: Inducible NO synthase (iNOS/NOS2), interferon-γ (IFN-γ), macrophages, monocytic MDSCs (M-MDSCs), myeloid-derived suppressor cells (MDSCs), nitric oxide (NO), toll-like receptor 2 (TLR2)
Introduction
Activation of toll-like receptors (TLRs) in myeloid-derived cells, such as dendritic cells (DCs), macrophages, and myeloid-derived suppressor cells (MDSCs), has both positive and negative impacts on acute and chronic inflammation and tumor growth.1-3 TLR signaling critically affects the cell type-specific responses of these myeloid-derived cells. Here we investigate how TLR signaling in these myeloid cells leads to differential regulation of tumor growth.
Administration of TLR2 ligands leads to inhibition of tumor growth in mice implanted with cancer cell lines.4,5 TLR2 forms a heterodimer with TLR1 or TLR6 for recognizing bacterial lipoproteins such as triacylated lipoproteins or diacylated lipopeptides, respectively.6 Activation of TLR2 by Pam2CSK4 initiates intracellular signaling through an adaptor protein, myeloid differentiation primary response gene 88 (MyD88). In DCs, TLR2-MyD88 signaling induces the production of pro-inflammatory cytokines and accelerates cross-presentation of tumor-associated antigens (TAAs), leading to activation and proliferation of TAA-specific CD8+ T cells and stimulation of anti-tumor natural killer (NK) cells.5,7,8 Thus, activation of TLR2 signaling in DCs is important for initiating anti-tumor effector mechanisms. However, recent studies highlight the tumor-promoting roles of the TLR2 signaling pathway in cancer. Endogenous TLR2 ligands promote tumor development in gastric cancer or accelerate tumor metastasis into lung by modulating myeloid cell function.9,10 TLR2-dependent induction of regulatory T cells (Tregs) inhibits anti-tumor immune responses triggered by TLR2 ligands.11 Thus, TLR2 signaling contributes to both inhibition and promotion of tumor growth. The differences in TLR responses appear to be dependent on the types of responding cells, as well as the intracellular signaling pathways downstream of MyD88. Whether TLR2 activation in myeloid cells, especially MDSCs and macrophages, affects the therapeutic efficacy of TLR2 ligands is unclear.
MDSCs are a heterogeneous population of immature myeloid cells that suppress the functions of T cells, NK cells, and DCs by secreting suppressive molecules.12 MDSCs negatively regulate excessive immune responses to maintain homeostasis disrupted by infection. However, in cancer development, MDSCs accumulate in tumors, as well as peripheral tissues, providing immunosuppressive conditions favorable for tumor cell growth. In mice, CD11b+Gr1+ MDSCs are divided into two populations: CD11b+Ly6G−Ly6Chigh cells and CD11b+Ly6G+Ly6Clow cells, which are called monocytic MDSCs (M-MDSCs) and polymorphonuclear/granulocytic MDSCs (PMN/G-MDSCs), respectively. M-MDSCs contain immature myeloid cells that are capable of differentiating in the tumor into mature cells including macrophages and DCs.13 By contrast, G-MDSCs have a terminally differentiated phenotype largely similar to that of neutrophils.14 Although iNOS is a critical factor for the induction of M-MDSC and G-MDSC suppressive activity on T cell proliferation, the immunosuppressive effectors of MDSC subsets are distinct. Nitric oxide (NO), generated by iNOS, is a major effector of M-MDSC-mediated suppression, whereas peroxynitrite (PNT), generated by the reaction of NO with reactive oxygen species (ROS) derived from the NADPH oxidase complex, plays a major role in the suppressive activity of G-MDSCs.15,16 These effector molecules derived from M-MDSCs or G-MDSCs act on T cells to suppress their growth via distinct modes of action.
Recent reports have suggested that the survival, differentiation, and suppressive activity of MDSCs are influenced by TLR signaling.17 As MDSCs express TLRs and accumulate in cancer, they, as well as DCs, appear to be primary targets of TLR ligands when administered into tumor-bearing hosts. However, the modulation of MDSC function by TLR signaling in cancer immunotherapy, and its effects on tumor growth, have not been well characterized. TLR2 activation by Pam2CSK4 lipopeptide leads to accumulation of CD11b+Gr1+ MDSCs in the tumors, as well as peripheral tissues, of tumor-bearing mice.18 Therefore, we hypothesized that systemic administration of TLR2 ligands for cancer treatment potentiates immunosuppression by MDSCs and, therefore, negatively affects the anti-tumor response mediated by DCs and CTLs. In the present study, we investigated the effects of TLR2 activation on G-MDSCs and M-MDSCs. TLR2 ligand administration did not enhance the suppressive activity of G-MDSCs, but did enhance that of M-MDSCs by inducing their differentiation into iNOS+ macrophages that inhibit T cell proliferation through NO production. IFN-γ produced by T cells was a critical regulator of this process. These effects of TLR2 signaling on M-MDSCs have a negative impact on the therapeutic efficacy of TLR2 ligands in tumor-bearing mice and could be an additional target for adjuvant immunotherapy for cancer.
Results
Pam2CSK4 sustains the survival of MDSCs
When tumor-bearing mice were treated with Pam2CSK4, the proportion of CD11b+Gr1+ cells was increased (Fig. 1A), as previously reported.18 To assess if TLR2 activation affects their viability, we isolated CD11b+Gr1+ cells from EG7 tumor-bearing mice and cultured them in the presence of Pam2CSK4. As shown in Fig. 1B, the viability of MDSCs was increased after a 24-h incubation with Pam2CSK4 (Fig. 1B). Conversely, the proportion of propidium iodide (PI)-stained dead cells was decreased by Pam2CSK4 (Fig. 1C). This effect was abrogated in MDSCs isolated from TLR2−/− mice bearing tumors (TLR2−/− MDSCs), suggesting that TLR2 activation increased the viability of MDSCs (Fig. 1B). IL-6 has been implicated in the development of MDSCs.19 Although Pam2CSK4 induced IL-6 production by MDSCs (Fig. S1), IL-6−/− MDSCs still showed prolonged survival in response to Pam2CSK4 (Fig. 1B). Thus, these results suggested that Pam2CSK4 prolonged survival of MDSCs through activation of TLR2 signaling, which is independent of autocrine production of IL-6 by MDSCs. Since TLR2 activation up-regulates the expression of several genes involved in cell cycle progression or cell survival/anti-apoptosis, leading to the proliferation of cancer cells,9 we analyzed mRNA expression in MDSCs of anti-apoptotic genes such as Bcl2l1 and Bcl3, and cell cycle progression genes such as Ccnd1, Ccnd2, Ier3, and c-Myc. Expression of these genes was increased in MDSCs after Pam2CSK4 treatment for 4 h. This effect was abrogated in TLR2−/− MDSCs (Fig. 1D). G-MDSCs and M-MDSCs expressed TLR2 at a similar level (Fig. 1E). Both of the MDSC subsets showed prolonged survival with Pam2CSK4 treatment in vitro (Fig. 1F). Thus, activation of TLR2 signaling enhanced the survival of both MDSC subsets, which may be responsible, at least in part, for their accumulation in tumor-bearing mice treated with Pam2CSK4.
Figure 1.

Pam2CSK4 sustains the survival of CD11b+Gr1+ MDSCs. (A) EG7 tumor-bearing mice were subcutaneously injected twice with PBS or 50 nmol Pam2CSK4 and OVA protein every 4 days. After 24 hours from last injection, the proportion of CD11b+Gr1+ cells in the spleen was analyzed by flow cytometry. Numbers adjacent to outlined areas indicate the percentage of relevant population. (B) CD11b+Gr1+ cells were isolated from EG7 tumor-bearing B6 WT, TLR2−/−, or IL-6−/− mice, and cultured in the presence of PBS or Pam2CSK4. After 24 h, cell viability was measured by WST-1 assay. (C) CD11b+Gr1+ cells treated with PBS (thin line histogram) or Pam2CSK4 (bold line histogram) for 24 hours were analyzed by flow cytometry after staining with PI. (D) Real-time PCR analysis of transcripts for Bcl2l1, Bcl3, Ccnd1, Ccnd2, ler3, and c-Myc in CD11b+Gr1+ cells isolated from tumor-bearing B6 WT or TLR2−/− mice after in vitro treatment with Pam2CSK4 or PBS for 4 h. (E) Flow cytometric analysis of Ly6G and Ly6C expression in CD11b+Gr1+ cells (left). TLR2 expression in G-MDSCs or M-MDSCs (right). (F) G-MDSCs and M-MDSCs were isolated from tumor-bearing mice and incubated with Pam2CSK4 or PBS for 24 h. Cell viability was measured by WST-1 assay. Data represent means ± standard deviation (SD) in graph. n = 3. **P < 0.005. *P < 0.05. All data shown are representative of more than 2 independent experiments.
Pam2CSK4 promotes differentiation of M-MDSCs into CD11b+F4/80+CD115+ macrophages
We tested whether the frequency of MDSC differentiation into macrophages was affected by Pam2CSK4 treatment, given that MDSCs have the potential to differentiate into macrophages and TLR2 ligands induce macrophage differentiation from monocytes.20 CD11b+Gr1+ cells isolated from tumor-bearing mice were labeled with fluorescent dye to trace their fate in vivo, then adoptively transferred into tumor-bearing mice that were injected with PBS or Pam2CSK4. A small proportion of the CD11b+Gr1+ MDSCs up-regulated macrophage markers, F4/80 and CD115 (M-CSFR), and decreased Gr1 expression in PBS-treated mice (Fig. 2A). Interestingly, Pam2CSK4 treatment increased the frequency of F4/80+ and CD115+ cells derived from adoptively transferred CD11b+Gr1+ cells (Fig. 2A). To determine which MDSC subset had the potential to differentiate into macrophages, we isolated each subset and cultured the cells in the presence of Pam2CSK4. F4/80+ and CD115+ cells were generated from M-MDSCs, but not G-MDSCs, and this response was enhanced by Pam2CSK4 (Fig. 2B). These results suggested that Pam2CSK4 promoted macrophage differentiation of M-MDSCs, in addition to prolonging their survival.
Figure 2.
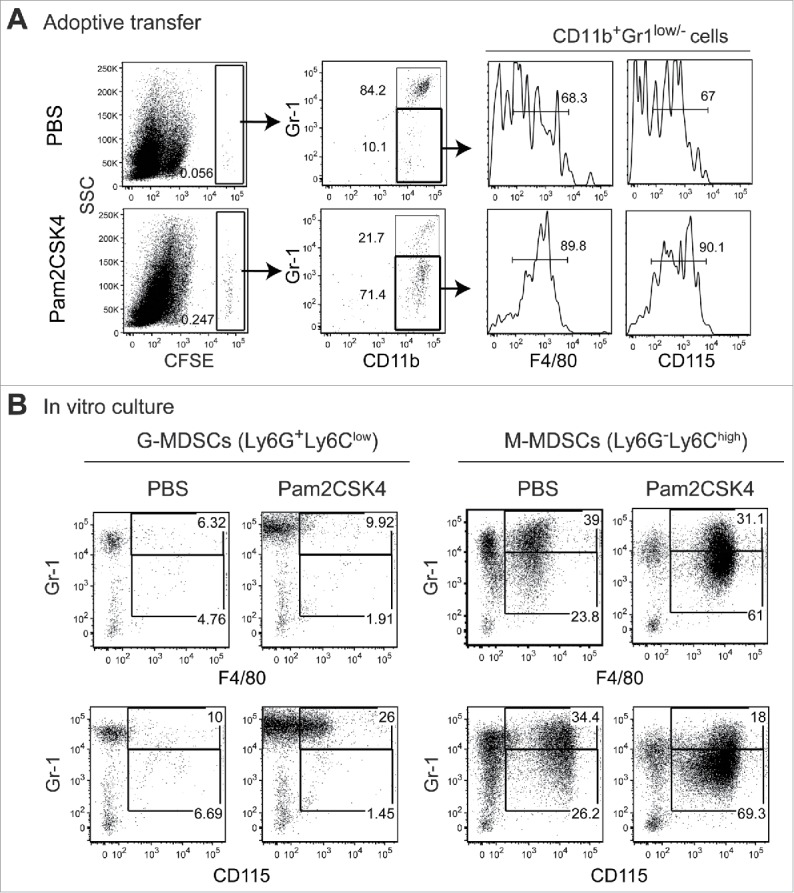
CD11b+Gr1+ cells differentiate into F4/80+/CD115+ macrophages in vivo and in vitro. (A) CFSE-labeled CD11b+Gr1+ cells (1 × 107) isolated from tumor-bearing mice were adoptively transferred into tumor-bearing mice. After 1 h, the mice were injected intravenously with PBS or 50 nmol Pam2CSK4. After 24 h, splenocytes were analyzed by flow cytometry. CFSE+ cells were gated and examined for CD11b and Gr1 expression. CD11b+Gr1− cells were further gated (bold line) and analyzed for F4/80 or CD115 expression. Numbers adjacent to outlined areas or above the brackets indicate the percentage of relevant population. (B) CD11b+Ly6G+Ly6Clow cells or CD11b+Ly6G−Ly6Chigh cells isolated from tumor-bearing mice were cultured in the presence or absence of Pam2CSK4. After 24 h, cells were analyzed for expression of Gr1, F4/80, and CD115. Numbers represent the percentage of gated cells. All data shown are representative of 2 independent experiments.
Pam2CSK4 enhances the immunosuppressive activity of M-MDSCs, but not G-MDSCs, through TLR2 signaling
Next, we investigated the effect of Pam2CSK4 treatment on the immunosuppressive activity of MDSCs. When ovalbumin (OVA)-specific CD8+ T cells from T cell receptor (TCR)-transgenic mice (OT-I mice) were cocultured with CD11c+ DCs from C57BL/6 wild type (B6 WT) mice, CD11b+Gr1+ MDSCs from tumor-bearing mice inhibited antigen-specific T cell proliferation in a dose-dependent manner (Fig. 3A). Interestingly, MDSC-induced T cell suppression was augmented by Pam2CSK4 (Fig. 3A). Pam2CSK4 failed to enhance the suppressive activity of TLR2−/− MDSCs, suggesting that TLR2 activation was essential for this effect (Fig. 3B).
Figure 3.
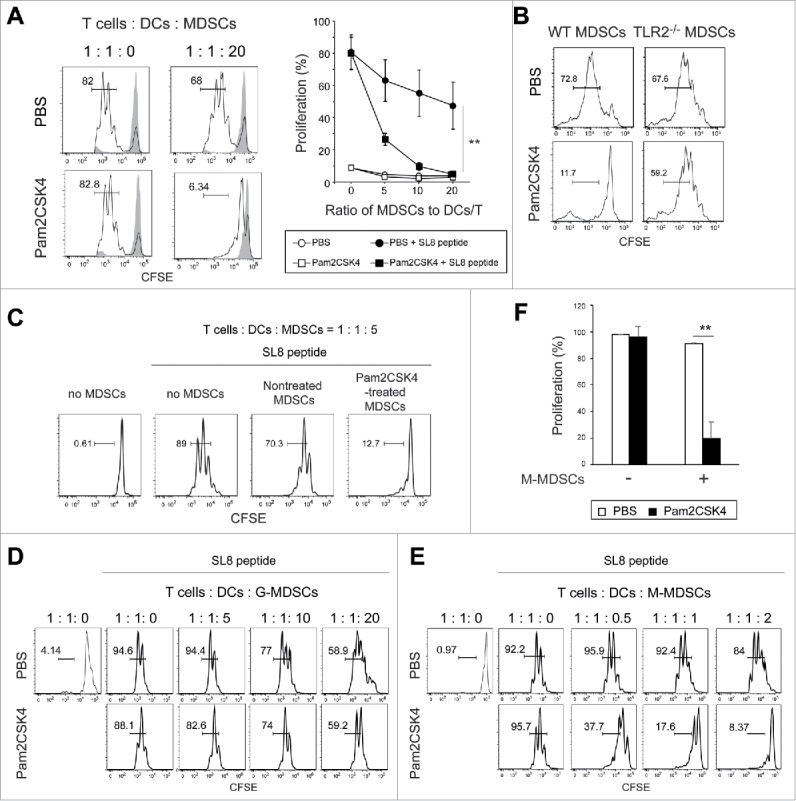
Pam2CSK4 enhances immunosuppressive activity of M-MDSCs but not G-MDSCs in vitro. (A and B) CD11b+Gr1+ cells isolated from EG7 tumor-bearing mice were cultured with CFSE-labeled CD8+ OT-I T cells (0.5 × 105), CD11c+ DCs (0.5 × 105), and SL8 peptide in the presence of 100 nM Pam2CSK4 or PBS. CD11b+Gr1+ cells were added to the cultures at the indicated ratios. After 3 days, CFSE fluorescence of CD8+ T cells was analyzed by flow cytometry to measure antigen-specific T cell proliferation. CD11b+Gr1+ cells were isolated from B6 WT mice (A and B) or TLR2−/− mice (B) implanted with EG7 tumors. Numbers near the brackets indicate the percentage of relevant population (thin line). Gray histograms indicate non-proliferated T cells in the absence of SL8 peptide. Each plot represents mean ± SD. n = 4. **P < 0.005. (right panel of A) (C) CD11b+Gr1+ cells isolated from tumor-bearing mice were pretreated with 100 nM Pam2CSK4 for 4 h, washed with the culture medium, and cultured with T cells and DCs as described in A. (D and E) G-MDSCs (D) or M-MDSCs (E) were cultured with CD8+ OT-I T cells (0.5 × 105) and CD11c+ DCs (0.5 × 105) as described in (A). (F) M-MDSCs (1 × 105) were cultured with CD8+ OT-I T cells (0.5 × 105) and CD11c+ DCs (0.5 × 105). Data represent means ± SD. n = 4. **P < 0.005. All data shown are representative of more than 2 independent experiments.
DCs and CD8+ T cells also express TLR2, implying that activation in those cells might result in T cell suppression. To examine whether TLR2 ligand directly act on MDSCs and leads to increasing their suppressive activity, they were pretreated with Pam2CSK4 for 4 h, thoroughly washed to remove the ligand, then cocultured with DCs, CD8+ OT-I T cells, and OVA257-264 SIINFEKL (SL8) peptides for 3 days. We found that MDSCs increased suppressive activity on CD8+ OT-I T cell proliferation by pretreatment with Pam2CSK4 (Fig. 3C). We examined the possibility that TLR2 activation in MDSCs leads to the inhibition of DC maturation, thereby impairing T cell proliferation. We analyzed the expression levels of co-stimulatory molecules on CD11c+ DCs in the co-cultures. Pam2CSK4 increased the expression levels of CD80, CD86, and CD40 on CD11c+ DCs, which were not reduced in the presence of MDSCs (Fig. S2). These results suggested that Pam2CSK4-stimulated TLR2 activation in MDSCs enhanced their suppressive activity without inhibiting DC maturation.
We determined which MDSC subset was responsible for the enhanced immunosuppressive activity observed in CD11b+Gr1+ MDSCs. M-MDSCs, but not G-MDSCs, exhibited the enhanced suppressive activity induced by Pam2CSK4 (Fig. 3D, E, and F). We observed a similar outcome with M-MDSCs isolated from mice implanted with OVA-expressing Lewis lung carcinoma (LLC-OVA), suggesting that this response was generally induced in M-MDSCs generated in tumor-bearing mice (Fig. S3). Thus, Pam2CSK4 enhanced the suppressive activity of M-MDSCs.
Sequestration restores proliferation of CD8+ T cells suppressed by TLR2 signal-activated M-MDSCs
Enhanced inhibition of T cell proliferation in the presence of Pam2CSK4 and M-MDSCs might be due to decreased T cell viability. To test this possibility, we stained cells with 7AAD to evaluate the viability of CD8+ T cells after 3-day coculture with M-MDSCs, peptide, and Pam2CSK4. We did not detect a decrease in CD8+ T cell viability when proliferation was largely suppressed by Pam2CSK4-activated M-MDSCs (Fig. S4A), implying that impaired T cell proliferation was not due to cytotoxic effects of M-MDSCs. Therefore, we tested whether non-proliferating CD8+ T cells regained their proliferative capacity upon separation from Pam2CSK4-activated M-MDSCs as illustrated in Fig. S4B. Firstly, CD8+ T cells were cocultured for 3 days with DCs in the presence or absence of M-MDSCs and Pam2CSK4 (1st culture). As shown in Fig. 3E and F, CD8+ T cells did not well proliferate after 3-day coculture with M-MDSCs, CD11c+ DCs, and SL8 peptide in the presence of Pam2CSK4 (condition 3), but they increased in cell volume, similar to proliferating CD8+ T cells observed in the absence of M-MDSCs (condition 2) (Fig. S4C). Secondly, CD8+ T cells were purified and cultured again in the absence of M-MDSCs for 3 days (2nd culture). As expected, CD8+ T cells that had been precultured with CD11c+ DCs, but not with SL8 peptide (condition 1), proliferated well upon stimulation with the peptide and CD11c+ DCs (condition b) (Fig. S4D). Interestingly, non-proliferating CD8+ T cells that had been cocultured with M-MDSCs, CD11c+ DCs, SL8 peptide, and Pam2CSK4 (condition 3) underwent proliferation when they were sorted and cultured alone (i.e., without antigen stimulation, condition a) (Fig. S4D). Therefore, Pam2CSK4-activated M-MDSCs strongly suppressed T cell proliferation for at least 3 days; however, T cell proliferation was restored by sequestering the T cells from the M-MDSCs.
iNOS activity is critical for the TLR2 signal-enhanced suppressive activity of M-MDSCs
To clarify the mechanism that enhances the suppressive activity of M-MDSCs in response to TLR2 stimulation, we examined the effects of Pam2CSK4 on the production of immunosuppressive factors from M-MDSCs. NO is a critical molecule responsible for the suppressive activity of M-MDSCs and macrophages, and T cell proliferation is reversibly down-regulated by NO.16,21 To examine the possibility that NO or an NO-related pathway is critical for the Pam2CSK4-enhanced suppressive activity of M-MDSCs, we tested the impact of inhibition of iNOS enzymatic activity on CD8+ T cell proliferation. As shown in Fig. 4A, the increase in suppressive activity mediated by Pam2CSK4 was largely abrogated by N6-(1-Iminoethyl)-L-lysine (L-NIL), an iNOS inhibitor. Similar results were observed using other iNOS inhibitors, NG-Nitro-L-arginine methyl ester (L-NAME) or NG-Monomethyl-L-arginine (L-NMMA). In contrast, N-acetyl-L-cysteine (NAC), a ROS scavenger, did not affect the suppressive activity of M-MDSCs (Fig. S5). We confirmed that NO was present in the conditioned media of M-MDSCs when they were cocultured with CD8+ OT-I T cells in the presence of SL8 peptide, and the levels were enhanced by Pam2CSK4 (Fig. 4B). These results suggested that NO was a critical molecule for T cell suppression by Pam2CSK4-activated M-MDSCs, as well as untreated M-MDSCs. Interestingly, NO was hardly produced in the absence of SL8 peptide, indicating that antigen presentation by M-MDSCs to CD8+ T cells was essential for NO production by M-MDSCs.
Figure 4.
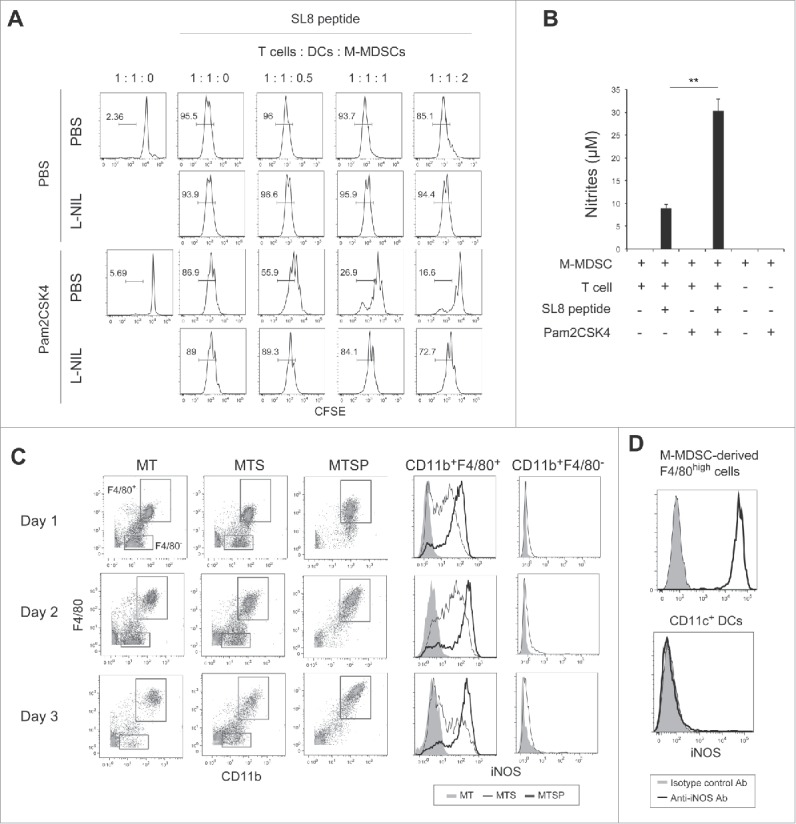
iNOS activity is essential for Pam2CSK4-enhanced suppressive activity. (A) L-NIL (50 nM) was added to cultures of M-MDSCs, CD8+ OT-I T cells, and CD11c+ DCs. T cell proliferation was analyzed by flow cytometry. Numbers above the brackets indicate the percentage of relevant population. (B) Nitrites in the conditioned media of the cocultures were measured by Griess reagent. Data represent means ± SD. n = 3. **P < 0.005. (C) M-MDSCs (1 × 105) were cultured with CD8+ OT-I T cells (0.5 × 105) in the presence or absence of SL8 peptide or Pam2CSK4. iNOS expression in F4/80+ and F4/80− cells was analyzed by flow cytometry. MT: M-MDSCs and T cells; MTS: M-MDSCs, T cells, and SL8 peptide; MTSP: M-MDSCs, T cells, SL8 peptide, and Pam2CSK4. (D) iNOS expression in F4/80+ cells and CD11c+ DCs in the cocultures was analyzed by flow cytometry. All data shown are representative of more than 2 independent experiments.
Next, we asked whether M-MDSCs or macrophages expressed iNOS when cocultured with CD8+ T cells in the presence of antigen, given that M-MDSCs differentiated into macrophages (Fig. 2B). We found that M-MDSC-derived CD11b+F4/80+ macrophages, but not M-MDSCs (CD11b+F4/80−), expressed iNOS for 2 days (Fig. 4C). Interestingly, iNOS expression in macrophages was increased and sustained for more than 3 days by Pam2CSK4 treatment. In contrast, iNOS was not detected in CD11c+ DCs (Fig. 4D). In the absence of peptide, iNOS was not detected in macrophages or M-MDSCs, suggesting that TCR-mediated activation of CD8+ T cells was required for iNOS expression. We have previously demonstrated that polyI:C, a ligand for TLR3, and melanoma differentiation-associated protein 5 (MDA5), modulate immunosuppressive myeloid cells including MDSCs.22-24 In contrast to Pam2CSK4, polyI:C did not increase the frequency of M-MDSC differentiation into F4/80+ macrophages. PolyI:C also did not enhance iNOS expression in M-MDSC-derived macrophages when cocultured with CD8+ OT-I T cells (Fig. S6). Thus, iNOS expression was induced in M-MDSC-derived macrophages by activated CD8+ T cells which was sufficiently induced by antigen-presenting M-MDSC and/or macrophages; this response was augmented by the signaling from Pam2CSK4-stimulated TLR2, but not polyI:C-stimulated TLR3 or MDA5.
CD8+ T cell-derived IFN-γ induces iNOS expression in macrophages differentiated from M-MDSCs
Macrophages express iNOS in response to IFN-γ.25 Antigen-dependent stimulation of T cells by antigen-presenting cells results in IFN-γ production. Thus, we tested whether IFN-γ is also a critical factor for the elevated iNOS expression in M-MDSC-derived macrophages. In parallel to NO production (Fig. 4B), the concentration of IFN-γ was increased in the conditioned media of M-MDSC and CD8+ T cell cultures in the presence of SL8 peptide. IFN-γ concentration was increased with the frequency of IFN-γ-producing CD8+ T cells (Fig. 5A and Fig. S7). These results suggested that M-MDSCs and/or macrophages were capable of presenting antigen to CD8+ T cells, leading them to secrete IFN-γ into the conditioned medium. Although Pam2CSK4 increased the production of IL-12, which induces IFN-γ from CD8+ T cells, IFN-γ production did not appear to be related to IL-12 production in this condition (Fig. 5A). As shown in Fig. 5B, anti-IFN-γ neutralizing antibody largely abrogated iNOS expression in M-MDSC-derived macrophages induced by SL8 peptide in the presence and absence of Pam2CSK4. Recombinant IFN-γ induced iNOS expression in F4/80+ macrophages, which was augmented by Pam2CSK4 (Fig. 5C). We confirmed the expression of IFNGR1, a subunit of IFN-γ receptor, on the surface of F4/80high macrophages differentiated from M-MDSCs (Fig. 5D). By contrast, IFN-γ did not induce iNOS expression in G-MDSCs (Fig. 5E). IFNGR1 was not detected on G-MDSCs (Fig. 5F). Pam2CSK4 enhanced macrophage differentiation and survival but did not enhance IFNGR1 expression (Fig. 5D). These results suggested that naïve CD8+ T cells were stimulated by antigen-presenting M-MDSC/macrophages and produced IFN-γ, which induced iNOS expression in macrophages differentiated from M-MDSCs. These responses were augmented by Pam2CSK4 through the activation of TLR2 signaling which increased the differentiation of M-MDSCs into macrophages.
Figure 5.
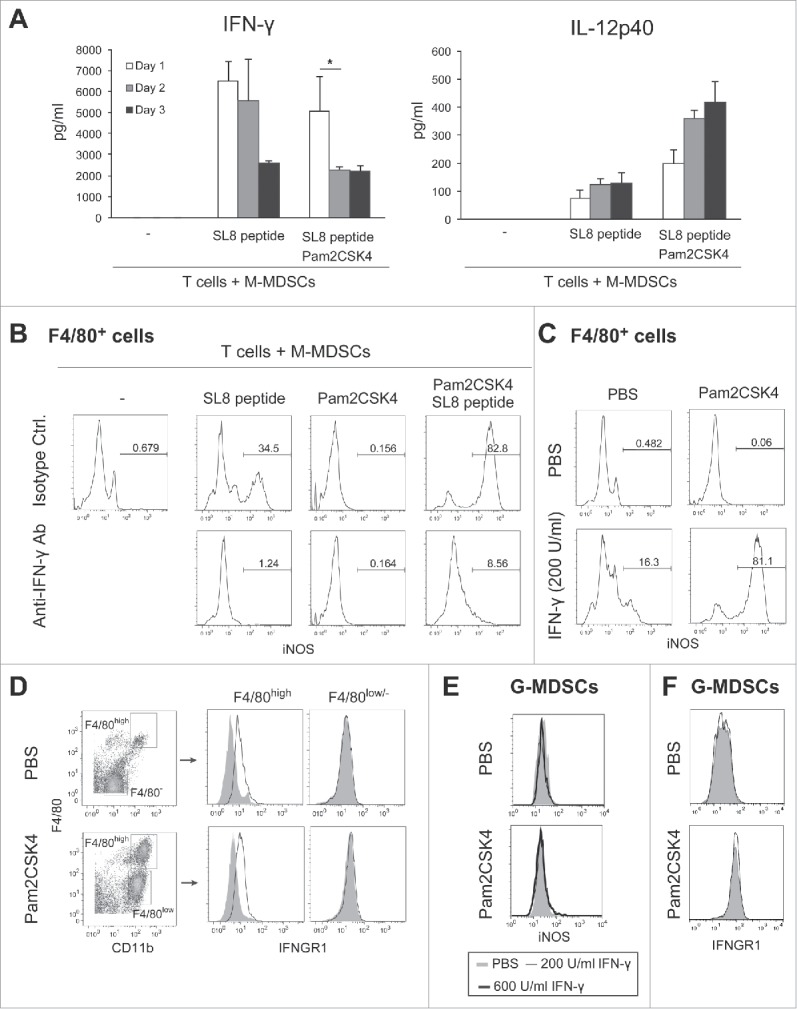
CD8+ T cell-produced IFN-γ induces iNOS expression in M-MDSC-derived macrophages. (A) The concentrations of IFN-γ (left panel) or IL-12p40 (right panel) in the conditioned media of CD8+ T cells cultured with M-MDSCs (ratio 1:2) were determined by ELISA. Data are shown as means ± SD. n = 3. *P < 0.05. (B) iNOS expression in F4/80+ cells after coculture of M-MDSCs with CD8+ OT-I T cells for 24 h in the presence of Pam2CSK4, anti-IFN-γ antibody, and/or isotype control antibody (isotype Ctrl.). Numbers above the brackets indicate the percentage of relevant population. (C) iNOS expression in F4/80+ cells derived from M-MDSCs cultured for 24 h in the presence of recombinant IFN-γ (200 U/mL) and Pam2CSK4. (D) IFNGR1 expression was determined in F4/80+ or F4/80− cells after 24-h culture in the presence of Pam2CSK4. Cells were stained with anti-IFNGR1 Ab (black line histogram) or isotype control Ab (gray-filled histogram). (E) iNOS expression in G-MDSCs after 24-h culture in the presence of IFN-γ (200 or 600 U/mL) and Pam2CSK4. (F) IFNGR1 expression in G-MDSCs. Cells were stained with anti-IFNGR1 Ab (black line histogram) or isotype control Ab (gray-filled histogram). All data shown are representative of more than 2 independent experiments.
Activation of TLR2 signaling increases iNOS-expressing macrophages in tumors
Tumor-infiltrating M-MDSCs and macrophages contribute to the generation of immunosuppressive conditions in the tumor microenvironment, where M-MDSCs rapidly differentiate into macrophages.26,27 We tested whether Pam2CSK4 treatment increased the number of iNOS-expressing macrophages in the tumor and prevented CTL-mediated anti-tumor immunity induced by TLR2-activated DCs. We examined the frequency of iNOS-expressing macrophages in tumors after the treatment of EG7 tumor-bearing mice with Pam2CSK4 and OVA protein. The proportion of iNOS-expressing CD11b+F4/80+ macrophages was increased with treatment in WT but not in TLR2−/− mice (Fig. 6A). This effect was not observed in monotherapy with OVA protein (Fig. S9). Co-administration of Pam2CSK4 and OVA protein increased the concentration of IFN-γ in tumors (Fig. 6B). This response was largely abrogated in CD8+ T cell-depleted mice, generated by anti-CD8β antibody treatment. These data indicated that treatment with Pam2CSK4 and antigen led to production of IFN-γ by activated CD8+ T cells, which induced iNOS expression in tumor-infiltrating macrophages. Next, we tested whether blockade of iNOS activity with an iNOS inhibitor enhanced the inhibitory effect of Pam2CSK4 treatment on tumor growth in mice. As shown in Fig. 6C, L-NAME treatment slightly but significantly enhanced the inhibition of tumor growth induced by Pam2CSK4 and OVA protein (Fig. 6C). OVA protein and/or L-NAME did not affect the tumor growth (Fig. S8A). We also tested if the iNOS inhibitor affected tumor growth in other tumor implant models than EG7 to examine the general sharing of this phenomenon with cancer cell lines. Similar results were obtained by L-NAME treatment in LLC-OVA tumor implant model (Fig. 6D, and Fig. S8B). L-NIL also potentiated tumor growth inhibition by Pam2CSK4 treatment (Fig. 6E and Fig. S8B). Thus, these results indicate that, through the induction of IFN-γ production by CD8+ T cells, Pam2CSK4 plus antigen treatment increased iNOS expression in macrophages in tumors, which may negatively regulate anti-tumor immunity induced by DC-mediated CTL activation.
Figure 6.
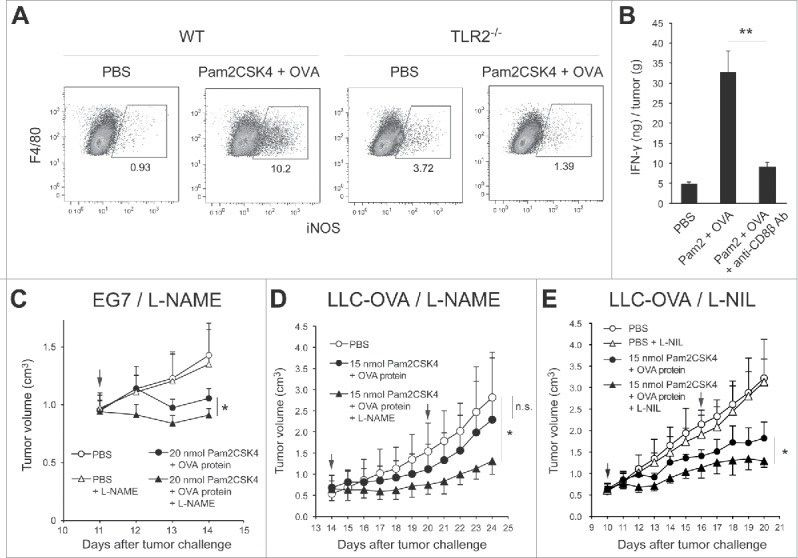
Blocking of iNOS activity augments the therapeutic potential of Pam2CSK4 in tumor-bearing mice. (A) B6 WT or TLR2−/− mice bearing EG7 tumors were treated with Pam2CSK4 and OVA protein. After 24 h, tumors were excised and iNOS expression in CD11b+F4/80+ cells was analyzed by flow cytometry. Numbers adjacent to outlined areas indicate the percentage of relevant population. (B) EG7 tumor-bearing mice were pretreated with anti-CD8β antibody for 24 h. Pam2CSK4 (Pam2) and OVA protein were injected into the mice. After 24 h, tumor lysate was prepared and IFN-γ concentration was measured by ELISA. IFN-γ levels were represented as IFN-γ (ng) /tumor (g). (C, D and E) B6 WT mice bearing EG7 (C) or LLC-OVA (D and E) tumors were treated with Pam2CSK4 and OVA protein as indicated by arrows. Mice were treated daily with L-NAME (2 mg) (C and D), L-NIL (0.5 mg) (E), or PBS. Data are shown as means ± SD. n = 4-5. *p < 0.05. All data shown are representative of more than 2 independent experiments.
Discussion
Our data demonstrated that TLR2 activation in M-MDSCs resulted in enhanced suppressive activity on T cell proliferation. M-MDSCs/and M-MDSDC-derived macrophages were capable of presenting antigen to CD8+ T cells, leading to transient induction of IFN-γ production. IFN-γ induced iNOS expression in macrophages. This mechanism is important for the suppressive activity of M-MDSCs both in the presence and absence of TLR2 ligand. Thus, our findings provide insight into a fundamental mechanism of T cell suppression by M-MDSCs. We propose that the mechanism of Pam2CSK4-enhanced suppression of T cell proliferation by M-MDSCs is largely divided into three steps (Fig. 7A). Firstly, Pam2CSK4 increases the viability of M-MDSCs and the frequency of differentiation into macrophages through TLR2 signaling. Secondly, M-MDSCs and/or macrophages present antigen to naïve CD8+ T cells and activate them to produce IFN-γ. Finally, IFN-γ induces iNOS expression in macrophages, which is increased by Pam2CSK4, leading to NO-mediated suppression of CD8+ T cell proliferation induced by DCs. Pam2CSK4 and antigen treatment increased IFN-γ production from CD8+ T cells and iNOS-expressing macrophages in tumors. iNOS inhibitors potentiated the therapeutic effect. Taken together, these data indicate that administration of TLR2 ligands leads to prolonged survival and enhanced suppressive activity of M-MDSCs largely dependent on NO production. TLR2 activation in M-MDSCs leads to dampening of tumor growth inhibition induced by TLR2-activated DCs. Consequently, targeting iNOS activity could overcome the limitations of current treatment using TLR2 ligands (Fig. 7B).
Figure 7.
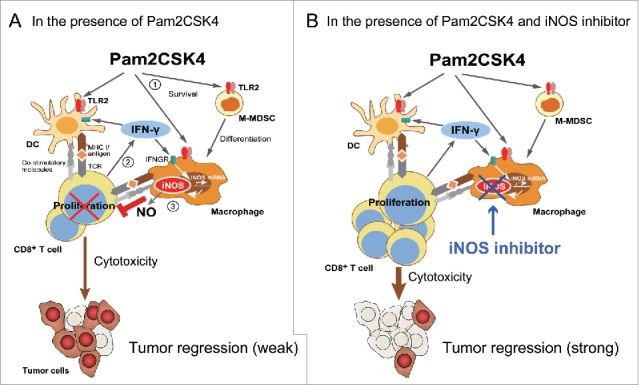
Putative mechanism of Pam2CSK4-enhanced immunosuppression by M-MDSCs. (A) M-MDSCs differentiate into macrophages. Pam2CSK4 increases their survival and differentiation through TLR2 signaling (Step 1). M-MDSCs and/or macrophages present peptide to CD8+ T cells, and transiently activate them to produce IFN-γ (Step 2). IFN-γ induces iNOS expression in macrophages differentiated from M-MDSCs. iNOS-generated NO inhibits T cell proliferation induced by DCs (Step 3). In contrast to M-MDSCs, DCs undergo maturation and activation in the presence of Pam2CSK4 and IFN-γ, leading to T cell proliferation. (B) By using iNOS inhibitor, NO-mediated T cell suppression by Pam2CSK4-activated M-MDSCs/macrophages is abrogated, thereby DCs can strongly induces the activation/proliferation of anti-tumor CD8+ T cells, leading to tumor regression.
Both subsets of MDSCs expressed TLR2 and showed prolonged survival by Pam2CSK4 treatment. Thus, administration of TLR2 ligand enhances accumulation of MDSCs. Intracellular TLR2 signaling appears to be sufficient for inducing prolonged survival of MDSCs. In addition, TLR2 activation may promote differentiation of M-MDSCs into macrophages, which may involve IL-15 or other molecules.20 Further study is required to identify the genes critical for inducing prolonged survival and differentiation of TLR2-activated MDSCs.
TLR2 signaling enhanced the suppressive activity of M-MDSCs but not G-MDSCs. This difference is probably attributable to the reactivity of each MDSC subset to the IFN-γ produced by antigen-stimulated T cells. Selective expression of iNOS in macrophages may be, in part, due to high expression of IFNGR1 on the macrophages compared with undifferentiated M-MDSCs and G-MDSCs. In contrast, splenic CD11c+ DCs express IFNGR but did not show iNOS expression in the presence of IFN-γ. These differential responses of myeloid cells to IFN-γ may be important in the regulation of tumor growth. The intracellular signaling pathway that increases Nos2 promoter activity in M-MDSC-derived macrophages through STAT125 may be absent from CD11c+ DCs. Alternatively, IFN-γ-induced STAT1 signaling may be negatively regulated by PIAS1 and STAT3, as observed in IL-15-induced DCs.28 Analyzing the differential responses of M-MDSC-derived macrophages and CD11c+ DCs to IFN-γ could help us identify a critical molecule for the regulation of immunosuppression by M-MDSCs.
M-MDSC-mediated T cell suppression is reportedly dependent on the production of NO and Arg1, as well as immunosuppressive cytokines including IL-10 and TGF-β.29 In our experimental condition, iNOS activity was critical for the in vitro suppressive activity of Pam2CSK4-treated, as well as untreated, M-MDSCs. In the presence of Pam2CSK4, M-MDSCs, at a ratio of 2:1 with T cells/DCs, almost completely inhibited CD8+ T cell proliferation induced by DCs. The non-proliferative state of CD8+ T cells induced by M-MDSCs appears to be reversible because T cells proliferated well after sequestration from Pam2CSK4-activated M-MDSCs. This observation was in agreement with evidence that NO reversibly inhibits T cell proliferation through inhibition of Jak3/STAT5 signaling downstream of the IL-2 receptor.21 We demonstrated that CD8+ T cells transiently proliferated and produced IFN-γ on day 1 of coculture in the presence of M-MDSCs and Pam2CSK4, but those responses were inhibited on days 2 and 3. Therefore, our data suggest that once the TCR signaling pathway is activated in T cells, the resulting NO production by M-MDSC-derived macrophages contributes to dampening the proliferation of CD8+ T cells. NO-mediated suppression of T cell proliferation is distinct from TCR modification by G-MDSC-derived PNT, which leads to T cell tolerance.30 Co-administration of an iNOS inhibitor and TLR2 ligand is likely to be effective for eliminating the suppression of CD8+ T cell proliferation by M-MDSC-derived macrophages expressing iNOS, leading to an increase in therapeutic efficacy without influencing the function of DCs.
TLR signaling pathways differentially modulate the function of MDSCs. Similar to Pam2CSK4, endogenous TLR2 ligand, Hsp72, derived from cancer cells promote tumor growth by inducing generation and systemic accumulation of MDSCs.19 S100A8/9 induces a similar effect through TLR4.31,32 By contrast, activation of TLR3, TLR7/8, or TLR9 by administration of an appropriate ligand leads to loss of the suppressive activity of MDSCs, resulting in the inhibition of tumor growth by restoring anti-tumor T cell responses.23,33-35 Notably, we observed that the frequency of iNOS expression in M-MDSC-derived macrophages was not increased by polyI:C treatment. Thus, augmentation of the suppressive activity of M-MDSCs appears to be specific to TLR2 signaling. This difference may be caused by an inability of polyI:C to activate or sustain survival of MDSCs. In addition, polyI:C and CpG ODN induces the production of type-I IFNs, which directly stimulate M-MDSCs to lose their immunosuppressive activity.24,33 In contrast, polyI:C-induced activation of the TLR3-TIR domain-containing adapter molecule-1 (TICAM-1)-IFN-α/β signaling axis induces the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) by CD11b+Ly6G+ G-MDSCs, resulting in inhibition of EL4 and B16 tumor growth.24 In this case, M-MDSCs are not involved in tumor regression induced by polyI:C. Taken together, MDSC subsets have potential to become both a positive and negative regulator of tumor growth, which can be modulated by TLR ligands.
TLR signaling pathways are promising targets for cancer immunotherapy.36,37 TLR2 signaling induces a potent anti-tumor response, which is primarily mediated through maturation of DCs.4,8 The proportion of immunosuppressive myeloid cells, including MDSCs, is generally increased as a tumor grows, and they accumulate dramatically in patients with late-stage cancer.38 Therefore, TLR2-enhanced immunosuppressive activity of myeloid cells may limit the clinical application of TLR2 ligands for cancer treatment. We propose that, in addition to enzymatic activity of iNOS, TLR2 signal transduction in MDSCs/macrophages, or the mechanism of M-MDSC differentiation into macrophages are possible targets for potentiating the therapeutic efficacy of TLR2 ligands (Fig. 7A and B). Alternatively, artificial TLR2 ligands that are modified to specifically activate CD11c+ DCs may improve clinical outcomes in cancer therapy, as they appear to stimulate M-MDSCs to a lesser degree.39 We and other groups have uncovered that TLR2 activation also promotes tumor growth and disables cancer treatment by different mechanisms.11,40,41 Therefore, combinational use of iNOS inhibitor and blocking agents which abrogate other suppressive effects of TLR2 signaling might further improve the therapeutic efficacy of the ligands.
Emerging evidence obtained from mouse models have suggested that tumor-supportive phenotype of tumor-associated myeloid cells could be a target of cancer immunotherapy.2,3 This concept is now applicable to the human innate immune system. Repolarization of tumor-associated macrophages in human colorectal cancer (CRC) orchestrates anti-tumor immunity.42 We provide evidence that therapeutic intervention by manipulating innate immune signaling pathway may sometimes potentiate tumor-supporting phenotype of myeloid cells, thereby accelerating tumor growth.
In summary, this study revealed a negative impact of TLR2 signaling in MDSCs on CD8+ T cell proliferation, thereby inhibiting anti-tumor immunity. When selecting TLR ligands for the development of cancer immunotherapy, we should consider the effects of TLR activation on immunosuppressive myeloid cells, which will influence the clinical response of cancer patients to the adjuvant therapy.
Materials and methods
Mice and cell lines C57BL6/J mice (6- to 8-week-old, female) were purchased from Clea Japan (Tokyo, Japan). TLR2−/− mice were kindly provided by Dr. S. Akira (Osaka University, Osaka, Japan). IL-6−/- mice were purchased from Charles River (Yokohama, Japan). OT-I TCR-transgenic mice were provided by Dr. Ishii (Tohoku University, Sendai, Japan). Mice were maintained under specific-pathogen-free conditions in the Hokkaido University Animal Facility (Sapporo, Japan). Mice aged 8- to 12-week-old were used in all experiments, which were performed according to the animal experimental ethics committee guidelines of Hokkaido University. All mice were used according to the guidelines of the Institutional Animal Care and Use Committee of Hokkaido University, which approved this study under ID numbers 08–0290, 13–0043, and 16–0045.
EG7 cells, purchased from ATCC (American Type Culture Collection, Manassas, VA, USA), were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 55 µM 2-mercapto-ethanol (2-ME), 1 mM sodium pyruvate, 100 U/mL penicillin, and 100 µg/mL streptomycin (Life Technologies, Carlsbad, CA, USA). LLC-OVA cells43 kindly provided by Dr. T. Nishimura and Dr. H. Kitamura (Hokkaido University, Sapporo, Japan), were cultured in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% FBS, 2 mM L-glutamine, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, 55 µM 2-ME, 100 U/mL penicillin, 100 µg/mL streptomycin (Life Technologies), and 50 µg/ml G418 (Roche, Basel, Switzerland).
Tumor challenge Mice were shaved at the back and subcutaneously injected with EG7 cells (2 × 106) or LLC-OVA cells (1 × 106) suspended in 200 µl phosphate buffered saline (PBS). Tumor size was measured using calipers. Tumor volume was calculated using the following formula: tumor volume (cm3) = (long diameter) × (short diameter)2 × 0.4. Mice were injected subcutaneously with Pam2CSK4 and purified EndoGrade® endotoxin-free OVA protein (Hyglos, Bernried, Germany) when tumor size reached 0.6 to 0.8 cm3. Pam2CSK4 and OVA protein (100 µg) were suspended in 200 µl PBS and 100 µl of the mixture was injected at 2 different sites around the tumors (200 µl/mouse). In some experiments, tumor-bearing mice were treated i.p. daily with L-NAME (2 mg/200 µl) or L-NIL (0.5 mg/200 µl) to block iNOS activity.
Antibodies and reagents FITC-anti-CD45 (30-F11); Alexa Fluor® 700- or APC-anti-CD45.2 (104); Alexa Fluor® 700-, FITC-, or PE-anti-CD11b (M1/70); biotin-, PE-, or APC-anti-Gr-1 (RB6-8C5); biotin-, FITC-, or APC-anti-Ly6G (1A8); FITC- or APC-anti-Ly6C (HK1.4); FITC- or PE-anti-CD8a (53-6.7); APC-anti-CD3 (145-2C11); PE-anti-TCRvβ5.1/5.2 (MR9-4); PE-anti-CD80 (16-10A1); PE-anti-CD86 (GL-1); FITC-, PE-, or APC-anti-CD11c (N418); PE-anti-H-2Kb/Db (28-8-6); Biotin-anti-IFNGR1 (2E2); purified anti-CD16/CD32 (2.4G2); and isotype antibodies were purchased from BioLegend (San Diego, CA, USA). PE-anti-CD40 (1C10), and PE-anti-NOS2 (CXNFT) antibodies were purchased from eBioscience (San Diego, CA, USA). 2,3-bis(palmitoyl) propyl Cys-Ser-Lys-Lys-Lys-Lys (Pam2CSK4) was synthesized by Biologica Co. Ltd (Nagoya, Japan). NG-Monomethyl-L-arginine, monoacetate salt (L-NMMA), and NG-Nitro-L-arginine methyl ester hydrochloride (L-NAME) were purchased from Merck KGaA (Darmstadt, Germany) or Sigma-Aldrich (St. Louis, MO, USA). L-N6-(1-Iminoethyl) lysine dihydrochloride (L-NIL) and N-acetyl-L-cysteine (NAC) was purchased from Cayman Chemical (Ann Arbor, MI) and Sigma-Aldrich, respectively. Carboxyfluorescein diacetate succinimidyl ester (CFSE) was purchased from Life Technologies (Carlsbad, CA, USA). OVA257–264 peptide (SIINFEKL, SL8 peptide) was purchased from MBL (Nagoya, Japan). PI was purchased from Sigma-Aldrich. NO in the conditioned media was measured by using the NO2/NO3 Assay Kit-C II (Colorimetric)-Griess Reagent Kit according to the manual (Dojindo, Kumamoto, Japan). WST-1 assay was performed according to the manufacturer's instructions (Dojindo). Ready-set-Go ELISA kits (eBioscience) were used to determine the concentration of mouse IFN-γ and IL-12p40.
Isolation of cells CD11b+Gr1+ cells were isolated by MACS® using biotinylated anti-Gr-1 antibody and streptavidin microbeads (Miltenyi Biotec, Gladbach, Germany) from single-cell suspension of spleens from EG7 tumor-bearing mice 2 to 3 weeks after tumor challenge. To isolate M-MDSCs, single-cell suspensions of splenocytes from tumor-bearing mice were stained with biotin-anti-Ly6G antibody and streptavidin microbeads. The labeled cells were applied to an LD column and the flow-through fraction was collected, then applied to an LS column. Ly6G− cells in the flow-through were collected, stained with biotin-anti-Gr-1 antibody (RB6-8C5) and streptavidin microbeads, and applied sequentially to an LS and MS column. The positive cell fraction was collected and used as CD11b+Ly6G−Ly6Chigh M-MDSCs. To isolate G-MDSCs, single-cell suspensions of splenocytes isolated from tumor-bearing mice were stained with biotin-anti-Ly6G antibody and streptavidin microbeads. The labeled cells were applied to an LS column, followed by an MS column. The positive cell fraction was collected and used as CD11b+Ly6G+Ly6Clow G-MDSCs. The purity and CD11b expression of cells in each fraction was confirmed by flow cytometry. Splenic CD11c+ DCs were purified from splenocyte suspensions from naïve B6 WT mice using anti-CD11c microbeads (Miltenyi Biotec). CD8+ OT-I T cells were purified from the spleens of OT-I TCR-transgenic mice by using anti-CD8α microbeads or a CD8α+ T Cell Isolation Kit (Miltenyi Biotec). Cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 55 µM 2-ME, 100 U/mL penicillin, and 100 µg/mL streptomycin (Life Technologies).
Flow cytometric analysis Single-cell suspensions from in vitro cultures or from tumors were incubated with anti-CD16/32 antibody, then stained with fluorescent antibodies. Intracellular staining of iNOS was performed using the BD Cytofix/Cytoperm™ kit (BD Biosciences, San Jose, CA, USA). Samples were analyzed on a FACS Calibur or FACS Aria II (BD Biosciences) and data analysis was performed with FlowJo® software (Tree Star, Ashland, OR, USA).
T cell proliferation assay T cell proliferation was measured by changes in fluorescence intensity using CFSE. CD8+ OT-I T cells were labeled with 1 μM CFSE for 10 min, washed twice with culture medium, then mixed with splenic CD11c+ DCs and/or MDSCs isolated from tumor-bearing mice. The mixture was cultured in a round bottom 96-well plate in the presence or absence of 50 nM SL8 peptide and/or 100 nM Pam2CSK4. After 3 days, cells were harvested, stained with APC-anti-CD8α, PE-anti-TCRvβ5.1/5.2, and Alexa Fluor® 700-anti-CD3ε antibodies. The CFSE signal of gated lymphocytes was analyzed with a FACS Calibur or FACS Aria II. The extent of cell proliferation was quantified by FlowJo® software (Tree Star).
Adoptive transfer CD11b+Gr1+ cells were isolated from the spleens of tumor-bearing mice, labeled with 1 µM CFSE for 10 min, and washed twice with culture medium. CFSE-labeled CD11b+Gr1+ MDSCs (1 × 107) were injected into EG7 tumor-bearing mice. After 1 h, the mice were injected intravenously with 50 nmol Pam2CSK4 or PBS. After 24 h, single-cell splenocyte suspensions were incubated with anti-CD16/32 antibody to block Fc receptors. Cells were then stained with fluorescent antibodies. Samples were analyzed by flow cytometry using a FACS Aria II (BD Bioscience); data analysis was performed using FlowJo® software (Tree Star).
Quantitative PCR Total RNA was prepared using an RNeasy Mini Kit (QIAGEN, Venlo, Netherlands) and used for cDNA synthesis with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™, Foster City, CA, USA). Quantitative PCR was performed with the StepOne™ Real-Time PCR system (Applied Biosystems™). Primer sequences for murine gene products were as shown in Supplementary Table 1.
Statistics Statistically significant differences between two groups were determined using the Student's t-test.
Supplementary Material
Acknowledgment
We are grateful to our laboratory members for their invaluable discussions. We gratefully acknowledged Drs. T. Nishimura and H. Kitamura (Hokkaido University, Sapporo Japan) for the gift of the LLC-OVA cell line. This work was supported by JSPS KAKENHI Grant Number, 16K08704, Grants from The Ministry of Health, Labour and Welfare (MHLW) and the Research on Development of New Drugs, the Japan Agency for Medical Research and Development (AMED) (16ak0101010h0005), the Takeda Science Foundation, and The Suhara Memorial Foundation.
References
- 1.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014-22. doi: 10.1038/ni.2703. PMID:24048123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Engblom C, Pfirschke C, Pittet MJ. The role of myeloid cells in cancer therapies. Nat Rev Cancer. 2016;16:447-62. doi: 10.1038/nrc.2016.54. PMID:27339708 [DOI] [PubMed] [Google Scholar]
- 3.Seya T, Shime H, Matsumoto M. Functional Alteration of Tumor-infiltrating Myeloid Cells in RNA Adjuvant Therapy. Anticancer Res. 2015;35:4385-92. PMID:26168476 [PubMed] [Google Scholar]
- 4.Akazawa T, Inoue N, Shime H, Kodama K, Matsumoto M, Seya T. Adjuvant engineering for cancer immunotherapy: Development of a synthetic TLR2 ligand with increased cell adhesion. Cancer Sci. 2010;101:1596-603. doi: 10.1111/j.1349-7006.2010.01583.x. PMID:20507323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawahata R, Shime H, Yamazaki S, Inoue N, Akazawa T, Fujimoto Y, Fukase K, Matsumoto M, Seya T. Failure of mycoplasma lipoprotein MALP-2 to induce NK cell activation through dendritic cell TLR2. Microbes Infect. 2011;13:350-8. doi: 10.1016/j.micinf.2010.12.003. PMID:21172450 [DOI] [PubMed] [Google Scholar]
- 6.Seya T, Shime H, Takeda Y, Tatematsu M, Takashima K, Matsumoto M. Adjuvant for vaccine immunotherapy of cancer – focusing on TLR2 and TLR3 agonists for safely enhancing antitumor immunity. Cancer Sci. 2015;106:1659-68. doi: 10.1111/cas.12824. PMID:26395101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen K-Y, Song Y-C, Chen I-H, Leng C-H, Chen H-W, Li H-J, Chong P, Liu S-J. Molecular mechanisms of TLR2-mediated antigen cross-presentation in dendritic cells. J Immunol. 2014;192:4233-41. doi: 10.4049/jimmunol.1302850. PMID:24683188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azuma M, Sawahata R, Akao Y, Ebihara T, Yamazaki S, Matsumoto M, Hashimoto M, Fukase K, Fujimoto Y, Seya T. The peptide sequence of diacyl lipopeptides determines dendritic cell TLR2-mediated NK activation. PLoS ONE. 2010;5(9):e12550. doi: 10.1371/journal.pone.0012550. PMID:20824059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, Dev A, Sievert W, Ooi CH, Ishikawa T-O, et al.. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22:466-78. doi: 10.1016/j.ccr.2012.08.010. PMID:23079657 [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, Luo J-L, Karin M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102-6. doi: 10.1038/nature07623. PMID:19122641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamazaki S, Okada K, Maruyama A, Matsumoto M, Yagita H, Seya T. TLR2-dependent induction of IL-10 and Foxp3+ CD25+ CD4+ regulatory T cells prevents effective anti-tumor immunity induced by Pam2 lipopeptides in vivo. PLoS ONE. 2011;6:e18833. doi: 10.1371/journal.pone.0018833. PMID:21533081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125:3356-64. doi: 10.1172/JCI80005. PMID:26168215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791-802. doi: 10.4049/jimmunol.181.8.5791. PMID:18832739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol. 2012;91:167-81. doi: 10.1189/jlb.0311177. PMID:21954284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol. 2002;168:689-95. doi: 10.4049/jimmunol.168.2.689. PMID:11777962 [DOI] [PubMed] [Google Scholar]
- 16.Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, Ochoa AC, Fletcher M, Velasco C, Wilk A, et al.. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. 2014;134:2853-64. doi: 10.1002/ijc.28622. PMID:24259296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skabytska Y, Wölbing F, Günther C, Köberle M, Kaesler S, Chen K-M, Guenova E, Demircioglu D, Kempf WE, Volz T, et al.. Cutaneous Innate Immune Sensing of Toll-like Receptor 2–6 Ligands Suppresses T Cell Immunity by Inducing Myeloid-Derived Suppressor Cells. Immunity. 2014;41:762-75. doi: 10.1016/j.immuni.2014.10.009. PMID:25456159 [DOI] [PubMed] [Google Scholar]
- 18.Maruyama A, Shime H, Takeda Y, Azuma M, Matsumoto M, Seya T. Pam2 lipopeptides systemically increase myeloid-derived suppressor cells through TLR2 signaling. Biochem Biophys Res Commun. 2015;457:445-50. doi: 10.1016/j.bbrc.2015.01.011. PMID:25596131 [DOI] [PubMed] [Google Scholar]
- 19.Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin J-P, Boireau W, Rouleau A, Simon B, Lanneau D, et al.. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010;120:457-71. PMID:20093776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krutzik SR, Tan B, Li H, Ochoa MT, Liu PT, Sharfstein SE, Graeber TG, Sieling PA, Liu Y-J, Rea TH, et al.. TLR activation triggers the rapid differentiation of monocytes into macrophages and dendritic cells. Nat Med. 2005;11:653-60. doi: 10.1038/nm1246. PMID:15880118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729-34. PMID:9637481 [PubMed] [Google Scholar]
- 22.Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, Tahara H, Inoue N, Seya T. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci USA. 2012;109:2066-71. doi: 10.1073/pnas.1113099109. PMID:22308357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shime H, Kojima A, Maruyama A, Saito Y, Oshiumi H, Matsumoto M, Seya T. Myeloid-Derived Suppressor Cells Confer Tumor-Suppressive Functions on Natural Killer Cells via Polyinosinic:Polycytidylic Acid Treatment in Mouse Tumor Models. J Innate Immun. 2014;6:293-305. doi: 10.1159/000355126. PMID:24192491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shime H, Matsumoto M, Seya T. Double-stranded RNA promotes CTL-independent tumor cytolysis mediated by CD11b(+)Ly6G(+) intratumor myeloid cells through the TICAM-1 signaling pathway. Cell Death Differ. 2017;24:385-96. doi: 10.1038/cdd.2016.131. PMID:27834952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol. 2005;174:4880-91. doi: 10.4049/jimmunol.174.8.4880. PMID:15814715 [DOI] [PubMed] [Google Scholar]
- 26.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn J-I, Cheng P, Cho H-I, Celis E, Quiceno DG, Padhya T, et al.. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439-53. doi: 10.1084/jem.20100587. PMID:20876310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V, Cheng P, Condamine T, Mony S, Languino LR, McCaffrey JC, Hockstein N, Guarino M, Masters G, Penman E, et al.. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity. 2016;44:303-15. doi: 10.1016/j.immuni.2016.01.014. PMID:26885857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanke NT, LaCasse CJ, Larmonier CB, Alizadeh D, Trad M, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N. PIAS1 and STAT-3 impair the tumoricidal potential of IFN-γ-stimulated mouse dendritic cells generated with IL-15. Eur J Immunol. 2014;44:2489-99. doi: 10.1002/eji.201343803. PMID:24777831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208-20. doi: 10.1016/j.it.2016.01.004. PMID:26858199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraj S, Schrum AG, Cho H-I, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol 2010;184:3106-16. doi: 10.4049/jimmunol.0902661. PMID:20142361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666-75. doi: 10.4049/jimmunol.181.7.4666. PMID:18802069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235-49. doi: 10.1084/jem.20080132. PMID:18809714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zoglmeier C, Bauer H, Nörenberg D, Wedekind G, Bittner P, Sandholzer N, Rapp M, Anz D, Endres S, Bourquin C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin Cancer Res. 2011;17:1765-75. doi: 10.1158/1078-0432.CCR-10-2672. PMID:21233400 [DOI] [PubMed] [Google Scholar]
- 34.Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol. 2012;188:1592-9. doi: 10.4049/jimmunol.1101304. PMID:22231700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee M, Park C-S, Lee Y-R, Im S-A, Song S, Lee C-K. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch Pharm Res. 2014;37:1234-40. doi: 10.1007/s12272-014-0379-4. PMID:24748512 [DOI] [PubMed] [Google Scholar]
- 36.Aranda F, Vacchelli E, Obrist F, Eggermont A, Galon J, Sautès-Fridman C, Cremer I, Henrik Ter Meulen J, Zitvogel L, Kroemer G, et al.. Trial Watch: Toll-like receptor agonists in oncological indications. Oncoimmunol. 2014;3:e29179. doi: 10.4161/onci.29179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pol J, Bloy N, Buqué A, Eggermont A, Cremer I, Sautès-Fridman C, Galon J, Tartour E, Zitvogel L, Kroemer G, et al.. Trial Watch: Peptide-based anticancer vaccines. Oncoimmunol. 2015;4:e974411-4. doi: 10.4161/2162402X.2014.974411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells—An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunol. 2015;4:e954829-11. doi: 10.4161/21624011.2014.954829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akazawa T, Ohashi T, Nakajima H, Nishizawa Y, Kodama K, Sugiura K, Inaba T, Inoue N. Development of a dendritic cell-targeting lipopeptide as an immunoadjuvant that inhibits tumor growth without inducing local inflammation. Int J Cancer. 2014;135:2847-56. doi: 10.1002/ijc.28939. PMID:24789268 [DOI] [PubMed] [Google Scholar]
- 40.Geisel J, Kahl F, Müller M, Wagner H, Kirschning CJ, Autenrieth IB, Frick J-S. IL-6 and maturation govern TLR2 and TLR4 induced TLR agonist tolerance and cross-tolerance in dendritic cells. J Immunol. 2007;179:5811-8. doi: 10.4049/jimmunol.179.9.5811. PMID:17947654 [DOI] [PubMed] [Google Scholar]
- 41.Tang M, Diao J, Gu H, Khatri I, Zhao J, Cattral MS. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep. 2015;13:2851-64. doi: 10.1016/j.celrep.2015.11.053. PMID:26711349 [DOI] [PubMed] [Google Scholar]
- 42.Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F, et al.. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell. 2016;29:587-601. doi: 10.1016/j.ccell.2016.03.005. PMID:27070705 [DOI] [PubMed] [Google Scholar]
- 43.Yokouchi H, Chamoto K, Wakita D, Yamazaki K, Shirato H, Takeshima T, Dosaka-Akita H, Nishimura M, Yue Z, Kitamura H, et al.. Combination tumor immunotherapy with radiotherapy and Th1 cell therapy against murine lung carcinoma. Clin Exp Metastasis. 2007;24:533-40. doi: 10.1007/s10585-007-9090-x. PMID:17653821 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.