

ABSTRACT
Successful development of immunotherapeutic strategies for hepatocellular cancer (HCC) has been impeded by limited understanding of tumor-induced profound tolerance and lack of a clinically faithful HCC model. Recently, we developed a novel model that recapitulates typical features of human HCC. Using this clinically relevant model, we demonstrate that tumor growth impairs host immunity and causes a profound exhaustion of tumor antigen-specific (TAS) CD8+ T cells. Increase in frequency and suppressive function of regulatory T cells (Tregs) is critically involved in this tumor-induced immune dysfunction. We further demonstrate that sunitinib suppresses Tregs and prevents tumor-induced immune tolerance, allowing TAS immunization to activate endogenous CD8+ T cells. As a result, this combinational strategy delays tumor growth. Importantly, the additional integration of exogenous naïve TAS CD8+ T cells by adoptive cell transfer (ACT) leads to the elimination of the established tumors without recurrence and promotes long-term survival of the treated mice. Mechanistically, sunitinib treatment primes the antitumor immune response by significantly decreasing Treg frequency, reducing TGF-β and IL-10 production by Tregs, and also protecting TAS CD8+ T cells from tumor-induced deletion in the setting of HCC. Taken together, sunitinib quantitatively and qualitatively modifies Tregs to overcome tumor-induced immune deficiency, suggesting the potential of sunitinib as a therapeutic immune activator for HCC control.
KEYWORDS: Hepatocellular Cancer (HCC), sunitinib, immunotolerance, immunotherapy, regulatory t cell (Treg), murine model, CD8+ T cells, IFN-γ, TNF-α
Introduction
Hepatocellular cancer (HCC) is the second leading cause of cancer death worldwide and over 700,000 patients die from this lethal disease annually.1,2 Most cases are diagnosed at a late stage and the current standards of care only provide limited benefit. Even when diagnosis is early, the presence of liver fibrosis/cirrhosis in more than 80% of HCC patients compromises therapeutic options.1,3 In 2007, sorafenib was approved by the Food and Drug Administration (FDA) as the first systemic treatment for patients with unresectable HCC;4 unfortunately, this treatment only increases the overall median lifespan of patients from 7.9 to 10.7 months.5,6 This statistically significant but modest improvement emphasizes that more effective therapies are urgently needed. A growing body of studies have demonstrated that immunotherapies offer great promise for HCC treatment,7-10 but none of these have been successfully translated into clinical application, as HCCs develop multiple immune escape mechanisms that thwart effective anti-tumor immune responses.11-14 Uncovering these underlying mechanisms will likely point to novel and clinically effective immunotherapeutic strategies for HCC.
Immune escape mechanisms in HCC patients include, but are not restricted to, production of immunosuppressive cytokines and prostaglandins, impairment of antigen-presenting cells, generation of inhibitory macrophages, increase of regulatory T cells (Tregs), and accumulation of myeloid-derived suppressor cells (MDSCs).15 Crosstalk of these factors orchestrates the immunosuppressive network within the tumor microenvironment (TME) to promote angiogenesis, tumor survival, and metastasis. Studying 84 HCC patients with chronic infection of hepatitis B and hepatitis C virus, or non-viral liver cirrhosis, Ormandy et al. found increased CD4+CD25+Tregs in the peripheral blood and tumor-infiltrating lymphocytes (TILs) in patients with HCCs.16 They found that the in vitro proliferation and cytokine production in CD4+CD25−T cells was potently suppressed by Tregs isolated from these patients.16 Lin et al. demonstrated that the 5-year survival rate is significantly lower in HCC patients with high numbers of tumor-infiltrating Tregs than patients with low numbers of tumor-infiltrating Tregs.17 In HCC-bearing mice, Tregs down-regulated the expression of costimulatory molecules, CD80/CD86, and inhibited production of TNF-α and IL-12 by dendritic cells (DCs); subsequently, these impaired DCs induced immune suppression.18 These results suggest that Tregs represent one of the primary tumor immune-escape mechanisms in HCC, and may be a dominant obstacle to successful tumor immunotherapy.19,20
Clinically, 90% of human HCCs occur in the setting of fibrosis, as chronic liver injury predisposes the affected liver to oncogenic mutations.21 We recently created a clinically realistic murine model of HCC in which tumors arise in the setting of liver fibrosis in immunocompetent C57BL/6 mice. This model mimics initiation and progression of human HCC, and reflects its typical histologic, biologic, and immunologic features.22 Characterization of this model demonstrated that the frequency of CD4+CD25+FoxP3+Tregs is significantly increased during late-stage tumor development which contributes to tumor-induced immunotolerance. This novel and clinically relevant model provides an ideal platform to study the critical role and the underlying mechanisms of Tregs in tumor-induced immunotolerance in the setting of HCC.22
Sunitinib is a multi-targeted receptor tyrosine kinases inhibitor that received FDA approval in 2006 as a standard of care for both clear cell renal cell carcinoma (ccRCC) and gastrointestinal stromal tumors (GIST).23 The drug is being investigated as a possible therapy for other cancers,24,25 and showed antitumor activity in patients with advanced HCC.26 Using our previous orthotopic murine model without liver fibrosis/cirrhosis, we demonstrated that sunitinib treatment alone promoted transient reduction in tumor size, but its combination with immunotherapy resulted in tumor regression.27 This provocative finding drives us to further explore sunitinib's immunomodulatory function in the setting of fibrotic HCC.22 Using our clinically relevant model, we now demonstrate that Tregs critically contribute to profound immunotolerance in late stage HCC development. Sunitinib treatment represses Tregs quantitatively and qualitatively, and also protects TAS T cells from tumor-induced deletion in the context of HCC. As a result, sunitinib treatment enables adoptive transfer of TAS CD8+ T cells plus immunization to prime a therapeutic immune response to destroy established tumors. These results reveal the potency of sunitinib in preventing tumor-induced tolerance, and that sunitinib-immunotherapy may represent a promising therapeutic modality in HCC control.
Materials and methods
Mice
Male C57BL/6 mice and B6.SJL mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Line MTD2 transgenic mice that express full-length SV40 T antigen (TAg) driven by the major urinary protein (MUP) promoter have been previously described.22,28,29 Line 416 mice served as the source of TAg-specific CD8+ T cells (TCR-I T cells) as described previously.28,30 All experiments with mice were performed under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the Penn State College of Medicine and the University of Missouri. All mice received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals”.
Peptides, reagent and antibodies
Peptides were synthesized at the Penn State Hershey Macromolecular Core Facility and solubilized in DMSO. Sunitinib was purchased from Pfizer (New York City, NY) and prepared as a 20 mM stock solution in DMSO and diluted to a 1% (wt/vol) working solution with a viscous liquid (0.5% Polysorbate 80, 10% polyethylene glycol 300 and 19.2% (vol/vol) 0.1 N hydrochloric acid). CCl4 and corn oil were purchased from Sigma (St Louis, MO). Unlabeledrat anti-mouse CD16/CD32 and fluorochrome-conjugated antibodies against CD3, CD8a, CD4, CD25, FoxP3, CD45.1, TNF-α and IFN-γ were purchased from eBioscience (San Diego, CA).
Cell line and medium
TAg-transformed B6/WT-19 cells have been described previously2,8 and were authenticated by DNA profiling with PCR (forward primer: GCTCATCAACCTGACTTTGGAGGC; reverse primer: GTAGCCTCATCATCACTAGATGGC) and passaged for fewer than 2 months after recovery from frozen low passage stocks. The cell line was maintained in DMEM (Cellgro, Manassas, VA) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 100 μg/mL kanamycin, 2 mM L-glutamine, 10 mM HEPES, 0.075% (w/v) NaHCO3, and 10% FBS at 37°C in a 5% CO2 humidified atmosphere.
Murine HCC model
The mouse model of HCC was prepared as described.22 Briefly, young male C57BL/6 mice received intraperitoneal injection of 10% CCl4 (v/v) solution in corn oil twice/week at a dose of 8 ml/kg body weight for four weeks.31,32 Two weeks after the last injection, TAg-transgenic hepatocytes isolated from young male MTD2 mice were administered via intrasplenic (ISPL) injection.27
Sunitinib administration, adoptive cell transfer (ACT), and immunization
Sunitinib was administered orally (0.2 mL) every other day for two weeks (40 mg/kg body weight). For ACT, 1 × 106 TCR-I T cells isolated from spleens and lymph nodes of 416 mice were suspended in 0.2 mL of HBSS and injected via tail vein as described.22 For immunization, 3 × 107 B6/WT-19 cells suspended in 0.2 mL of PBS were injected intraperitoneally into mice at the indicated time in each experiment.
Lymphocyte isolation
Isolation of lymphocyte populations from spleens and lymph nodes were performed as previously described.27 To isolate lymphocytes from liver or tumor tissue, a liver perfusion was performed via the portal vein with 15 mL PBS and 15 mL collagenase (Sigma, Saint Louis, MO) at 1 mg/mL22,33. The harvested tumor or liver tissues were cut into small pieces and incubated in collagenase solution for 20 mins at 37°C. After that, the solution was quenched by RPMI 1640 with 10% FBS and passed through a 70 μm cell strainer (BD Falcon, San Jose, CA) to remove the tissue debris. The collected solution was spun down by centrifugation at 100 × g for 1 min. The supernatant was harvested and centrifuged at 400 × g for 5 mins. The cell pellet was resuspended in a 15% Histodenz solution (Sigma, Saint Louis, MO) followed by centrifugation for 25 min at 800 × g at 4°C, mononuclear cells were harvested at the interface and washed twice. Single cell suspensions were maintained in RPMI 1640 medium (Cellgro, Manassas, VA) supplemented with 100 U/mL penicillin, 2 mM L-glutamine, 10 mM HEPES, 50 μM 2-mercaptoethanol and 10% FBS until analysis.
Flow cytometry
Ex vivo staining of lymphocytes with fluorochrome-labeled antibodies was performed on single-cell suspensions.27 Stained cells were analyzed using a FACScan flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star). Staining of intracellular IFN-γ and TNF-α was performed as described previously.27 Staining of FoxP3 was performed with a buffer set from eBioscience.
Isolation of TCR-I T cells, CD4+CD25+Tregs, and CD4+CD25−T cells
According to the manufacturer's instructions, TCR-I T cells were enriched from the lymphocytes isolated from spleen and lymph nodes of 416 mice by MACS sorting with CD8+ magnetic microbeads (Miltenyi Biotech, Auburn, CA). CD4+CD25−T cells and CD4+CD25+Tregs were isolated from whole lymphocytes using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotech, Auburn, CA). Briefly, CD4+ T cells were first isolated by negative selection followed by positive selection with microbeads conjugated to anti-CD25 antibody.
Generation of bone marrow-derived dendritic cells (DCs)
Bone marrow was harvested from femurs and tibias of C57BL/6 mice to prepare DCs as we previously described.34 In brief, red blood cells (RBCs) in the bone marrow were lysed with RBC lysis buffer (eBioscience, San Diego, CA) to prepare single cell suspension. 3 × 106 cells were then plated in a 100 mM petri dish and cultured in 10 mL DMEM medium (cellgro, Manassas, VA) supplemented with 10% FBS, 10 mM HEPES, 1:100 (v/v) non-essential amino acids (Sigma, St Louis, MO), 1 mM sodium pyruvate (Sigma, St Louis, MO), and 20 ng/mL granulocyte-macrophage colony-stimulating factor (mGM-CSF) (R&D system, Minneapolis MN). 5 mL fresh complete medium containing 20 ng/mL mGM-CSF was added to the culture on day 4. Nonadherent cells were harvested on day 7 as a source of BMDCs and pulsed with 2 μM peptide for 3 hours.
Cell proliferation assay
1 × 105 cells were seeded into anti-CD3 coated 96-well plates and cultured for 2 days. Proliferation assays were performed using CellTiter 96 Aqueous One Solution Cell Proliferation Assay Kit (Promega, Madison, WI) according to the manufacturer's instructions. Absorbance was measured at 490 nm using SpectraMax Plus384 from Molecular Devices. For some experiments, lymphocytes were labeled with 1 µM carboxy fluorescein diacetate succinimidyl ester (CFSE) (Invitrogen, Carlsbad, CA) to analyze T cell proliferation.22 CFSE-labeled cells were incubated in the presence or absence of the indicated stimulations and the dilution of CFSE measured by flow cytometry to evaluate cell proliferation.
ELISA
Cell culture supernatants were harvested at the indicated time points and cell debris was removed by gentle centrifugation at 1,500 rpm for 10 min at 4°C. IFN-γ, TGF-β, and IL-10 levels were measured with mouse Quantikine ELISA Kits specific for IFN-γ (Cat # DY485), IL-10 (Cat # DY417) and TGF-β (Cat # 1679) (R&D systems, Minneapolis, MN) according to the manufacturer's directions.
In vivo cytotoxicity assay
3 × 106 splenocytes from sex-matched B6.SJL (CD45.1+) mice were suspended in complete RPMI medium and pulsed with 2 μM peptides for 1 hour at 37°C corresponding to: Influenza (Flu) nucleoprotein (NP) (366ASNENMETM374), epitope I (206SAINNYAQKL215), and epitope IV/411L (404VVYDFLKL411). Pulsed-cells were washed and re-suspended in PBS containing 0.1% BSA at a concentration of 1 × 106/mL, then labeled with different concentrations of CFSE for 10 min at 37°C: 0.25 μM for Flu-pulsed cells, 0.5 μM for epitope I-pulsed cells and 5 μM for epitope IV-pulsed cells. After washing twice with PBS, the CFSE-labeled cells were mixed at an equal proportion. 1 × 106 total mixed cells were i.v. injected into each mouse with the indicated treatment. 16 hours after injection, lymphocytes isolated from each mouse were stained with anti-CD45.1 antibody for flow cytometry analysis. Percentage of eliminated target cells was determined using the following formula: [ratio of CFSEhi(site IV) or CFSEmed(site I) cells to CFSElow cells in control unimmunized mice – ratio of CFSEhi(site IV) or CFSEmed(site I) cells to CFSElowcells in experimental mice]/[ratio of CFSEhi(site IV) or CFSEmed(site I) cells to CFSElowcells in control unimmunized mice].
Statistics and survival analysis
Paired data were analyzed using a 2-tailed paired Student's t test. A P value of less than 0.05 was considered significant. For lifespan analysis, mice were monitored for the development of ascites and the impairment of gait and breathing, all of which are indicative of end-stage liver tumors. Survival curves were constructed following the Kaplan-Meier method using GraphPad Prism software (La Jolla, CA). Significance was determined by single-factor analysis of variance, and validated using the log-rank test. P values of <0.05 were considered significant.
Results
Tregs function as a critical immune cell in tumor-induced immune tolerance in HCC
We recently developed and reported a novel and clinically relevant murine model of HCC.22 Using this model, we analyzed the immune response in late stage tumor-bearing mice following TAS immunization with TAg-transformed B6/WT-19 fibroblasts. On day seven, splenic lymphocytes were isolated from tumor-free and tumor-bearing mice (Supplementary Fig. 1) and subjected to stimulation. We detected a significant decrease in the frequency of CD8+ T cells producing IFN-γ upon TAg epitope-I stimulation in tumor-bearing mice compared to tumor-free mice (Fig. 1A). We also detected a two-fold reduction in the expansion of splenic lymphocytes in tumor-bearing mice in response to anti-CD3 stimulation compared to that from tumor-free mice (Fig. 1B). Correspondingly, approximately 200 pg/ml of IFN-γ was detected in the culture supernatant from splenic lymphocytes in tumor-bearing mice, which was substantially less than the IFN-γ production (>1500 pg/ml) seen in tumor-free mice (Fig. 1B). These results indicate that late-stage tumors induce TAS CD8+ T cell exhausted.
Figure 1.

Tregs function as a critical immune cell in tumor-induced immune tolerance. (A) Suppression of IFN-γ production in CD8+ T cells in tumor-bearing mice in response to TAS immunization. Seven days after immunization with 3 × 107 of B6/WT-19 cells, splenic lymphocytes were isolated from immunized tumor-free and tumor-bearing mice, and in vitro stimulated with 2 μM of peptide I or control peptide flu for 4 hours. Subsequently, cultured cells were stained with fluorochrome-conjugated antibodies against CD8 and IFN-γ for conducting flow cytometry analysis. Representative and accumulated frequency of CD8+ T cells producing IFN-γ are shown. (B) In vitro depletion of CD4+CD25+ Tregs restores the proliferation and activity of lymphocytes. Lymphocytes were isolated from the spleen of tumor-free and tumor-bearing mice and depleted CD4+CD25+Tregs. 1 × 105 of lymphocytes with or without Tregs depletion were seeded in anti-CD3 coated 96-well plates and cultured for 2 days. Lymphocyte proliferation was analyzed with Cell'Titer 96 Aqueous One Solution Cell Proliferation Assay Kit. The IFN-γ level in supernatants was measured by ELISA. (C) The increased frequency of Tregs in tumor-bearing mice. Lymphocytes were isolated from the spleen, lymph node, and liver of tumor-free and tumor-bearing mice, stained with fluorochrome-conjugated antibodies for CD3, CD4, CD8, CD25, FoxP3 for conducting flow cytometry analysis. The representative and mean frequency of CD4+CD25+FoxP3+Tregs in the gated CD4+ T cells was shown. (D-F) CD4+CD25−T cells were isolated from normal or tumor-bearing mice and named normal response cells (nRCs) and tumor response cell (tRCs), respectively); CD4+CD25+Tregs isolated from tumor-free and tumor-bearing mice were termed as normal Treg (nTreg) and tumor Treg (tTreg), respectively. Isolated nRCs or tRCs were labeled with 1 µM of CFSE and cultured with nTregs or tTregs at a ratio of 1:1 and 5:1 in the presence of 1 µg/ml of anti-CD3 Ab plus 2 µg/ml of anti-CD28 Ab. 48 hours later, the cells were harvested to perform flow cytometry after staining with fluorochrome-conjugated antibodies. IFN-γ production was measured in supernatants as in Methods. (D) Quantitative data depicting the CFSE dilution profile of nRC and tRC in the presence of nTreg or tTreg are shown. (E) IFN-γ production in nRC in the presence of nTregs or tTreg is shown. (F) IL-10 production in nRC in the presence of nTreg or tTreg. n = 3, the error bar represented mean ± SD. Asterisks represented statistically significant differences (p < 0.05).
Next, we investigated how tumor growth impacts the frequency of distinct immune cell subsets.35,36 Consistent with our previous results, there was a significant increase in CD4+CD25+Foxp3+ Tregs in the spleen, draining lymph nodes (DLNs), and livers from late tumor-bearing mice relative to tumor-free mice (Fig. 1C). A significant change in CD8+ T cells was found only in the DLNs, while slight increases in spleen and liver were detected (Supplemental Fig. 2A). No significant change was found in CD4+ T-cell frequency from all three sites between tumor-free and tumor-bearing mice (supplemental Fig. 2B).
To investigate whether increased Tregs contribute to the observed reduced lymphocyte response, CD4+CD25+Tregs were depleted from splenic lymphocytes prior to in vitro stimulation. When the Treg-depleted lymphocytes were stimulated with anti-CD3, there was a 2-fold increase (p < 0.05) in the proliferation of lymphocyte for tumor-free mice and a 10-fold (p < 0.05) increase for tumor-bearing mice; furthermore, the resulting absolute number of lymphocytes was almost equivalent between normal mice and tumor-bearing mice after Treg depletion (Fig. 1B). Additionally, lymphocytes derived from tumor-bearing and tumor-free mice, respectively, generated a 10-fold (p < 0.05) and 2-fold (p < 0.05) increase in IFN-γ production after Treg depletion (Fig. 1B). These results suggest that tumor progression dramatically enhances Tregs' immune suppressive function on lymphocyte activation and expansion. However, depletion of CD4+CD25+Treg only partially restores IFN-γ production, suggesting that additional suppressive mechanisms contribute to HCC-induced immune suppression. One potential additional mechanism includes MDSCs, which are also increased in HCC. We have found that MDSCs are reduced in HCC-bearing mice following treatment with sunitinib.22,37,38 It is likely that both of these suppressive cell types contribute to the overall immune suppression observed. The fact that sunitinib can reverse both of these suggests that an overall resetting of the immune suppressive environment is occurring.
To further define the functional discrepancy of Tregs in tumor-free and tumor-bearing mice, responder CD4+CD25−T cells and suppressive CD4+CD25+Tregs were enriched from normal mice and tumor-bearing mice, respectively and designated normal responder cells (nRCs), tumor responder cells (tRCs), normal Tregs (nTregs), and tumor Tregs (tTregs). nRCs and tRCs were labeled with CFSE, then co-cultured with isolated Tregs at a ratio of 1:1 in the presence of soluble anti-CD3 and anti-CD28. The extent of proliferation inhibition mediated by the indicated Tregs was determined by CFSE dilution. As shown in Fig. 1D, more CFSE dilution was detected in nRCs than tRCs in the absence of nTreg and tTreg, suggesting the reduced expansion of tRCs from tumor-bearing mice. We previously demonstrated that CD4+ T cells from HCC-bearing mice significantly increase expression of PD-1 and CTLA-4.22 Together these results suggest that tRCs from HCC-bearing mice are exhausted. In addition, tTregs more potently inhibited proliferation of RCs from either donor than nTregs. Meanwhile, the addition of nTregs at a ratio of either 1:5 or 1:1 significantly reduced IFN-γ production in nRCs (Fig. 1E, p < 0.05), but increased IL-10 production (Fig. 1F, p < 0.05). The addition of the tTregs resulted in a greater reduction of IFN-γ (p < 0.01) and an increase of IL-10 in nRCs (p < 0.01). These results suggest that tTregs have stronger suppressive function on T cells compared to nTregs, similar to observations in human HCC.39
Sunitinib treatment overcomes CD8+ T cell tolerance in HCC and improves its tumoricidal activity
We previously demonstrated that administration of sunitinib activates a therapeutic antitumor immune response in HCC-bearing mice.22,27 To explore the underlying mechanism, ex vivo and in vivo experiments were used to define how sunitinib impacts the function of effector CD8+ T cells. Sunitinib was orally administered to late tumor-bearing mice every other day for two consecutive weeks followed by immunization with TAg-transformed B6/WT-19 cells. Seven days after immunization, splenic lymphocytes were isolated from sunitinib-treated or control vehicle-treated tumor-bearing mice, and subsequently in vitro stimulated with TAg epitope I or IV peptide for 5 hours, followed by evaluation of cytokine production. As shown in Fig. 2A-D, sunitinib treatment significantly increased the frequency of CD8+ T cells producing IFN-γ and TNF-α compared to vehicle-treated tumor-bearing mice. Consistent with our previous results, these ex vivo experiments indicate that sunitinib treatment promotes the differentiation of cytokine-producing CD8+ T cells in tumor-bearing mice.
Figure 2.

Sunitinib treatment prevents endogenous CD8+T cell tolerance. Tumor-bearing mice were treated with sunitinib or vehicle for 2 weeks followed by immunization with 3 × 107 B6/WT-19 cells. Seven days after immunization, half of the mice were used to isolate splenic lymphocytes for detecting cytokine production. The other half was used to measure in vivo cytotoxicity. To measure cytokine production, the splenic lymphocytes were isolated from the indicated mice and cultured with 2 μM of peptide I, IV or Flu in the presence of 3 µg/ml brefeldin. 4 hours later, cells were labeled with antibodies to detect CD8, IFN-γ, and TNF-α and subjected to flow cytometry. A representative dot plot and accumulated data depicting the frequency of IFN-γ-producing CD8+T cells (A and B) and TNF-α-producing CD8+T cells are shown (C and D). An in vivo cytotoxic assay was performed using donor lymphocytes from the spleen of B6.SJL mice (CD45.1+) to differentiate host cells (CD45.2+) in tumor-bearing mice. Donor cells were pulsed with 2 μM of peptide I, IV or Flu followed by labeling with different concentrations of CFSE, and then mixed at equal proportions. 1 × 106 cells were injected intravenously into indicated mice. Normal mice with or without immunization were included as positive and negative controls. 16 hours after injection, lymphocytes isolated from each mouse were labeled with anti-CD45.1 antibody for flow cytometry analysis. Data from a representative experiment (E and combined data (F) for the frequency of remaining donor CD45.1+ lymphocytes pulsed with the indicated peptideare shown.. n = 3; error bars represented means ± SD. Asterisk represented significant difference (p < 0.05).
Next, we evaluated the effect of sunitinib in enhancing in vivo cytotoxic function on TAS target cells. In brief, CD45.1+ target cells were prepared from the spleen of congenic B6/SJL mice and pulsed with distinct peptides including control Flu, TAg epitope I, or TAg epitope IV followed by labeling with low, medium, and high concentrations of CFSE, respectively. A mixture of peptide-pulsed and CFSE-labeled target cells at equal proportions were i.v. injected into C57BL/6 (CD45.2+) tumor-bearing mice that were treated with sunitinib or control vehicle and immunized with B6/WT-19 cells. free mice with or without immunization were used for positive and negative controls. Only 48% and 7% of epitope I and epitope IV-pulsed targets, respectively, remained in immunized tumor-free mice (Fig. 2E), while 99.8% of epitope I-pulsed and 45% of epitope IV-pulsed target cells remained in vehicle-treated tumor-bearing mice, consistent with limited T cell activation in tumor-bearing mice. In contrast, only 52% of epitope I-pulsed and 5% of epitope IV-pulsed target cells were detected in sunitinib-treated tumor-bearing mice, similar to that observed in control immunized C57BL/6 mice. This result suggests that sunitinib treatment activates in vivo cytotoxic function among TAS T cells. Collectively, sunitinib treatment restores the antitumor immune response to immunization in tumor-bearing mice.
Sunitinib treatment reduces Treg frequency and normalizes suppressive function in tumor-bearing mice
To explore why sunitinib restores in vivo anti-tumor cytotoxic function in tumor-bearing mice, we investigated the impact of sunitinib treatment on the profile of major immune cell subsets. As shown in Fig. 3A-D, sunitinib treatment induced modest alteration in the frequencies of CD4+ T and CD8+ T cells within the spleens, lymph nodes, and livers of tumor-bearing mice. In contrast, a significant reduction in the frequency of Tregs was detected at these three sites in tumor-bearing mice after receiving sunitinib treatment (p < 0.01) (Fig. 3E and F).
Figure 3.

Sunitinib treatment systemically reduces Treg frequency in tumor-bearing mice. Tumor-bearing mice were treated with vehicle or sunitinib every other day for 2 consecutive weeks. Lymphocytes were isolated from DLN, spleen and tumor tissue in sunitinib-treated mice. Normal C57BL/6 mice and vehicle-treated tumor-bearing mice were used as controls. Lymphocytes were labeled with antibodies for CD3, CD4, CD8, and CD25 (surface staining), and Foxp3 (intracellular staining), followed by flow cytometry. The representative (A) and accumulated (B) frequency of CD4+ T cells in CD3-gated T cells from the spleen is depicted. The representative (C) and accumulated (D) frequency of CD8+ T cells in CD3-gated T cells from the spleen is depicted. The representative (E) and accumulated (F) frequency of CD4+CD25+FoxP3+Tregs in the CD4-gated T cells from DLN, spleen and the liver is depicted. Error bar represented mean ± SD, n = 3. Asterisk represented significant difference (p < 0.05).
Next, we investigated whether sunitinib treatment impacted Treg suppressive function in the setting of HCC. We isolated CD4+CD25+Tregs in normal and tumor-bearing mice with or without sunitinib treatment; and CD4+ T or CD8+ T cells in the spleens of normal C57BL/6 mice. CFSE-labeled CD4+ T or CD8+ T cells were mixed with nTregs or tTregs at a ratio of 1:1 and then stimulated with soluble anti-CD3/anti-CD28. tTregs significantly reduced CD8+ T cell proliferation in response to the stimulation compared to nTregs which only showed slight suppression (Fig. 4A and B). However, this increased suppression of tTreg was blocked by sunitinib since tTregs obtained from sunitinib-treated tumor-bearing mice achieved an equivalent CD8+ T-cell proliferation with nTregs. In contrast, both nTregs and tTregs suppressed CD4+ T cell proliferation with more inhibition observed with tTreg. Interestingly, this increased suppression with tTregs was eliminated by sunitinib since addition of tTregs from sunitinib-treated tumor-bearing mice allowed an even higher level of CD4+ T cell proliferation than with nTregs (Fig. 4C and D). These results indicate that Tregs in normal mice differently impact the proliferation of CD4+ T cells and CD8+ T cells. Tumor growth enhances Treg suppressive function which is effectively reversed by sunitinib treatment.
Figure 4.
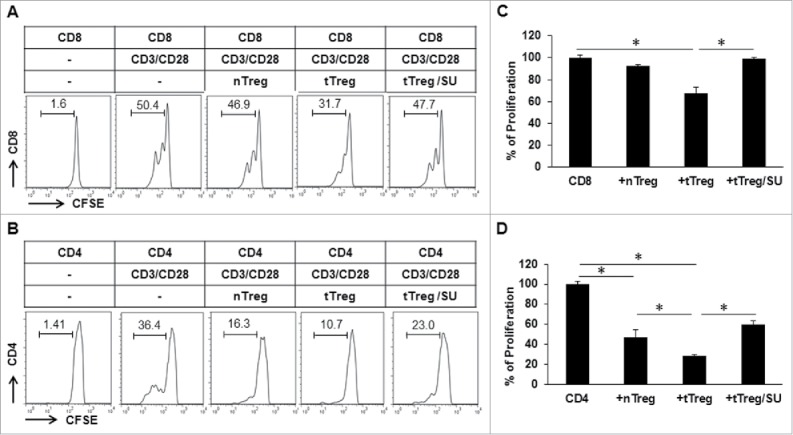
Sunitinib treatment blocks the suppressive function of Tregs in tumor-bearing mice. Tumor-bearing mice were treated with sunitinib or vehicle every other day for 2 weeks. Lymphocytes were isolated from the spleen of tumor-free mice, vehicle-treated and sunitinib-treated tumor-bearing mice to purify CD4+CD25+Tregs as described in the preceding experiments. Reactive CD8+ and CD4+ T cells were isolated from normal mice and labeled with 1 µM of CFSE as in Methods. 5 × 104 CFSE-labeled reactive CD8+ or CD4+ T cells were cultured with the indicated CD4+CD25+Tregs at a ratio of 1:1 for 48 hours in the presence of 1 µg/ml of anti-CD3 antibody and 2 µg/ml of anti-CD28 antibody, then subjected to flow cytometry. (A) Representative histograms depicting the CFSE profile of CD8+ T cells in the presence of the indicated Tregs are shown. (B) Representative histograms depicting the CFSE profile of CD4+ T cells in the presence of the indicated Tregs are shown. (C) The accumulated results for CD8+ T-cell proliferation in the presence of the indicated Tregs normalized to control CD8+ T cells in the absence of Tregs. (D) The accumulated results for CD4+ T-cell proliferation in the presence of the indicated Tregs normalized to control CD4+ T cells in the absence of Treg. n = 3; error bars represented means ± SD. Asterisk represented significant difference (p < 0.05).
To further understand the effect of sunitinib-educated Tregs on TAS CD8+ T cells, TCR-I T cells which specifically recognized TAg epitope-I were enriched from 416 mice and labeled with CFSE. CFSE-labeled TCR-I T cells were co-cultured with the indicated Tregs at a ratio of 1:1 for 72 hours in the presence of epitope-I-pulsed dendritic cells (DCs). Addition of tTregs from tumor-bearing mice significantly reduced TCR-I T-cell proliferation and IFN-γ production compared to nTregs obtained from normal mice (Fig. 5, p < 0.05); this increased suppression of TCR-I T-cells was not detected in the tTregs obtained from sunitinib-treated tumor-bearing mice. These findings suggest that Tregs from tumor-bearing mice exhibit potent suppressive activity on TAS CD8+ T cells, and that in vivo sunitinib treatment abrogates their increased suppressive effect. These data suggest that suppression of Tregs with sunitinib provides an approach to reduce tumor-induced immunotolerance in HCC.
Figure 5.
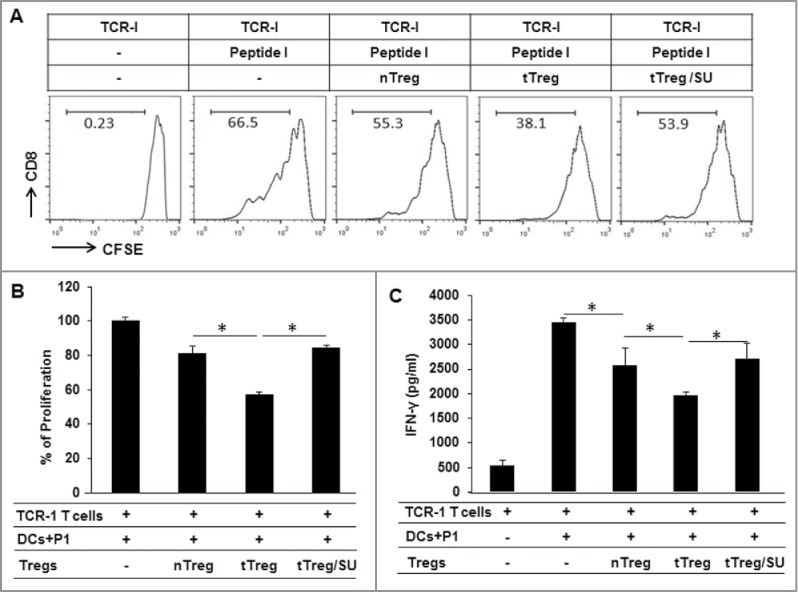
Sunitinib treatment reduces Tregs suppressive activity on TAS CD8+ T cells. Splenic lymphocytes were isolated from tumor-free mice, vehicle-treated tumor-bearing mice, and sunitinib-treated tumor-bearing mice, respectively, then used to purify CD4+CD25+Tregs as previously described. TAS TCR-I T cells were prepared from the spleen of TCR-I transgenic 416 mice by positive selection with microbeads conjugated to anti-CD8 antibody. 5 × 104 TCR-I T cells were labeled with 1 µM CFSE and co-cultured with 5 × 103 BMDC derived from normal mice in the presence or absence of the indicated Tregs at a ratio of 1:1, stimulated with 2 µM of peptide I for 72 hours, then harvested and subjected to flow cytometry; IFN-γ production in supernatants was measured by ELISA. (A) Representative histograms depicting the CFSE dilution profile of TCR-I T cells in presence of the indicated Tregs. (B) The accumulated results for TCR-I T-cell proliferation in the presence of the indicated Tregs which was normalized to control CD8+ T cells in the absence of Treg. (C) The accumulated IFN-γ production in TCR-I T cells in the presence of the indicated Tregs. n = 3; error bars represented means ± SD. Asterisk represented significant difference (p < 0.05).
In vivo treatment of tumor-bearing mice with sunitinib modulates cytokine secretion in Tregs
To understand whether sunitinib treatment affects Treg cytokine production, enriched Tregs from sunitinib-treated or vehicle–treated tumor-bearing mice were seeded into anti-CD3-coated plates and then stimulated with soluble anti-CD28 in the presence of rIL-2 for 72 h. nTregs from normal mice were used as controls. The levels of TGF-β and IL-10 in the culture supernatant were measured by ELISA. As shown in Fig. 6A and B, tTregs from tumor-bearing mice secreted significantly higher levels of TGF-β (2000 pg/ml) and IL-10 (220 pg/ml) than that nTregs (1300 pg/ml and 100 pg/ml); however, tTregs isolated from sunitinib-treated tumor-bearing mice secreted TGF-β and IL-10 at levels similar to nTregs.
Figure 6.

Suppressive function in tTregs is reduced by in vivo treatment with sunitinib and in vitro treatment with antibodies for TGF-β and IL-10. Splenic lymphocytes were prepared from tumor-free mice, vehicle-treated tumor-bearing mice, and sunitinib-treated tumor-bearing mice, respectively, and used to purify CD4+CD25+Tregs as described. To investigate cytokine production, 1 × 105 Tregs were seeded in 1 µg/ml of anti-CD3-coated 96-well plates in the presence of 2 µg/ml of anti-CD28 antibody and 5 µg/ml of rIL-2 for 72 hours. Supernatants were subjected to ELISA to measure TGF-β (A) and IL-10 (B). To investigate the role of TGF-β and IL-10 in tTreg-mediated suppression on TAS CD8+ T cells, TAg-specific TCR-I T cells were enriched from 416 mice and labeled with CFSE. The CSFE-labeled TCR-I T cells were co-cultured with tTregs at a ratio of 1:1 in the absence or presence of epitope-I-pulsed DCs (same as Figure 5) and antibodies for IL-10 and TGF-β for 72 hours. Supernatants were subjected to ELISA to measure IFN-γ, the cells were used to conduct flow cytometry for detecting dilution of CFSE in TCR-I T cells. (C) Representative histograms depicting the CFSE dilution profile of TCR-I T cells in presence of tTregs and antibodies for IL-10 and TGF-β. (D) The accumulated results for TCR-I T-cell proliferation in the presence of tTregs and antibodies for IL-10 and TGF-β which was normalized to P-I-stimulated TCR-I T cells in the absence of tTreg and antibodies. (E) The accumulated IFN-γ production in TCR-I T cells in the presence of tTregs and antibodies for IL-10 and TGF-β. To test the in vivo suppressive function of Tregs, 3 × 105 Tregs enriched from tumor-free and tumor-bearing mice were injected into wild type C57BL/6 mice. On the same day, the recipient mice were immunized with 3 × 107 B6/WT-19 cells. 7 days later, splenic lymphocytes were isolated and in vitro stimulated with TAg epitope-I or epitope-IV peptides, and the production of IFN-γ and TNF-α were measured using flow cytometry with fluorochrome-conjugated antibodies against CD8, IFN-γ, and TNF- α. IFN-γ (F) and TNF-α (G) production in CD8+T cells is shown. n = 3; error bars represented means ± SD. Asterisk represented significant difference (p < 0.05).
To investigate if IL-10 and TGF-β are responsible for the immune suppression of tTreg, TAg-specific TCR-I T cells were enriched from 416 mice and labeled with CFSE. The CSFE-labeled TCR-I T cells were co-cultured with tTregs at a ratio of 1:1 in the absence or presence of epitope-I-pulsed DCs (same as Figure 5) and antibodies for IL-10 and TGF-β for 72 hours. The results showed that addition of tTregs significantly suppressed TCR-I T-cell proliferation (Fig. 6C and D, p < 0.05) and its IFN- γ production (Fig. 6E, p < 0.05) in response to TAS stimulation; blockade of IL-10 and TGF-β with antibodies released tTreg-mediated suppression on TCR-I T-cells (Fig. 6C-E). These results suggest that tTregs exert the suppressive function through the production of IL-10 and TGF-β in the setting of HCC.
Sunitinib-educated tTregs display normalized immune suppression when transferred into wild type mice given vaccination
To investigate the in vivo suppressive effect of Tregs on immune activation, Tregs were isolated from normal mice, tumor-bearing mice, and sunitinib-treated tumor-bearing mice, and then infused into wild type mice by tail veil injection followed by immunization with B6/WT-19 cells. Infusion of tTregs from tumor-bearing mice into wild-type mice resulted in higher suppression of CD8+ T cell function. This effect was demonstrated by decreased frequency of IFN-γ and TNF-α-producing CD8+ T cells compared to infusion with nTregs from tumor-free mice; however, this increased suppressive function was abrogated by infusion of tTregs from sunitinib-treated tumor-bearing mice (Fig. 6F and G and supplementary Fig. 3). These results further validate that in vivo treatment with sunitinib eliminates the tumor-enhanced tTreg function.
Sunitinib enhances adoptive T-cell therapy to eradicate established HCC without recurrence
Given sunitinib's powerful effect on reducing Treg frequency and immunosuppressive function, we explored the potential of sunitinib in combination with adoptive T-cell therapy on established tumors in our novel clinically relevant HCC model. ACT of TCR-I T cells in immunized mice did not impact tumor progression as the tumor growth rate was similar to that observed in vehicle-treated mice (Fig. 7A and B). The combination of sunitinib treatment and immunization without ACT of TCR-I T-cells successfully slowed tumor growth, but failed to induce observable tumor regression. However, tumor regression was observed in tumor-bearing mice receiving sunitinib treatment plus ACT and immunization, as tumors resolved completely by week 10 (Fig. 7B). These tumors did not recur within a ten-week observation period. In addition, the magnitude of the detectable TCR-I T cells in the sunitinib-treated tumor-bearing mice was equivalent to that in normal control mice on day 6 post ACT; this population was higher than that observed in vehicle-treated tumor-bearing mice (Fig 7D). This result suggests that sunitinib protects tumor antigen-specific CD8+ T cells from deletion in the TME.
Figure 7.

Sunitinib treatment integrates ACT of TCR-I T cells to reject the established tumors. Tumor-bearing mice were divided into 4 groups: Group 1-Vehicle: vehicle treatment; Group 2-TCR-I: ACT of 1 × 106 of naive TAS TCR-I T cells; Group 3-SU: Sunitinib treatment every other day for 2 weeks; Group 4-SU+TCR-I: Sunitinib treatment every other day for 2 weeks followed by ACT of 1 × 106 of naïve TCR-I T cells. Mice in all four groups received immunization with 3 × 107 of B6/WT-19 cells one day after the indicated final treatments. (A) Representative MRI images reveal tumor volumes in the indicated tumor-bearing mice. (B) The accumulated results for tumor volumes in the indicated tumor-bearing mice over the treatment are shown (n = 8). (C) The number of surviving mice in each group was determined over time (n = 8). (D) Sunitinib prevented TCR-I T cell from tumor-induced deletion. Tumor-bearing mice were treated with sunitinib or vehicle as described above. 1 × 106 naïve TCR-I T cells were labeled with 5μM CFSE and subsequently injected intravenously into normal mice, vehicle-treated tumor-bearing mice, and sunitinib-treated tumor-bearing mice, respectively; 6 days after ACT, lymphocytes were isolated from indicated mice and stained with anti-CD8 antibody. A representative profile of CFSE revealed the frequency of the remaining TCR-I T cells in the indicated mice (n = 5).
Survival analysis revealed 100% mortality within five months in tumor-bearing mice that received vehicle or ACT with immunization (Fig. 7C). In contrast, the combination of sunitinib and ACT plus immunization resulted in 100% survival (up to 9 months post treatment with sunitinib, when animals were sacrificed), while sunitinib plus immunization without ACT led to only 60% survival ten weeks post sunitinib treatment. The data suggest that sunitinib promotes endogenous T-cell activity to limit tumor progression and synergizes with ACT to achieve durable tumor destruction.
Discussion
Here, we demonstrate that tumor progression causes the accumulation of Tregs and enhances their suppressive function in the setting of fibrotic HCC. Tregs represent a dominant immune cell contributing to tumor-induced immune tolerance. Sunitinib, an FDA-approved multiple tyrosine kinase inhibitor, prevents tumor-induced tolerance by decreasing Treg frequency and eliminating its excessive suppressive function. Therefore, sunitinib-mediated activation of endogenous and exogenous TAS CD8+ T cells synergizes with sunitinib's inherent tumoricidal function to destroy the established tumors. These results indicate that while profound immune tolerance is induced in late stage HCC development, re-activation of host immunity can be achieved to suppress tumor growth, supporting this approach as a potential of immune-based therapeutic strategy in the treatment of HCC.14
Immunotherapeutic interventions represent a novel and potentially attractive treatment option for patients with HCC. However, compared to other tumors, few immunotherapy trials for HCC have been conducted and have generated contrasting results.40 Clinical responses have been disappointing and indicate that overcoming profound exhaustion of effector CD8+ T cells in the TME remains a challenging task.41 Using in vivo and in vitro comprehensive experiments, we demonstrate that Tregs are key immune cells associated with profound immunotolerance in HCC. Moreover, we identify that sunitinib, an FDA-approved drug, is able to reduce Treg frequency and block its suppressive function. While one open-label, and phase III trial showed that overall survival with sunitinib monotherapy at a dose of 40 mg/kg was not superior or equivalent to sorafenib monotherapy with more frequent and severe adverse events,42 we have found that sunitinib at half of this dose significantly suppresses tumor growth and prevents tumor-induced immunotolerance.22 In a recent study, we showed that combination treatment of tumor-bearing mice with sunitinib (20 mg/kg) and anti-PD-1 promoted a significant therapeutic effect in our clinically relevant model.22 Thus, while sorafenib provides some single agent benefit, sunitinib at a reduced dose may be a preferable chemotherapeutic agent to use in combination with immunotherapy for the treatment of HCC.
We report, for the first time to our knowledge, that sunitinib treatment led to the partial recovery of endogenous TAS CD8+ T cell activity, evidenced by sunitinib-mediated increase in TAS CD8+ T-cell accumulation and IFN-γ and TNF-α production. As a result, treatment of tumor-bearing mice with sunitinib followed by TAS immunization delayed tumor growth. This partial therapeutic benefit could be explained by insufficient anti-tumor immunity, as endogenous T cells in tumor-bearing mice are functionally defective and unable to be fully restored by sunitinib treatment. Interestingly, sunitinib treatment allows the subsequently transferred exogenous TAS CD8+ T cells to exert strong cytotoxic effects, resulting in the eradication of established tumors. These results suggest sunitinib's potential to serve as an effective immune activator by modulating the TME. In accordance with this finding,27 a recent study on breast cancer demonstrated sequential sunitinib administration and tumor antigen vaccination boosted the immune response.43 Another group reported that the combination of sunitinib and vaccination generated superior anti-tumor therapeutic efficacy in patients with melanoma.44 In a murine model of colon carcinoma, continuous sunitinib treatment followed by vaccination increases intratumoral infiltration of TAS T lymphocytes, reduced tumor volumes, and increased survival of tumor-bearing mice.45
The potential of sunitinib as a powerful and clinically-useful immunomodulatory agent has encouraged us to explore the likely mechanisms. A significant finding is that sunitinib treatment prevents TAS CD8+ T cells from tumor-induced deletion. More fused TAS TCR-I T cells post ACT can be detected in sunitinib-treated tumor-bearing mice, compared to vehicle-treated mice. Importantly, this increase in TCR-I T-cell accumulation is associated with enhanced therapeutic efficacy, as the regression of established tumors is only found in tumor-bearing mice treated with sunitinib followed by TCR-I T-cell transfer and immunization. Mechanistically, sunitinib-mediated reduction in the frequency of Treg is implicated with this therapeutic effect. This result is consistent with our previous finding in an orthotopic murine model of HCC without liver fibrosis/cirrhosis,27 and supported by others' studies on colon cancer4,6 and head and neck squamous cell carcinoma (HNSCC).47 However, further studies are needed to elucidate the underlying mechanisms, such as whether in vivo treatment with sunitinib facilitates Treg apoptosis or blocks differentiation.
A second important finding of this study is that sunitinib treatment regulates Treg cytokine production. We demonstrate that Tregs from late-stage tumor-bearing mice secret a higher level of IL-10and TGF-β in comparison with Tregs from tumor-free mice. In vivo sunitinib treatment is able to reduce expression of these cytokine, decreasing their suppressive effect on CD8+ T cells to the basal level seen in Tregs from tumor-free mice. This may explain why in vivo sunitinib treatment normalizes the exaggerated immunosuppressive activity of Tregs in tumor-bearing mice. In agreement with our current findings, Strauss et al demonstrated that Tregs isolated from patients with HNSCC exhibited stronger immunosuppression to autologous CD4+ and CD8+ T cells.47 Studies on RCC patients showed that sunitinib induces a Th1 immune response (IFN-γ expression) while reducing Tregs in the blood.48,49 Using a murine model of colon cancer, Benedetto et al demonstrated that four weeks of sunitinib treatment altered Tregs in tumor-bearing mice both quantitatively, by decreasing their numbers, and qualitatively, by inhibiting their ability to suppress T-cell proliferation.45 Studies on human HCC suggested that tumor-infiltrating CD4+CD69+Tregs suppress antitumor immune response via expressing membrane-bound TGF-β1.50 Stewart et al. further demonstrated that Tregs are the predominant source of IL-10 in the TME that can restrain Th17 responses.51,52 These comprehensive studies suggest that the immunosuppressive function of Tregs in HCC-bearing mice is associated with the increased production of immune suppressive cytokines. Compelling clinical data suggest that different immune cell subsets including Th1, Th2, Th17, and Tregs, carry out specialized immunoregulatory functions to either enhance or inhibit immune responses in cancer5,3 and autoimmunity.54 Considering the association of Th17 cells with the fibrosis in the liver, sunitinib might also be altering the frequency or function of Th17 cells toward a Th1-like anti-tumor response consistent with our prior findings of increased CD8+ T cell activity. However, the effect on T cell subset skewing remains to be determined.
In conclusion, our study provides cellular and molecular mechanistic insight into sunitinib-mediated prevention of tumor-driven immune suppression in HCC. The current findings raise the possibility that a combination of sunitinib with active immunotherapy might be effective in patients with HCC and deserves to be evaluated in human clinical trials.
Supplementary Material
Biographies
Dai Liu, MD, PhD, Postdoc, Department of Bioengineering Vanderbilt University, 5824B Stevenson Center, VU Mailbox: PMB 351631, Nashville, TN 37235-163, Tel: 615-343-1099, Email: dai.liu@vanderbilt.edu
Guangfu Li, PhD, DVM, Assistant Professor, Department of Surgery, Department of Molecular Microbiology and Immunology, University of Missouri-Columbia, One Hospital Dr., Medical Sciences Building, M272, Columbia, MO 65212, Tel: 573-882-7124, Fax: 573-884-4585, E-mail: liguan@health.missouri.edu
Diego Avella, MD, Thoracic Surgeon, Assistant Professor of Surgery, University of Missouri, MC520A Mchaney Hall, Columbia, MO 65212, Tel: 7176791297, Email: diegoma142@yahoo.com
Eric T. Kimchi, MD, Associate Professor, Department of Surgery, University of Missouri, MC520A Mchaney Hall, Columbia, MO 65212, Tel: (573) 882–8131, Email: kimchie@health.missouri.edu
Jussuf T. Kaifi, MD, PhD, Assistant Professor, Department of Surgery, University of Missouri, MC419 Mchaney Hall, Columbia, MO 65212, Tel: (573) 882–6956, Email: kaifij@health.missouri.edu
Mark P. Rubinstein, PhD, Assistant Professor, Department of Surgery, Medical University of South Carolina, 86 Jonathan Lucas Street, Charleston, SC 29425, Tel: (843)792-1451, Email: rubinsmp@musc.edu
E. Ramsay Camp, MD, Associate Professor, Surgical Oncology, Department of Surgery, Medical University of South Carolina, 114 Doughty St; Rm 241, Charleston, SC 29425, Tel: 843-876-4420, Fax: 843-876-3046
Don C. Rockey, MD, Professor and Chairman, Department of Internal Medicine, Medical University of South Carolina, 96 Jonathan Lucas Street, Suite 803, Charleston, SC 29425, Tel: 843-792-2914, Fax: 843-792-5265, Email: rockey@musc.edu
Kevin F. Staveley-O'Carroll, MD, PhD, Professor and Chair of Surgery, Director of Ellis Fischel Cancer Center, One Hospital Drive, Mc501, University of Missouri-Columbia, Columbia, MO 65212, TEL: 573-882-4158, FAX: 573-884-4585, E-mail: ocarrollk@health.missouri.edu
Todd D. Schell, PhD, Professor of Microbiology and Immunology, Pennsylvania State University College of Medicine, Mailing address: PO Box, 850, Dept. Microbiology & Immunology H107, Hershey, PA 17033, Tel: 717-531-8169, Email: tschell@psu.edu
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest
Acknowledgments
Grant Support: 1 R01 CA164335-01A1 (K. F. Staveley-O'Carroll, PI) from the National Cancer Institute/National Institutes of Health, R01 DK057830 (D. C. Rockey, PI) from the National Institute of Diabetes and Digestive Kidney Diseases /National Institutes of Health. We thank the staff of the Penn State College of Medicine Flow Cyometry Core for assistance.
Abbreviations
- ACT
adoptive cell transfer
- BMDC
bone marrow-derived dendritic cells
- B6/WT-19 cells
mouse embryonic fibroblast transformed with SV40 T antigen
- CCl4
carbon tetrachloride
- DC
dendritic cell
- DLN
draining lymph node
- HCC
hepatocellular carcinoma
- IP
Intraperitoneal
- IV
Intravenous
- MLR
mixed lymphocyte response
- MRI
magnetic resonance imaging
- nRCs
normal responder cells
- nTregs
normal Tregs
- SU
sunitinib
- TAg
SV40 T antigen
- TAS
tumor antigen-specific
- TCR-I
SV40 T antigen epitope I-specific TCR transgenic
- TME
tumor microenvironment
- Treg
regulatory T cell
- tRC
tumor responder cell
- TSA
tumor-specific antigen
- tTreg
tumor Treg
References
- 1.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245-1255. doi: 10.1016/S0140-6736(11)61347-0. PMID:22353262 [DOI] [PubMed] [Google Scholar]
- 2.Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology. 2015;61:191-199. doi: 10.1002/hep.27388. PMID:25142309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mavros MN, Mayo SC, Hyder O, Pawlik TM. A systematic review: treatment and prognosis of patients with fibrolamellar hepatocellular carcinoma. J Am Coll Surg. 2012;215:820-830. doi: 10.1016/j.jamcollsurg.2012.08.001. PMID:22981432 [DOI] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al.. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-390. [DOI] [PubMed] [Google Scholar]
- 5.Palmer DH. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:2498; author reply 2498–2499. PMID:19065750 [PubMed] [Google Scholar]
- 6.Copur MS. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:2498; author reply 2498–2499. PMID:19065701 [PubMed] [Google Scholar]
- 7.Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, Duran SD, Hernandez J, Seja E, Potter DM, et al.. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin Cancer Res. 2006;12:2817-2825. doi: 10.1158/1078-0432.CCR-05-2856. PMID:16675576 [DOI] [PubMed] [Google Scholar]
- 8.Butterfield LH, Ribas A, Potter DM, Economou JS. Spontaneous and vaccine induced AFP-specific T cell phenotypes in subjects with AFP-positive hepatocellular cancer. Cancer Immunol Immunother. 2007;56:1931-1943. doi: 10.1007/s00262-007-0337-9. PMID:17522860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sprinzl MF, Galle PR. Facing the dawn of immunotherapy for hepatocellular carcinoma. J Hepatol. 2013;59:9-10. doi: 10.1016/j.jhep.2013.04.002. PMID:23571018 [DOI] [PubMed] [Google Scholar]
- 10.Ho M, Kim H. Glypican-3: a new target for cancer immunotherapy. Eur J Cancer. 2011;47:333-338. doi: 10.1016/j.ejca.2010.10.024. PMID:21112773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mapara MY, Sykes M. Tolerance and cancer: mechanisms of tumor evasion and strategies for breaking tolerance. J Clin Oncol. 2004;22:1136-1151. doi: 10.1200/JCO.2004.10.041. PMID:15020616 [DOI] [PubMed] [Google Scholar]
- 12.Willimsky G, Schmidt K, Loddenkemper C, Gellermann J, Blankenstein T. Virus-induced hepatocellular carcinomas cause antigen-specific local tolerance. J Clin Invest. 2013;123:1032-1043. doi: 10.1172/JCI64742. PMID:23454765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buschow C, Charo J, Anders K, Loddenkemper C, Jukica A, Alsamah W, Perez C, Willimsky G, Blankenstein T. In vivo imaging of an inducible oncogenic tumor antigen visualizes tumor progression and predicts CTL tolerance. J Immunol. 2010;184:2930-2938. doi: 10.4049/jimmunol.0900893. PMID:20142365 [DOI] [PubMed] [Google Scholar]
- 14.Liu D, Staveley-O'Carroll KF, Li G. Immune-based Therapy Clinical Trials in Hepatocellular Carcinoma. J Clin Cell Immunol. 2015;6:376. doi: 10.4172/2155-9899.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korangy F, Hochst B, Manns MP, Greten TF. Immunotherapy of hepatocellular carcinoma. Expert Rev Gastroenterol Hepatol. 2010;4:345-353. doi: 10.1586/egh.10.18. PMID:20528121 [DOI] [PubMed] [Google Scholar]
- 16.Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res. 2005;65:2457-2464. doi: 10.1158/0008-5472.CAN-04-3232. PMID:15781662 [DOI] [PubMed] [Google Scholar]
- 17.Lin GH, Wang J, Li SH, Xu L, Li SP. Relationship and clinical significance of TGF-beta1 expression with Treg cell infiltration in hepatocellular carcinoma. Chin J Cancer. 2010;29:403-407. doi: 10.5732/cjc.009.10628. PMID:20346216 [DOI] [PubMed] [Google Scholar]
- 18.Chen X, Du Y, Huang Z. CD4+CD25+ Treg derived from hepatocellular carcinoma mice inhibits tumor immunity. Immunol Lett. 2012;148:83-89. doi: 10.1016/j.imlet.2012.09.002. PMID:23000301 [DOI] [PubMed] [Google Scholar]
- 19.Sakaguchi S, Powrie F. Emerging challenges in regulatory T cell function and biology. Science. 2007;317:627-629. doi: 10.1126/science.1142331. PMID:17673654 [DOI] [PubMed] [Google Scholar]
- 20.Coghill JM, Fowler KA, West ML, Fulton LM, van Deventer H, McKinnon KP, Vincent BG, Lin K, Panoskaltsis-Mortari A, Cook DN, et al.. CC chemokine receptor 8 potentiates donor Treg survival and is critical for the prevention of murine graft-versus-host disease. Blood. 2013;122:825-836. doi: 10.1182/blood-2012-06-435735. PMID:23798714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michelotti GA, Xie G, Swiderska M, Choi SS, Karaca G, Kruger L, Premont R, Yang L, Syn WK, Metzger D, et al.. Smoothened is a master regulator of adult liver repair. J Clin Invest. 2013;123:2380-2394. PMID:23563311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li G, Liu D, Cooper TK, Kimchi ET, Qi X, Avella DM, Li N, Yang QX, Kester M, Rountree CB, et al.. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J Hepatol. 2017;66(1):75–85. doi: 10.1016/j.jhep.2016.07.044. Epub 2016 Aug 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman VL, Rock EP, Dagher R, Ramchandani RP, Abraham S, Gobburu JV, Booth BP, Verbois SL, Morse DE, Liang CY, et al.. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res. 2007;13:1367-1373. doi: 10.1158/1078-0432.CCR-06-2328. PMID:17332278 [DOI] [PubMed] [Google Scholar]
- 24.Zhao X, Meng XY, Sun B, Ding LJ, Jiang ZF, Song ST, Wu SK. [Clinical observation of sunitinib treatment for refractory advanced breast cancer ulcer]. Zhonghua Yi Xue Za Zhi. 2013;93:96-98. PMID:23648343 [PubMed] [Google Scholar]
- 25.Faivre SJ, Bouattour M, Dreyer C, Raymond E. Sunitinib in hepatocellular carcinoma: redefining appropriate dosing, schedule, and activity end points. J Clin Oncol. 2009;27:e248-250; author reply e251-242. doi: 10.1200/JCO.2009.25.0670. PMID:19901099 [DOI] [PubMed] [Google Scholar]
- 26.Zhu AX, Sahani DV, Duda DG, di Tomaso E, Ancukiewicz M, Catalano OA, Sindhwani V, Blaszkowsky LS, Yoon SS, Lahdenranta J, et al.. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: a phase II study. J Clin Oncol. 2009;27:3027-3035. doi: 10.1200/JCO.2008.20.9908. PMID:19470923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Avella DM, Li G, Schell TD, Liu D, Zhang SS, Lou X, Berg A, Kimchi ET, Tagaram HR, Yang Q, et al.. Regression of established hepatocellular carcinoma is induced by chemoimmunotherapy in an orthotopic murine model. Hepatology. 2012;55:141-152. doi: 10.1002/hep.24652. PMID:21898502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tevethia SS, Greenfield RS, Flyer DC, Tevethia MJ. SV40 transplantation antigen: relationship to SV40-specific proteins. Cold Spring Harb Symp Quant Biol. 1980;44Pt 1:235-242. doi: 10.1101/SQB.1980.044.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Schirmacher P, Held WA, Yang D, Biempica L, Rogler CE. Selective amplification of periportal transitional cells precedes formation of hepatocellular carcinoma in SV40 large tag transgenic mice. Am J Pathol. 1991;139:231-241. PMID:1649555 [PMC free article] [PubMed] [Google Scholar]
- 30.Staveley-O'Carroll K, Schell TD, Jimenez M, Mylin LM, Tevethia MJ, Schoenberger SP, Tevethia SS. In vivo ligation of CD40 enhances priming against the endogenous tumor antigen and promotes CD8+ T cell effector function in SV40T antigen transgenic mice. J Immunol. 2003;171:697-707. doi: 10.4049/jimmunol.171.2.697. PMID:12847236 [DOI] [PubMed] [Google Scholar]
- 31.Domenicali M, Caraceni P, Giannone F, Baldassarre M, Lucchetti G, Quarta C, Patti C, Catani L, Nanni C, Lemoli RM, et al.. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J Hepatol. 2009;51:991-999. doi: 10.1016/j.jhep.2009.09.008. PMID:19853952 [DOI] [PubMed] [Google Scholar]
- 32.Teixeira-Clerc F, Julien B, Grenard P, Tran Van Nhieu J, Deveaux V, Li L, Serriere-Lanneau V, Ledent C, Mallat A, Lotersztajn S. CB1 cannabinoid receptor antagonism: a new strategy for the treatment of liver fibrosis. Nat Med. 2006;12:671-676. doi: 10.1038/nm1421. PMID:16715087 [DOI] [PubMed] [Google Scholar]
- 33.Fang X, Du P, Liu Y, Tang J. Efficient isolation of mouse liver NKT cells by perfusion. PLoS ONE. 2010;5:e10288. doi: 10.1371/journal.pone.0010288. PMID:20422018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu JZ, Wu XZ, Goel S, Gowda NM, Kumar S, Krishnegowda G, Mishra G, Weinberg R, Li G, Gaestel M, et al.. MAPK-activated Protein Kinase 2 Differentially Regulates Plasmodium falciparum Glycosylphosphatidylinositol-induced Production of Tumor Necrosis Factor-alpha and Interleukin-12 in Macrophages. J Bio Chem. 2009;284:15750-15761. doi: 10.1074/jbc.M901111200. PMID:19359247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah W, Yan X, Jing L, Zhou Y, Chen H, Wang Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell Mol Immunol. 2011;8:59-66. doi: 10.1038/cmi.2010.56. PMID:21200385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng LS, Zhuang Y, Shi Y, Zhao YL, Wang TT, Chen N, Cheng P, Liu T, Liu XF, Zhang JY, et al.. Increased tumor-infiltrating CD8(+)Foxp3(+) T lymphocytes are associated with tumor progression in human gastric cancer. Cancer Immunol Immunother. 2012;61:2183-2192. doi: 10.1007/s00262-012-1277-6. PMID:22729557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4:e989764. doi: 10.4161/2162402X.2014.989764. PMID:25949902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, et al.. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15:2148-2157. doi: 10.1158/1078-0432.CCR-08-1332. PMID:19276286 [DOI] [PubMed] [Google Scholar]
- 39.Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol Lett. 2013;150:116-122. doi: 10.1016/j.imlet.2012.12.004. PMID:23261718 [DOI] [PubMed] [Google Scholar]
- 40.Tagliamonte M, Petrizzo A, Tornesello ML, Ciliberto G, Buonaguro FM, Buonaguro L. Combinatorial immunotherapy strategies for hepatocellular carcinoma. Curr Opin Immunol. 2016;39:103-113. doi: 10.1016/j.coi.2016.01.005. PMID:26851637 [DOI] [PubMed] [Google Scholar]
- 41.Pardee AD, Butterfield LH. Immunotherapy of hepatocellular carcinoma: Unique challenges and clinical opportunities. Oncoimmunology. 2012;1:48-55. doi: 10.4161/onci.1.1.18344. PMID:22720211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, Chung HC, Song X, Xu J, Poggi G, et al.. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J Clin Oncol. 2013;31:4067-4075. doi: 10.1200/JCO.2012.45.8372. PMID:24081937 [DOI] [PubMed] [Google Scholar]
- 43.Jaini R, Rayman P, Cohen PA, Finke JH, Tuohy VK. Combination of sunitinib with anti-tumor vaccination inhibits T cell priming and requires careful scheduling to achieve productive immunotherapy. Int J Cancer. 2014;134:1695-1705. doi: 10.1002/ijc.28488. PMID:24105638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2011;129:2158-2170. doi: 10.1002/ijc.25863. PMID:21170961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farsaci B, Higgins JP, Hodge JW. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int J Cancer. 2012;130:1948-1959. doi: 10.1002/ijc.26219. PMID:21633954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514-2522. doi: 10.1158/0008-5472.CAN-08-4709. PMID:19276342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301-6311. doi: 10.1158/1078-0432.CCR-07-1403. PMID:17975141 [DOI] [PubMed] [Google Scholar]
- 48.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506-2513. doi: 10.1158/0008-5472.CAN-08-4323. PMID:19244102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, et al.. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674-6682. doi: 10.1158/1078-0432.CCR-07-5212. PMID:18927310 [DOI] [PubMed] [Google Scholar]
- 50.Han Y, Yang Y, Chen Z, Jiang Z, Gu Y, Liu Y, Xu S, Lin C, Pan Z, Zhou W, et al.. Human hepatocellular carcinoma-infiltrating CD4(+)CD69(+)Foxp3(−) regulatory T cell suppresses T cell response via membrane-bound TGF-beta1. J Mol Med (Berl). 2014;92:539-550. doi: 10.1007/s00109-014-1143-4. PMID:24668348 [DOI] [PubMed] [Google Scholar]
- 51.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674. doi: 10.1016/j.cell.2011.02.013. PMID:21376230 [DOI] [PubMed] [Google Scholar]
- 52.Stewart CA, Metheny H, Iida N, Smith L, Hanson M, Steinhagen F, Leighty RM, Roers A, Karp CL, Müller W, et al.. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J Clin Invest. 2013;123:4859-4874. doi: 10.1172/JCI65180. PMID:24216477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cabrera R, Szabo G. Another Armed CD4(+) T Cell Ready to Battle Hepatocellular Carcinoma. Hepatology. 2013;58:1-3. doi: 10.1002/hep.26377. PMID:23475554 [DOI] [PubMed] [Google Scholar]
- 54.Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ-specific autoimmunity. J Autoimmunity. 2008;31:252-256. doi: 10.1016/j.jaut.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.