

ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) is often diagnosed when liver metastases already emerged. This study elucidated the impact of hepatic stromal cells on growth behavior of premalignant and malignant pancreatic ductal epithelial cells (PDECs). Liver sections of tumor-bearing KPC mice comprised micrometastases displaying low proliferation located in an unobtrusive hepatic microenvironment whereas macrometastases containing more proliferating cells were surrounded by hepatic myofibroblasts (HMFs). In an age-related syngeneic PDAC mouse model livers with signs of age-related inflammation exhibited significantly more proliferating disseminated tumor cells (DTCs) and micrometastases despite comparable primary tumor growth and DTC numbers. Hepatic stellate cells (HSC), representing a physiologic liver stroma, promoted an IL-8 mediated quiescence-associated phenotype (QAP) of PDECs in coculture. QAP included flattened cell morphology, Ki67-negativity and reduced proliferation, elevated senescence-associated β galactosidase activity and diminished p-Erk/p-p38-ratio. In contrast, proliferation of PDECs was enhanced by VEGF in the presence of HMF. Switching the micromilieu from HSC to HMF or blocking VEGF reversed QAP in PDECs. This study demonstrates how HSCs induce and maintain a reversible QAP in disseminated PDAC cells, while inflammatory HMFs foster QAP reversal and metastatic outgrowth. Overall, the importance of the hepatic microenvironment in induction and reversal of dormancy during PDAC metastasis is emphasized.
KEYWORDS: dormancy, pancreatic cancer, tumor stroma, metastases, inflammation
Abbreviations
- α-SMA
alpha-smooth muscle actin
- DTCs
Disseminated tumor cells
- HMF
M-HT, Hepatic myofibroblast cell line
- HMFs
hepatic myofibroblast
- HSC
M1-4HSC, Hepatic stellate cell line
- HSCs
hepatic stellate cells
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- PDECs
Pancreatic ductal epithelial cells
- QAP
quiescence-associated phenotype
- SABG
Senescence-associated beta-galactosidase
- TGF-β1
Transforming Growth Factor-beta1
- VEGF
Vascular Endothelial Growth Factor
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth most common cause of cancer-related deaths worldwide. Its high metastatic potential, profound chemoresistance and the absence of specific clinical symptoms manifest in a dismal prognosis. The 5-year survival rate is <8%.1 Median age at diagnosis is 71 years.2 Known risk factors for PDAC development are advanced age, lifestyle factors such as smoking or alcoholic abuse, long lasting chronic pancreatitis and type-2 diabetes mellitus (summarized in3). Surgical resection of the primary tumor currently represents the only curative therapy. However, ∼80% patients are diagnosed at an already advanced, often metastasized and hence inoperable stage. Commonly even patients undergoing successful R0 resection develop liver metastases shortly after surgery.3,4,5
PDAC may evolve directly from ductal cells progressing to PDAC via Pancreatic Intraepithelial Neoplasia (PanIN) or from acinar cells, which underwent acinar-to-ductal metaplasia before PanIN formation.3 PanIN progression is accompanied by successive accumulation of mutations in several oncogenes and tumor suppressor genes, e.g. KRAS, CDKN2 A, P53, with KRAS mutation being one of the earliest genetic alterations.6
The liver is the predominant target organ of PDAC metastasis. Recent studies provide evidence that dissemination of pancreatic ductal epithelial cells (PDECs) to the liver may already commence at PanIN stages.7 Outgrowth of early disseminating PDECs to overt metastases, however, demands acquisition of further pro-metastatic traits.7,8,9 Moreover, several studies demonstrated that factors deriving from tumor cells of primary lesions such as TIMP metallopeptidase inhibitor 1 (TIMP1) or exosomes containing chemokines or cytokines modulate the liver microenvironment and act to generate a pre-metastatic niche.10,11,12
The activation and transdifferentiation of hepatic stellate cells (HSCs) into hepatic myofibroblasts (HMFs) by e.g., transforming growth factor (TGF)-β1, or fibroblast growth factor-2 (FGF2) is considered a crucial component of this process.10,13 HSCs are of particular interest as they are – after activation and transdifferentiation into HMFs – key effectors of inflammatory processes in the liver.14 HMFs are characterized by elevated release of extracellular matrix (ECM) molecules and cytokines, e.g., Interleukin-6 (IL)-6 and TGF-β1 which foster pre-metastatic niche formation.10,15 Similar inflammation-mediated alterations of the hepatic microenvironment might be induced by Inflammaging, which is defined as aging-related smoldering inflammation.16,17,18
While a profound influence of inflammatory processes on primary PDAC development is well appreciated, the impact of the hepatic microenvironment on regulation of survival and growth behavior of disseminated PDECs is insufficiently understood. Several studies on other tumor entities support the view that disseminated tumor cells (DTCs) can persist in secondary sites in a viable but non-dividing state thereby remaining clinically unobtrusive and undetectable for extended time periods.19,20 This reversible state of quiescence is termed dormancy in which tumor mass dormancy can be distinguished from cellular dormancy, the latter implying a reversible growth arrest of solitary cells. Hallmarks of cellular dormancy are a flattened cell morphology, Ki67-negativity, reduced ratio of phosphorylated ERK (p-ERK) to phosphorylated p38 (p-p38) and increased p21 expression.19,20,21 All these features are also characteristics of senescent cells, which additionally exhibit an elevated senescence-associated β-galactosidase (SABG) activity.20,22 The acquisition of further mutations (e.g., in P53) as well as microenvironmental alterations may induce reawakening of dormant cells leading to local relapse and/or outgrowth of metastases even after curative therapy.23 A recent study strongly supports the role of inflammation in escape from tumor latency.24 However, underlying mechanisms leading to such microenvironmental alterations and favoring dormancy reversal in PDAC are poorly understood.
Using endogenous and syngeneic PDAC mouse models as well as in vitro coculture systems, we investigated the role of the hepatic microenvironment as a determinant for growth behavior of disseminated premalignant and malignant PDECs in the liver and identify mechanisms underlying the stroma-mediated re-induction of tumor cell proliferation. Overall, this study provides novel insights into the mechanisms underlying metastatic outgrowth of PDAC. This improved understanding of metastatic processes in PDAC is mandatory for the development of more effective screening and therapeutic strategies for this highly malignant tumor.
Results
Proliferative activity of PDAC cells in liver metastases correlates with the presence of HSCs or HMFs
To investigate whether the size and proliferative activity of liver metastases correlate with the abundance of HSCs or HMFs, liver sections of KPC mice were examined for the presence of micrometastases (lesion diameter ≤ 200 μm) and macrometastases (lesion diameter > 200 µm), their Ki67 status and the ratio of HMFs to HSCs in the direct lesion surrounding. Immunohistochemical stainings of serial liver tissue sections revealed the occurrence of micrometastases with a low proliferative activity of PDAC cells (Median score 2 ≙ 10–50% Ki67+ cells) predominantly in areas with low ratios of HMFs to HSCs as determined by staining of α-SMA (for HMFs) and desmin (for HSCs) (Median αSMA/desmin ratio score = 1) (Fig. 1, Supplementary Figure 1A). Concurrently, macrometastases containing a significantly higher amount of proliferative CK19+ cells (Median score 3 ≙ 50–100% Ki67+ cells) were mostly detected in liver areas with high presence of HMFs (Median αSMA/desmin ratio score = 3) (Fig. 1, Supplementary Figure 1B). Overall, these data suggest that the size and proliferative activity of PDAC liver metastases correlates with the abundance of HMFs in the metastasis stroma.
Figure 1.
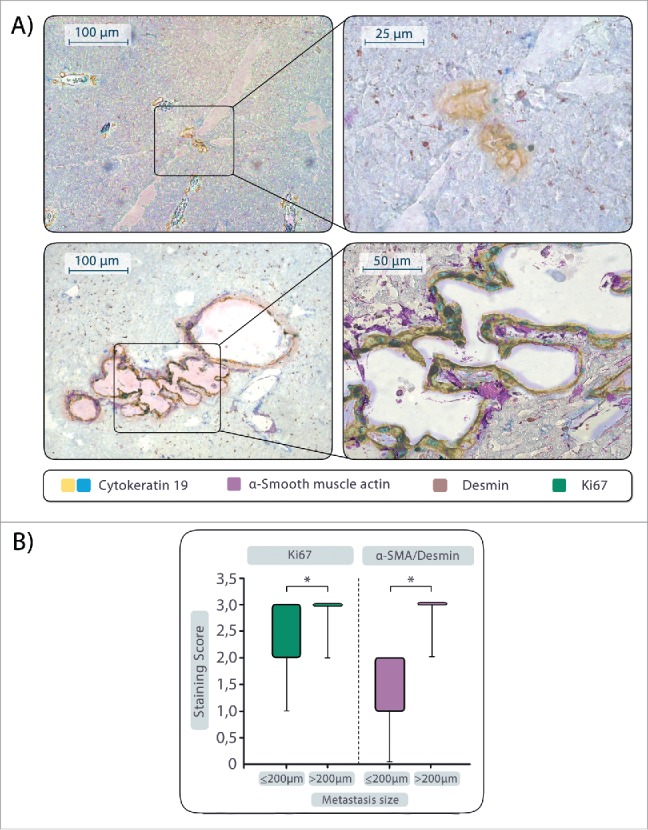
Proliferative activity of PDAC cells in liver metastases correlates with presence of hepatic stellate cells (HSCs) or hepatic myofibroblasts (HMFs). Liver sections of mice harboring endogenous advanced PDAC (n = 13) were examined for the presence of micrometastases (lesion diameter ≤ 200 μm) and macrometastases (lesion diameter > 200 µm) by staining of Cytokeratin-19, Ki67, α-SMA (for detection of HMFs) and desmin (for detection of HSCs). (A) Representative images of a micrometastasis (top) and a macrometastasis (bottom) are shown after overlay of all stainings performed in serial sections. B) Scoring of Ki67 and determination of the αSMA/desmin score in micro- and macrometastases. Data represent median values with quartiles (Q0,75 as upper, Q0.25 as lower deviation) of 7 micro- and 9 macrometastases. * = p< 0.05.
Aged livers exhibit enhanced outgrowth of PDAC metastases
To further elucidate whether growth behavior and metastatic outgrowth of PDAC cells is determined by the condition of the liver microenvironment, a syngeneic PDAC mouse model was deployed in which aging was chosen as a trigger of inflammation-associated hepatic alterations.25,26,27 R254 PDAC cells were inoculated into the pancreas of mice aged either 8 or 52 weeks, the latter being used to model higher age of humans at the time point of PDAC diagnosis. Two weeks after tumor cell inoculation, both groups exhibited similar outgrowth of primary tumors (7.2 mm3 in 8 weeks old mice versus 6.0 mm3 in 52 weeks old mice) (Fig. 2A). While similarly low numbers of DTCs were detected in liver tissues of both groups (0.4 vs. 0.5 DTCs/view field) (Fig. 2B), a significantly higher number of DTCs was Ki67+ in the livers of aging mice (23.2% in 8 weeks old mice vs. 62.1% in 52 weeks old mice, Fig. 2C). Due to their low abundance and thus the negligible impact of DTCs on the microenvironment, liver tissues were used to assess signs of aging-related inflammation. PCR based analysis demonstrated a slightly elevated αSMA/desmin ratio (1.4 in 8 weeks old mice vs. 1.7 in 52 weeks old mice)(Supplementary Fig. 2) along with elevated gene expression levels of FGF-2, IL-1β, TGF-β1 and significantly increased VEGF-A expression in aged livers (Fig. 2D). These factors are known to be released at higher levels by HMFs than by HSCs13,28,29 Surprisingly, livers of young mice expressed higher levels of the murine functional IL-8 homologues MIP-2 and LIX compared with old mice (Fig. 2D). Four weeks after tumor cell inoculation, aged mice showed slightly larger primary tumors compared with young mice (Fig. 2E), whereas significantly higher numbers of DTCs (2.1 vs. 4 DTCs/view field, Fig. 2F) and micrometasteses, defined as clusters of more than 5 tumor cells (0 vs. 3 micrometastases/liver section, Fig. 2G), were detected in livers of aging mice. Although proliferation of DTCs was comparable (Fig. 2H), micrometastases were only found in aged livers (Fig. 2G). Overall, these data support the view that aging-related smoldering inflammation fosters PDAC outgrowth in pancreas and liver with more pronounced effects in the hepatic microenvironment.
Figure 2.

Enhanced outgrowth of PDAC metastases in aged livers. GFP-expressing R254 PDAC cells were injected into pancreata of C57BL/6J mice aged either 8 or 52 weeks. Two and 4 weeks after injection, animals were analyzed for presence of primary and secondary lesions by ultrasound before pancreata and livers were resected and analyzed via immunofluorescence staining of GFP (to detect R254 cells) and Ki67. (A) Size of primary tumors, (B) number of DTCs/view field and (C) Ki67+GFP+ DTCs in livers and (D) expression of inflammation- and HMF-related genes 2 weeks after tumor cell inoculation. (E) Size of primary tumors, (F) number of GFP+ DTCs/view field and (G) number of GFP+ micrometastases (clusters of minimum 5 cells) in livers along with representative images of mouse livers showing GFP+ DTCs and micrometastasis (marked in white circles) at 400-fold magnification. H) number of Ki67+GFP+ DTCs 4 weeks after tumor cell inoculation. Data represent the median values with quartiles (Q0,75 as upper, Q0.25 as lower deviation) or mean ± SD in 10 animals/group. * = p < 0.05.
Presence of HSC promotes the enrichment of PDECs with a quiescence-associated phenotype (QAP)
To further unravel the mechanisms by which HSCs and their inflammatory counterpart HMFs impact on growth behavior of PDECs, an indirect coculture system was applied. For this purpose, premalignant H6c7-kras cells (resembling PDECs disseminating at an early stage of PDAC development) and malignant Panc1 cells (to model late dissemination from established PDAC) were cultivated in the presence of either M1–4HSC cells (HSC) or M-HT cells (HMF) for 6 d. The presence of HSC resulted in lower viable cell numbers in both PDEC lines compared with the respective HMF-coculture (Fig. 3A). Determination of caspase-3/7 activity and Ki67 staining demonstrated that these effects were not due to differences in apoptosis induction (Supplementary Fig. 3), but stromal cell-mediated changes in the proliferative activity of PDECs (Fig. 3B), which was significantly lower under HSC-coculture (15.9% vs. 42.9% for H6c7-kras and 19.2% vs. 49.8% for Panc1). Moreover, HSC-coculture resulted in a higher emergence of PDECs characterized by Ki67-negativity and a flattened enlarged morphology (Fig. 3C), features being associated with both cellular senescence and dormancy21,22,23 According with this QAP, the ratio of phosphorylated ERK (p-ERK) to phosphorylated p38 (p-p38) was found decreased in HSC-cocultured PDECs compared with HMF-cocultured PDECs (Fig. 3D). In addition, particularly Panc1 cells showed a markedly higher expression of the cell cycle inhibitor p21 further supporting HSC-mediated growth arrest (Fig. 3D). Moreover, a significantly higher abundance of SABG+ H6c7-kras and Panc1 cells was found after HSC-coculture (H6c7-kras: 69.2%, Panc1: 45.3%) compared with HMF-coculture (H6c7-kras: 30.2%, Panc1: 12.3%) (Fig. 3E). Of note, HSC- and HMF-mediated effects were also visible in comparison to monocultured PDECs, further supporting the differential impact of these hepatic stromal cells (Supplementary Fig. 4). Finally, key findings were confirmed in another coculture setting in which both PDEC lines were cocultured with human liver derived stellate cells (HHSteC) that were either stimulated with all-trans retinoic acid (ATRA) (HHSteC-HSC, resembling the HSC phenotype) or TGF-β1 (HHSteC-HMF, resembling the HMF phenotype) before coculture (Supplementary Fig. 5). Overall, these findings indicate that the presence of HSC promotes a QAP in PDECs including flattened morphology, Ki67-negativity, SABG-positivity and a decreased p-Erk/p-p38 ratio.
Figure 3.
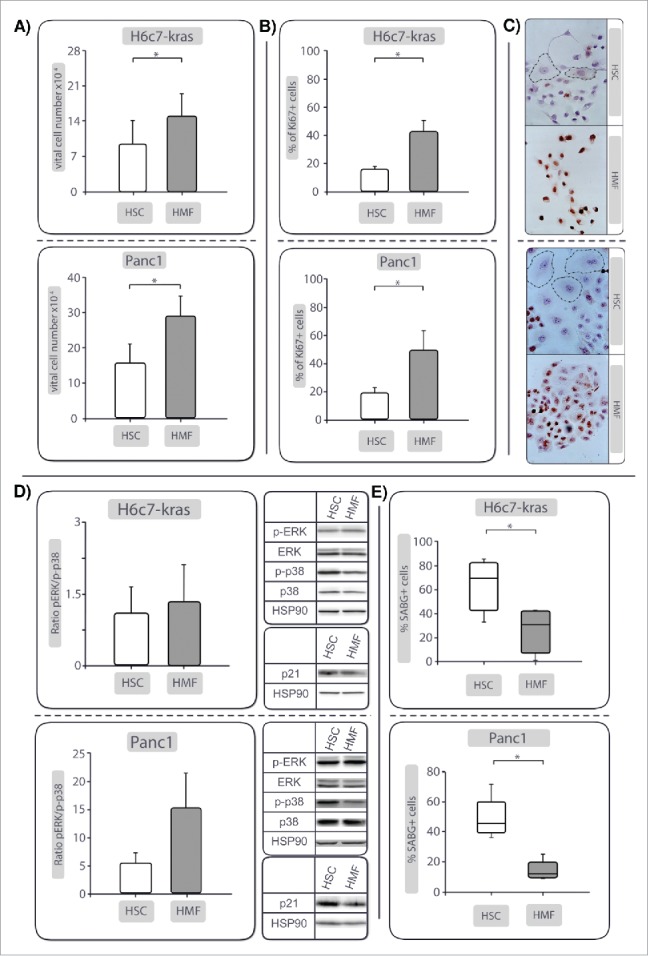
PDECs exhibit a reduced proliferative activity and quiescence-associated phenotype (QAP) in the presence of HSC compared with HMF. H6c7-kras and Panc1 cells were indirectly cocultured either in the presence of HSC or HMF. After 6 days, (A) viable cell numbers, and (B) percentage of Ki67+ cells were determined. (C) shows representative images of Ki67 stainings in H6c7-kras and Panc1 cells under different coculture conditions shown at 400-fold magnification (encircled: PDECs exhibiting flattened enlarged morphology). (D) Representative Western blots showing expression of total and phosphorylated Erk (Erk/p-Erk) and p38 (p38/p-p38) and p21. Hsp90 was used as loading control. Data of densitometric analysis of the ratio of p-Erk and p-p38 expression are presented. E) shows the percentage of SABG+ cells after coculture. Data shown in A, (B)and (D)are presented as mean ± SD of 4–5 independent experiments. Data shown in (E) represent median values with quartiles (Q0,75 as upper, Q0.25 as lower deviation) of 5 independent experiments. * = p< 0.05.
HSC-mediated QAP in H6c7-kras and Panc1 cells is reversed in the presence of HMF
To test whether the presence of inflammatory cells like HMFs reverts HSC-mediated QAP of PDECs, coculture of H6c7-kras and Panc1 cells was prolonged for further 6 d in the presence of fresh HSC or HMF, respectively. Proliferation of PDECs was surveilled by real-time Life Cell Imaging, particularly focusing on cells with QAP-associated flattened and enlarged morphology (Fig. 3C). While enlarged PDECs remained mainly non-proliferating under continuous HSC-coculture (HSC-HSC) (Fig. 4A, Supplementary Video 1 A), the number of dividing cells showing a flattened, enlarged QAP-morphology was clearly higher when conditions were changed to HMF-coculture. This effect was most pronounced in Panc1 cells (Fig. 4B, Supplementary Video1B). Subsequent Ki67-stainings showed the occurrence of PDECs maintaining an enlarged QAP-morphology but being clearly Ki67+ (Fig. 4A-C). Intriguingly, cells with QAP-morphology stained positive for SABG even after complete cell division (Supplementary Fig. 6). In the rare case of PDECs reacquiring proliferative capacity in the presence of HSC, these cell divisions occurred notably slower or were directly followed by cell death (Supplementary Video 1A). Overall, these results suggest that the temporary growth arrest of PDECs can be reversed in the presence of inflammatory hepatic stromal cells leading to enhanced proliferation and cell growth. Moreover, these data imply a role of the hepatic microenvironment in the maintenance and reversal of dormancy in PDAC.
Figure 4.
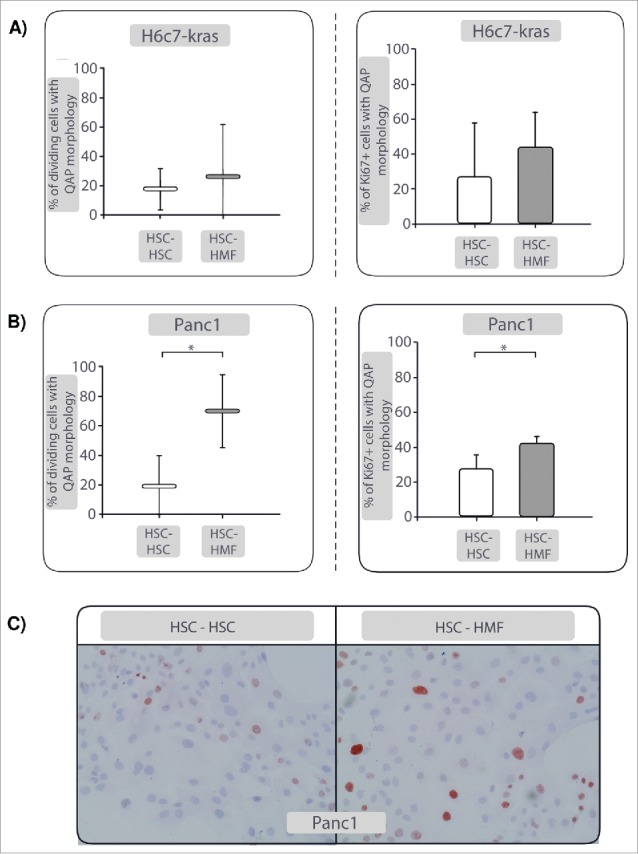
The quiescence-associated phenotype (QAP) of PDECs can be reversed in the presence of HMF. After 6 d coculture with HSC, coculture of H6c7-kras and Panc1 cells was prolonged for further 6 d in the presence of fresh HSC (HSC-HSC) or HMF (HSC-HMF). The percentage of dividing cells with a flattened, enlarged QAP-morphology as determined by real-time Life Cell Imaging and the percentage of Ki67+ dividing cells with flattened, enlarged QAP-morphology cells in (A) H6c7-kras and (B) Panc1 cells are depicted. (C) Representative images of Ki67 stainings in Panc1 cells after HSC-HSC- and HSC-HMF-coculture are shown at 400-fold magnification. Data in Aand (B) represent median values with quartiles (Q0,75 as upper, Q0.25 as lower deviation) or mean ± of 5 independent experiments. * = p < 0.05.
Identification of factors involved in hepatic stroma-mediated effects on PDECs growth behavior
To elucidate factors being involved in the stroma-mediated growth behavior of PDECs, supernatants of HSC- or HMF-cocultures were analyzed for the abundance of soluble inflammatory mediators by Luminex multiplex array. Factors showing a considerably higher abundance in supernatants of HSC-cocultured Panc1 cells were mIL-6 (1000.4 pg/ml under HSC vs. 124.8 pg/ml under HMF) and hIL-8 (244.8 pg/ml under HSC vs. 192.5 pg/ml under HMF) (Fig. 5A, B) pointing to a role of these factors in the mediation of QAP in Panc1 cells. In contrast, mVEGF (722,4 pg/ml under HSC vs. 1049,8 pg/ml under HMF) and hVEGF (348,8 pg/ml under HSC vs. 594,2 pg/ml under HMF) were elevated in supernatants of Panc1 cells cocultured with HMF (Fig. 5C, D) suggesting that these factors might be responsible for increased proliferation and QAP reversal mediated by HMF. Similar results were observed in supernatants of cocultured H6c7-kras cells (data not shown) supporting the role of these factors in induction and reversion of QAP in PDECs in the hepatic microenvironment.
Figure 5.
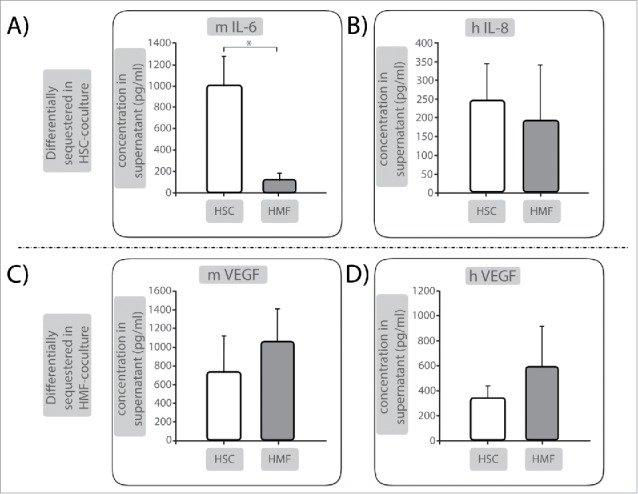
Identification of factors involved in hepatic stroma-mediated effects on PDECs growth behavior. Supernatants of Panc1 cells cocultured either in the presence of HSC or HMF for 6 d were analyzed by Luminex multiplex analysis. (A) Murine (m) IL-6 and (B) human (h) IL-8 as well as (C) m VEGF and (D) h VEGF were detected at differential levels in supernatants. Data are presented as mean ± SD of 3 independent experiments. * = p < 0.05.
IL-8 is important for HSC-mediated QAP in PDECs
Since mIL-6 and hIL-8 were elevated under HSC-coculture, the ability of either factor to mediate QAP in PDECs was investigated. While blocking of IL-6 signaling by different means (blocking human or murine IL-6 receptors, neutralizing soluble human and murine IL-6 and specific inhibition of IL-6 trans signaling) did not influence the QAP in Panc1 cells (data not shown), application of a neutralizing anti-IL-8 antibody during HSC-coculture increased the number of viable cells by 31% (Fig. 6A) and concomitantly decreased the percentage of SABG+ cells by 34% in Panc1 cells (Fig. 6B). This was associated with a clearly increased p-Erk/p-p38 ratio (Fig. 6C) compared with control treatment. In summary, these data identify IL-8 as QAP-mediating factor in PDECs.
Figure 6.
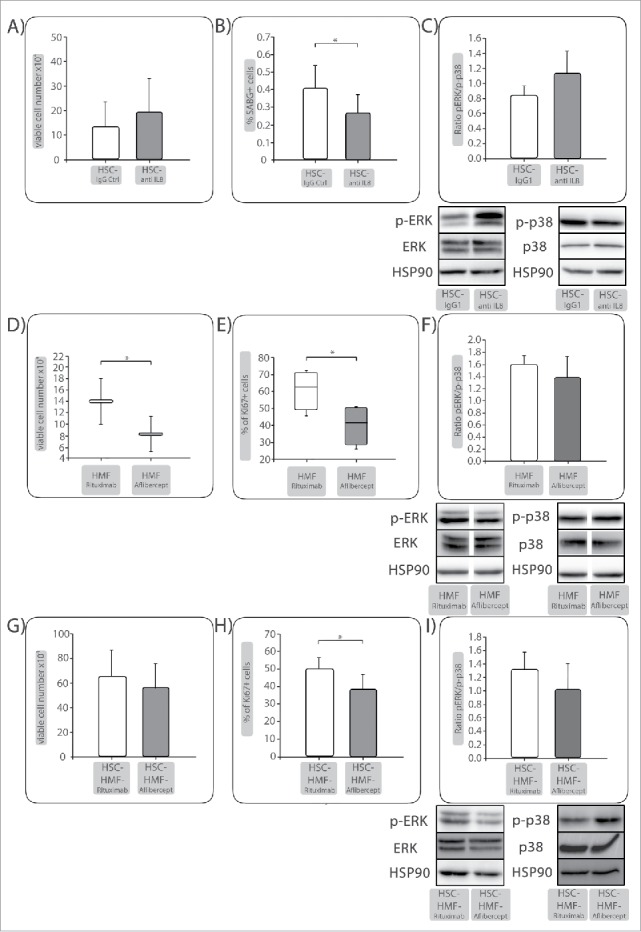
IL-8 is important for HSC-mediated QAP in PDECs while VEGF is a proliferation promoting factor under HMF-coculture. Panc1 cells were indirectly cocultured with (A-C) HSC and either treated with control IgG or an anti-IL-8 antibody. (D-F) Panc1 cells were indirectly cocultured with HMF and either treated with Rituximab as control or Aflibercept to neutralize VEGF. After 6 days, A&D) viable cell numbers and (B) percentage of SABG+ cells or (E) percentage of Ki67+ cells were determined, depicted as mean ± SD or median values with quartiles (Q0,75 as upper, Q0.25 as lower deviation) of 4–5 independent experiments. C+F) Representative Western blots showing expression of phosphorylated Erk (p-Erk) and p38 (p-p38). Hsp90 or Tubulin were used as loading control as appropriate. Data of densitometric analysis of the ratio of p-Erk and p-p38 expression are presented as mean ± SD of 3–4 independent experiments. After 6 d coculture with HSC, coculture of Panc1 cells was prolonged for further 6 d in the presence of HMF (HSC-HMF). (G) viable cell numbers and (H) percentage of Ki67+ cells were determined. I) Representative Western blots showing expression of phosphorylated Erk (p-Erk) and p38 (p-p38). Hsp90 were used as loading control as appropriate. Data of densitometric analysis of the ratio of p-Erk and p-p38 expression are presented as mean ± SD of 3 independent experiments. * = p < 0.05.
VEGF is a proliferation-promoting factor under HMF-coculture
Next, it was investigated whether elevated VEGF levels detected under HMF-coculture might account for enhanced proliferation of PDECs. VEGF activity was blocked during 6 d of HMF-coculture by Aflibercept (neutralizing both, murine and human VEGF) while Rituximab was used as a control. As shown in Fig. 6D, Aflibercept significantly reduced the number of viable Panc1 cells under HMF-coculture by 41%. Accordingly, Aflibercept treated cells exhibited significantly reduced proliferation (Fig. 6E) and reduced p-Erk/p-p38 ratio (Fig. 6F). Furthermore, Aflibercept treatment under HMF-coculture following HSC-coculture also reduced the number of viable (Fig. 6G) and proliferating (Fig. 6H) cells which was associated with a diminished p-Erk/p-p38 ratio compared with control treatment (Fig. 6I). Overall, these data show a role of VEGF as proliferation-promoting and QAP reverting factor of PDECs in the context of an inflamed hepatic microenvironment.
Discussion
This is the first study providing clear evidence that aging-related smoldering inflammation of the liver promotes the metastatic outgrowth of dormant pancreatic DTCs. These results are in line with recent studies on the pro-metastatic impact of inflammation in the secondary context.10,11 In an age-related syngeneic PDAC mouse model, 8 and 52 weeks aged mice exhibited comparable primary tumor sizes and numbers of DTCs in the liver 2 weeks after tumor cell inoculation. In contrast, significantly higher abundances of proliferating DTCs were observed in the livers of aging mice, which were also characterized by an elevated inflammatory cytokine expression. Four weeks post-injection, micrometastases were detected exclusively in livers of aged mice, whereas their primary tumors were only slightly larger. Furthermore, indirect coculture experiments revealed that HSC, representing a physiologic liver microenvironment, promoted a QAP in PDECs which was conferred by IL-8. In contrast, proliferation of PDECs was significantly enhanced in a VEGF-dependent manner in the presence of HMF – the inflammatory counterpart of HSC. Finally, real-time Life Cell Imaging showed that QAP can be reversed by changing hepatic stroma conditions from HSC to HMF as well as by blocking VEGF. These findings support the view that inflammation-associated alterations of the liver microenvironment promote reversal of dormancy as well as DTC proliferation and thereby foster metastatic outgrowth of PDAC as it has recently been shown in a corresponding breast cancer study.24
While De Cock et al. provoked inflammation of lungs by application of lipopolysaccharide leading to reversal of dormancy and metastasis formation, this study emphasizes the impact of aging as an inflammatory trigger for metastatic outgrowth. Inflammaging is defined as smoldering inflammation-associated with aging.16 Accordingly, analyses of C57BL/6J mice of different age, also used in our study, demonstrated that aged livers are characterized by elevated levels of inflammatory cytokines such as IL-6, IL-1β and TNF-α.26 Moreover, increased levels of immune cell attractant chemokines along with higher numbers of macrophages, T- and B lymphocytes as well as neutrophils can be detected in aged livers, too.25 Livers of aged mice used in our study were even older than in the above mentioned studies and predominantly characterized by elevated levels of FGF-2, IL-1β, TGF-β1 and VEGF-A, while the young mice showed higher expression levels of the IL-8 homologues MIP-2 and LIX. Two recent studies demonstrated that the release of exosomes or factors such as TIMP1 by tumor cells of the primary tumor led to comparable alterations, thereby initiating the pre-metastatic niche.10,11 Moreover, macrophages seem to play a central role in this process as they essentially promote HSC-activation10,12 which represents the ultimate step in remodeling of the hepatic microenvironment/pre-metastatic niche formation independent of the inflammatory trigger. Based on latter 2 studies, initiation of the pre-metastatic niche seems not to occur before primary PDAC establishment while the study by Grünwald et al. supports the view that modification of the hepatic microenvironment may commence earlier. They showed that TIMP1, one essential factor for HSC-activation and thereby niche preparation, is elevated in plasma of patients with PDAC but also with chronic pancreatitis and in KPC mice at PanIN stages.11 Furthermore, these studies also indicate that inflammatory alterations in the secondary context precede seeding of DTCs and are thus critical for their homing into the liver. However, since DTCs do not immediately outgrow to overt metastases after seeding, the question arises which factors trigger the reversion of dormancy. Thus, our findings extend these studies identifying aging as an important inflammatory trigger to promote outgrowth of PDAC metastases in the hepatic microenvironment. Our results also support the view that disseminated PDECs seed to the liver and are kept dormant by a physiologic liver microenvironment. Then, their awakening and outgrowth to visible metastases is caused by a switch to a smolderingly inflamed status of the liver caused by aging. This inflammatory switch, however, may also be mediated by exposure to certain lifestyle factors (e.g., alcohol, cigarette smoke) or factors derived from the primary context. Several studies describe that even patients diagnosed with resectable PDAC most likely exhibit a disseminated stage which cannot be detected by current imaging modalities.30,31,32 Accordingly, we detected DTCs in the liver when primary tumors were still quite small. Moreover, our study identifies aging as a promoter of PDAC outgrowth with minor effects in the primary context and stronger impact on the hepatic microenvironment. The fact that not the number of DTCs but only the proliferation of seeded DTCs was higher in livers of aged mice 2 weeks after tumor cell inoculation indicates that aging-related alterations of the liver did not impact homing to the secondary context but rather growth behavior of DTCs after having seeded into the organ. This view is in line with findings of Rhim et al. demonstrating in the KC mouse model that PDECs (solely exhibiting mutated KRAS but no other PDAC-related mutations) acquire the ability to disseminate from the pancreas and seed to the liver already at PanIN stages but fail to grow out to metastases.7 Outgrowth appears to require the acquisition of further genetic and epigenetic alterations (e.g., in TP53, CDKN2 A or SMAD4) and/or alterations in the microenvironment.7,8,9,33 In line with these findings, HSC-mediated acquisition of QAP was observed in H6c7-kras and Panc1 cells and was thus independent of the TP53 status. In contrast, HMF-mediated QAP reversal appears to depend on the mutational status of TP53 as HMF mediated regain of proliferative activity was most pronounced and fastest in Panc1 cells and slower or incomplete in H6c7-kras or Colo357 cells (data not shown). The fact that PDECs with QAP-morphology still stained positive for SABG even after complete cell division supports the view that this stromal-mediated QAP is a reversible form of senescence. In contrast to most other markers that are common in both, dormancy and senescence, SABG activity is considered a characteristic of senescent cells only.34 Moreover, these findings imply the possibility that the temporary growth arrest of PDECs which enables them to survive in the hepatic microenvironment could also represent a state of reversible senescence.
Elevated levels of proinflammatory cytokines can be part of the senescence-associated secretory phenotype (SASP).35 Our studies identified IL-8 not only as part of the SASP but also as an inducer of the QAP in PDECs, since IL-8 neutralization during HSC-coculture significantly increased the number of viable cells while preventing the appearance of cells with QAP. Accordingly, young mice showed higher expression levels of the murine IL-8 homologues MIP-2 and LIX. In contrast to studies which report tumor promoting effects of these factors in PDAC36,37 our study suggests that they may also have a dormancy maintaining impact on disseminating PDECs in the liver. Interestingly, HSC-cocultured PDECs also exhibited stem cell properties as indicated by enhanced stem cell marker expression and increased colony formation (unpublished observations). Finally, in search for the proliferation-promoting factor under HMF-coculture we identified VEGF (murine and human derived) being highly elevated under these conditions. In line with a previous study in which VEGF inhibition increased cellular senescence in colorectal cancer cells,38 application of Aflibercept (neutralizing both murine and human VEGF) significantly reduced the number of viable and proliferating PDECs cells under HMF-coculture, pointing to a role of VEGF as proliferation promoting trigger in an inflamed hepatic microenvironment. Accordingly, we detected significantly elevated VEGF-A RNA levels in livers of aged mice compared with young mice. It was previously shown that abdominal surgery increases systemic levels of inflammatory mediators thereby promoting local relapses and formation of PDAC metastases.39 Thus, we cannot rule out that the observed elevated gene expression of inflammatory mediators in aged livers is solely caused by higher age of mice, as the observed changes may reflect a stronger inflammatory reaction caused by the surgery performed for orthotopic inoculation of tumor cells. It has to be mentioned that clinical application of neither Bevacizumab40,41 nor Aflibercept42 improved overall survival of patients with advanced PDAC. However, our data are not contradictory to these clinical findings as they indicate a potential benefit for such treatments (e.g., as adjuvant therapy) at earlier stages of tumorigenesis to prevent metastatic outgrowth rather than affecting the established metastatic disease as suggested by others.43
Overall, this study provides novel insights into the mechanisms underlying metastatic outgrowth of PDAC. It supports the view that in disseminating PDECs, which have successfully seeded into the liver, a QAP is induced and maintained as long as a physiologic microenvironment implying the presence of HSCs is given. This HSCs-mediated QAP is characterized by Ki67-negativity (cell cycle arrest) and a reduced p-Erk/p-p38 ratio and essentially depends on IL-8. At this stage, metastatic lesions are dormant and thereby occult. Aging but also other inflammatory triggers, such as exposure to certain lifestyle factors or cancer therapies, may favor a smoldering inflammation leading to activation/transdifferentiation of HSCs into HMFs. This remodeling of the hepatic microenvironment is associated with further elevation of inflammatory mediators including VEGF. Elevated VEGF levels can in turn promote QAP reversal in PDECs leading to enhanced proliferation and metastatic outgrowth (Fig. 7).
Figure 7.
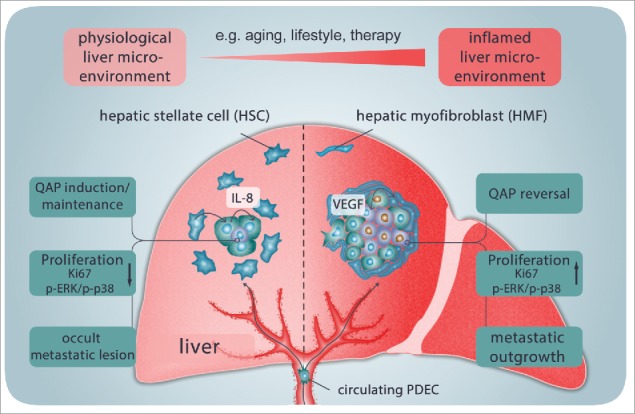
Model of QAP induction and reversal in PDECs disseminated in the liver, contributing to liver metastases in PDAC. After seeding the liver, a quiescence-associated phenotype (QAP) is induced and maintained in disseminated PDECs as long as a physiologic microenvironment implying the presence of HSCs is given. The HSCs-mediated QAP is characterized by the absence of Ki67 (cell cycle arrest) and reduced p-Erk/p-p38 ratio and essentially depends on IL-8. At this stage, metastatic lesions are dormant and occult. Aging, exposure to certain lifestyle factors or cancer therapies may favor a smoldering inflammation leading to activation/transdifferentiation of HSCs to HMFs and remodeling of the hepatic microenvironment which is associated with an increase in inflammatory mediators such as VEGF. Elevated VEGF levels in turn promote QAP reversal in PDECs leading to enhanced proliferation and metastatic outgrowth.
Importantly, inflammatory alterations defined as smoldering inflammation are not detected by current measures in clinical routine. However, as shown here and by others, even those low concentrated inflammatory triggers – most relevant, when persisting over long time – seem potent enough to drive tumor progression. Certainly, more studies are needed to further deepen our understanding of the role of subclinical inflammation and dynamics underlying PDAC metastasis, which is pivotal in improving screening and diagnostic measures as well as designing more effective therapies of this devastating disease.
Materials and methods
Mouse strains and tumor models
All animal studies were executed in compliance with European guidelines for the care and use of laboratory animals and approved by local authorities (Az. 55.2–1–54–2532–31–11) and (V242–77326/2015 (123–10/11). For initial screening of liver metastases, liver tissues of KPC mice (Pdx1-Cre;LSL-KrasG12D/+;LSL-Trp53R172H/+) were used.44 For examining age-related metastasis formation, 3 × 103 R254 cells (fLuc/EGFP, Ptf1aCre/+;LSL-KrasG12D/+;LSL-Trp53R172H/+)45 diluted in 25 µl NaCl were injected into the pancreas head of C57BL/6J mice aged either 8 or 52 weeks. To model the higher age of human PDAC patients at time point of diagnosis, age of aged mice (under consideration of pancreatic carcinoma) was calculated by using The human-to-mouse age map search tool (http://rubinlab.bgu.ac.il/mouseAPK/H2M/). Laparotomy and postoperative care was performed as described previously.39 Tumor screening was performed 2 and 4 weeks post-injection via abdominal ultrasound examination using a Vevo770 (FujiFilm VisualSonics Inc.). After mouse sacrifice, pancreata and livers were bisected and either snap-frozen in liquid nitrogen for PCR analysis or fixed in 4.5% PBS buffered formalin for immunohistological staining.
Immunhistochemistry
Serial 3 µm sections from formalin-fixed and paraffin-embedded tissue specimens were deparaffinized and rehydrated with xylene and graded ethanol baths. Endogenous peroxidases were quenched using 1.5% H2O2 (#8070.1; Roth). Antigen retrieval was performed in citrate buffer (pH 6.0) for 20 min, followed by 60 min blocking in 4% BSA/PBS + 0.3% Triton. For IHC-stainings, mouse anti-α-smooth muscle actin (α-SMA) (#A2547; Sigma-Aldrich) or mouse anti-Ki67 (#556003; BD Biosciences) antibodies were mixed with rabbit anti-Cytokeratin-19 (CK19)-antibody (#10712–1-AP; Proteintech Europe) or rabbit anti-desmin (#D93F5; Cell Signaling) diluted in 1% BSA/PBS + 0.3% Triton, unless indicated otherwise. Antibodies were diluted according to manufacturer's instructions and incubated overnight at 4 °C. For visualization and mounting, the Lab Vision™ MultiVision Polymer Detection System-Kit (#TL-012-MHRA; Thermo Fisher) was used following manufacturer's instructions. All stainings were verified performing parallel negative-control stainings with corresponding IgG-control antibodies. Sections were evaluated using a Zeiss Axio Scope-A1 microscope equipped with an Axiocam-503 color (Zeiss). Ki67-scoring of CK19+ cells/areas was done as described previously46 and α-SMA- and desmin-immunostaining was assessed using a 4-tiered system: no immunostaining (0 = negative), weak (grade 1), medium (grade 2) and strong immunostaining (grade 3).
RNA isolation and RT-qPCR
Total RNA was isolated with total RNA kit peqGOLD (#12–6834–02; Thermo Fisher) and subjected to reversed transcription using the RevertAid First Strand cDNA Synthesis Kit (#K1621; Thermo Fisher) according to manufacturer's instructions. Primers were purchased from Realtime Primers (via Biomol) or Eurofins (via MWG). Primer sequences are provided in Table 1. PCR was performed as duplicate analysis with a LightCycler 480 (Roche) for maximum 50 cycles and melting curve analysis as quality control. The expression of genes of interest was normalized to gene expression of reference GAPDH and β-actin.
Table 1.
Primer sequences used for RT-qPCR.
| Target |
Primer sequences |
| mouse α-SMA (Acta2) | F: 5′- ATG CAG AAG GAG ATC ACA GC -3′ |
| R: 5′- CAG CTT CGT CGT ATT CCT GT -3′ | |
| mouse β-Actin | F: 5′- AAG AGC TAT GAG CTG CCT -3′ |
| R: 5′- TAC GGA TGT CAA CGT CAC -3′ | |
| mouse Desmin | F: 5′- CAG GAG ATG GAA TAC CG -3′ |
| R: 5′- GGC CAT CTC ATC CTT TAG GT -3′ | |
| mouse Gapdh | F: 5′- TCC ATG ACA ACT TTG GTA TCG TGG -3′ |
| R: 5′- GAC GCC TGC TTC ACC ACC TTC T -3′ | |
| mouse IL-1β | F: 5′- ATC CTC TGT GAC TCA TGG GAT G -3′ |
| R: 5′- GAT CCA CAC TCT CCA GCT GCA -3′ | |
| mouse LIX | F: 5′- CAG AAG GAG GTC TGT CTG GA-3′ |
| R: 5′- TGG TTT CCC TTT TCT CT CA-3′ | |
| mouse MIP-2 | F: 5′- CAG ACT CCA GCC ACA CTT CA -3′ |
| R: 5′- TTC AGG GTC AAG GCA AAC TT-3′ | |
| mouse TGF-β1 | F: 5′- GCT GAA CCA AGG AGA CGG AA-3′ |
| R: 5′- AGA AGT TGG CT GGT AGC CC -3′ | |
| mouse VEGF-A | F: 5′- ACT GGA CCC TGG CTT TAC TG -3′ |
| R: 5′- TCT GCT CTC CTT CTG TCG TG -3′ |
Immunofluorescence
For detection and characterization of GFP-labeled R254 DTCs and liver metastases, 3 µm liver sections were deparaffinized, blocked as described above and subsequently incubated with rabbit anti-GFP (#A-11122; Thermo Fisher) and mouse anti-Ki67 (see above) antibodies diluted in 1% BSA/PBS + 0.3% Triton and incubated overnight at 4 °C. Following blocking in 0.1% Sudan Black-B (#199664; Sigma-Aldrich) in 70% ethanol for 20 min and subsequent washing, visualization was done using Alexa Fluor 488 goat anti-mouse IgG H+L (#A11029; Life Technologies) and goat anti-rabbit IgG H+L DyLight 594 (#35560; Thermo Fisher) diluted in 1% BSA/PBS + 0.3% Triton. Nuclei were stained with Hoechst 33258 (#861405; Sigma-Adrich) and sections were mounted in FluorSave Reagent (#345789; Merck Millipore). One entire liver section per animal was screened for the presence of GFP+ metastatic lesions. For characterization of DTCs, 10 view fields were photographed at 200-fold magnification and median number of GFP+ DTCs and their Ki67 status determined.
Cell lines and cell culture
The human premalignant PDEC line H6c7-kras (kindly provided by M.S. Tsao, Ontario Cancer Institute, Toronto, Canada)47 was cultured in H6c7-medium (50% RPMI-1640 medium (#F1215; Biochrom) and 50% KSF-medium (#17005–034; Thermo Fisher)) supplemented with 5% fetal calf serum (#S0115), 0.5% L-glutamine (#K0283)(both Biochrom), 50 µg/ml bovine pituitary extract (#13028–014; Thermo Fisher), 5 ng/ml epidermal growth factor (#PHG0311; Thermo Fisher) + 0.5 µg/ml puromycin (#ant-pr-1; InvivoGen). The human malignant PDEC line Panc1 ( = human PDAC cell line) was obtained from ATCC (LGC Standards) and kept in RPMI-1640 supplemented with 10% FCS, 1% L-glutamine, 1% sodium pyruvate (#L0473; Biochrom). The murine PDAC cell line R254 was isolated from PDAC tissue derived from a Pdx1-Cre;LSL-KrasG12D;LSL-Trp53R172 H/+ mouse and cultured in DMEM supplemented with 10% FCS, 1% Pen/Strep and 1% L-glutamine.45 Routine cell culture and all experiments were conducted at 37 °C, 5% CO2, 86% humidity and regularly checked for the absence of Mycoplasma. The genotype of the cell lines was confirmed by Short Tandem Repeats Analysis. The murine hepatic stellate cell line M1-4HSC was used to model a physiologic liver microenvironment.48 M1–4HSC were cultured in DMEM high glucose containing 10% FCS, 1% Pen/Strep and 1% L-glutamine. To avoid transactivation of the cells to HMFs, a passage number of 10 and a confluence of 80% were not exceeded. M-HT cells, resulting from long-term exposure of M1–4HSC to 1 ng/ml TGF-β1 (#580702; BioLegend)30 were used to model an inflamed hepatic microenvironment. TGF-β1 exposure occurred during routine cell culture which was performed under the same conditions as for M1–4HSC. TGF-β1 treatment was stopped after seeding for cocultures. The phenotypes of M1–4HSC and M-HT were regularly examined by evaluating α-SMA, desmin and collagen-1A1 expression.
Indirect coculture of hepatic stromal cells and PDECs and real-time Life Cell Imaging
One day before coculture, 1 × 104 H6c7-kras or Panc1 cells were seeded in 6-well-plates in 2 ml medium/well while 5 × 104 M1–4HSC or M-HT in 1.5 ml medium/well were seeded into corresponding transwell inserts with 0.4 µm diameter pores (#657641; Greiner Bio-One). After 24 h, medium in all wells and transwells was exchanged for fresh coculture medium (Panc1 coculture: 1:1 (v/v) mix of DMEM high glucose with 10% FCS, 1% L-glutamine, 1% sodium pyruvate and RPMI-1640 with 10% FCS, 1% L-glutamine, 1% sodium pyruvate; H6c7-kras coculture: RPMI-1640 with 10% FCS, 1% L-glutamine, 1% sodium pyruvate). Afterwards, transwells were inserted into respective wells with epithelial cells. After 6 d of coculture, cells were prepared for further analysis. For 12 day cocultures, 0.5 × 104 H6c7-kras or Panc1 PDECs were seeded. Transwells were substituted after 6 days for transwells with fresh M1-4HSCs (HSC-HSC-coculture) or M-HTs (HSC-HMF-coculture), prepared as described above and PDECs were surveilled in a JuLI™ Br Life cell analyzer (VWR). For examination of QAP reversion, proliferation of Panc1 cells with a flattened, enlarged morphology (QAP-morphology) was particularly monitored.
Determination of viable cell number
After detachment with Accutase (#P10–21100; PanBiotech), cells were stained with trypan blue (#93535; Sigma-Aldrich) and counted using Neubauer counting chambers. For quantification of viable cells, cells exposing blue staining were excluded from counting.
Determination of inflammatory mediators in cell culture supernatants
Luminex Multiplex Screening Assay (#A3C8CE39, R&D Systems) was used to determine protein levels of 27 human and 6 murine cytokines in culture supernatants according to the manufacturer's instructions after 6 d of coculture and centrifugation of supernatants at 1200 RPM for 5 min. Fluorometric measurements were performed in a Bio-Plex 200 reader (Bio-Rad) using the corresponding Bio-Plex manager software (by courtesy of the Department of Immunobiology, Research Center Borstel, Borstel, Germany). Protein values of simultaneously cultured media controls were subtracted from protein values of coculture supernatants.
Blocking of IL-8 and VEGF during coculture
Blocking agents were added to medium of cocultures set up as described above upon start of coculture and again after 72 h. Soluble IL-8 was blocked using 2.5 µg/ml neutralizing human CXCL8/IL-8 antibody (#MAB208; R&D Systems) while monoclonal mouse IgG1 (#MAB002; R&D Systems) was used as isotype control. VEGF was blocked using 10 µg/ml Aflibercept (Bayer Vital) and anti-CD-20 antibody Rituximab (Roche) was used as isotype control.
Immunocytochemistry
For immunocytochemical stainings, cells were grown on glass coverslips. After washing in PBS, cells were fixed with 4.5% paraformaldehyde solution for 10 min at room temperature (RT) and washed again. Endogenous peroxidases were quenched in 0.3% H2O2 in methanol at -20 °C for 10 min followed by blocking in 4% BSA/PBS for 60 min. Fixed cells were incubated in mouse anti-Ki67 (see above) antibody in 1% BSA/PBS for 1 h at RT. For detection, Envision+ System HRP anti-mouse or anti-rabbit (#K4001, #K3464; Dako) was applied for 30 min at RT. After washing, immunostaining was visualized with AEC Substrate (#K3464, Dako) for 10 min and a brief hematoxilin staining was affiliated.
Determination of SABG-activity
To visualize the lysosomal compartment of senescent cells, the Senescence β-Galactosidase Staining Kit (#9860; Cell Signaling) was used according to manufacturer's instructions.
Western Blotting
Electrophoresis and Western blotting of whole cell lysates were performed as described previously49,50 The following antibodies were used according to manufacturer´s recommendations: rabbit-anti-HSP90 α/β (#sc-7947; Santa Cruz), rabbit-anti-Erk1/Erk2 (#9102 S), rabbit-anti-phospho Erk1/Erk2 (T204/Y209)(#9109 S), rabbit-anti-p38 (#9212), mouse-anti-phospho-p38 (T180/Y182)(#4511 S)(all from Cell Signaling), mouse-anti-p21 (#610233; BD Biosciences). Primary antibodies were incubated overnight at 4° C. Anti-rabbit- (#7074 S) or anti-mouse (#7076 S) HRP-linked antibodies (both Cell Signaling) served as secondary antibodies (RT; 1 h). After washing in TBST, blots were developed with SuperSignal West Dura Extended Duration Substrate (#1859024, #1859025; Perbio Sciences). Average band intensities were determined by densitometry using ImageJ1.47v software (National Institutes of Health, USA). Values of proteins of interest were normalized to values of the corresponding HSP90-loading control.
Statistical analysis
For statistical analysis, the SigmaPlot Software 12.5 (Systat Software GmbH) was used. The Shapiro-Wilk test was applied to test for normal distribution. Parametric data were analyzed by t-test or one-way RM ANOVA, while non-parametric data were analyzed by Kruskal-Wallis one-way ANOVA on ranks test. P-values < 0.05 were regarded as statistically significant and are indicated with an asterisk (*).
Supplemental Material. The supplementary material contains further images of immunohistostainings of CK19 and Ki67 as well as α-SMA and desmin in liver micrometastases and macrometastases of PDAC bearing KPC mice (Supplementary Figure 1). Furthermore, the α-SMA/desmin-ratio of livers extracted 2 weeks after R254 application as determined via qPCR is depicted (Supplementary Figure 2).
The effect of HSC and HMF on apoptosis induction (Supplementary Figure 3) and the proliferative behavior of H6c7-kras and Panc1 cells in comparison to monocultured cells (Supplementary Figure 4) is pictured. Stainings showing the phenotypes of HHSteC-HSC and HHSteC-HMF as well as their effect on H6c7-kras and Panc1 cells upon coculture are depicted in Supplementary Figure 5. The corresponding methodological approach is provided. Furthermore, videos obtained from real-time Life Cell Imaging showing Panc1 cells during HSC-HSC-coculture (Supplementary Video 1A) or HSC-HMF-coculture (Supplementary Video 1B) are included. Supplementary Figure 6 shows SABG-stainings of Panc1 cells in comparison to phase contrast images obtained after HSC-HMF-coculture.
Supplementary Material
Conflict of interest disclosure
The authors disclose no conflict of interest.
Acknowledgments
The authors thank Dagmar Leisner and Sandra Krüger for excellent technical assistance. We thank Alexa Klettner for providing Aflibercept, Matthias Peipp for providing Rituximab, Daniela Wesch for anti-IL-8 agents; Christoph Garbers, Georg Waetzig and Conaris Research Institute AG for IL-6 agents. Moreover, we thank Frank Leypoldt and Martina Jansen for the opportunity to use their microscopic devices.
Financial support
This work was supported by the Medical Faculty of Christian-Albrechts-University Kiel and the Deutsche Forschungsgemeinschaft (SE1831/2–3) and the Cluster of Excellence Infammation at interfaces.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. eng. doi: 10.3322/caac.21254. PMID:25559415. [DOI] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. eng. doi: 10.1056/NEJMra1404198. PMID:25207767. [DOI] [PubMed] [Google Scholar]
- 3.Neuzillet C, Tijeras-Raballand A, Bourget P, Cros J, Couvelard A, Sauvanet A, Vullierme M-P, Tournigand C, Hammel P. State of the artand future directions of pancreatic ductal adenocarcinoma therapy. Pharmacol Ther. 2015;155:80–104. eng. doi: 10.1016/j.pharmthera.2015.08.006. PMID:26299994. [DOI] [PubMed] [Google Scholar]
- 4.Sperti C, Pasquali C, Piccoli A, Pedrazzoli S. Recurrence after resection for ductal adenocarcinoma of the pancreas. World J Surg. 1997;21:195–200. doi: 10.1007/s002689900215. PMID:8995078. [DOI] [PubMed] [Google Scholar]
- 5.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, et al. . A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–1210. eng. doi: 10.1056/NEJMoa032295. PMID:15028824. [DOI] [PubMed] [Google Scholar]
- 6.Kanda M, Matthaei H, Wu J, Hong S-M, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, Goggins M. Presence of somatic mutations in most early-stage pancreatic intraepithelial neo-plasia. Gastroenterology. 2012;142:730–733.e9. eng. doi: 10.1053/j.gastro.2011.12.042. PMID:22226782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, et al. . EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. eng. doi: 10.1016/j.cell.2011.11.025. PMID:22265420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, Word AE, Carrer A, Salz TH, Natsume S, et al. . Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–376. eng. doi: 10.1038/ng.3753. PMID:28092686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roe J-S, Hwang C-I, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, Maiorino L, Tiriac H, Young CM, Miyabayashi K, et al. . Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. 2017. eng. doi: 10.1016/j.cell.2017.07.007. PMID:28757253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et al. . Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. eng. doi: 10.1038/ncb3169. PMID:25985394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grunwald B, Harant V, Schaten S, Fruhschutz M, Spallek R, Hochst B, Stutzer K, Berchtold S, Erkan M, Prokopchuk O, et al. . Pancreatic premalignant lesions secrete tissue inhibitor of metalloproteinases-1, which activates hepatic stellate cells via cd63 signaling to create a premetastatic niche in the liver. Gastroenterology. 2016;151:1011–1024. e7. eng. doi: 10.1053/j.gastro.2016.07.043. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, Ireland L, Sakai T, Sakai K, Kim Y-S, et al. . Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18:549–560. eng. doi: 10.1038/ncb3340. PMID:27088855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman SL. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. eng. doi: 10.1152/physrev.00013.2007. PMID:18195085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. eng. doi: 10.1172/JCI24282. PMID:15690074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim D-H, Provenzano PP, Smith CL, Levchenko A. Matrix nanotopography as a regulator of cell function. J Cell Biol. 2012;197:351–360. eng. doi: 10.1083/jcb.201108062. PMID:22547406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. eng. doi: 10.1016/j.cell.2013.05.039. PMID:23746838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheedfar F, Di Biase S, Koonen D, Vinciguerra M. Liver diseases and aging: Friends or foes? Aging Cell. 2013;12:950–954. eng. doi: 10.1111/acel.12128. PMID:23815295. [DOI] [PubMed] [Google Scholar]
- 18.Tajiri K, Shimizu Y. Branched-chain amino acids in liver diseases. World J Gastroenterol. 2013;19:7620–7629. eng. doi: 10.3748/wjg.v19.i43.7620. PMID:24282351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38 (SAPK). Cancer Res. 2003;63:1684–1695. eng. [PubMed] [Google Scholar]
- 20.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. eng. doi: 10.1038/nrc2256. PMID:17957189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee M, van Golen KL. Farnesyl transferase inhibitor treatment of breast cancer cells leads to altered RhoA and RhoC GTPase activity and induces a dormant phenotype. Int J Cancer. 2011;129:61–69. eng. doi: 10.1002/ijc.25655. PMID:20824700. [DOI] [PubMed] [Google Scholar]
- 22.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. eng. doi: 10.1101/gad.1971610. PMID:21078816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat Rev Cancer. 2014;14:611–622. eng. doi: 10.1038/nrc3793. PMID:25118602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation Triggers Zeb1-dependent escape from tumor latency. Cancer Res. 2016;76:6778–6784. eng. doi: 10.1158/0008-5472.CAN-16-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh P, Coskun ZZ, Goode C, Dean A, Thompson-Snipes L, Darlington G. Lymphoid neogenesis and immune infiltration in aged liver. Hepatology. 2008;47:1680–1690. eng. doi: 10.1002/hep.22224. PMID:18395842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tung BT, Rodriguez-Bies E, Thanh HN, Le-Thi-Thu H, Navas P, Sanchez VM, Lopez-Lluch G. Organ and tissue-dependent effect of resveratrol and exercise on antioxidant defenses of old mice. Aging Clin Exp Res. 2015;27:775–783. eng. doi: 10.1007/s40520-015-0366-8. PMID:25952010. [DOI] [PubMed] [Google Scholar]
- 27.White RR, Milholland B, MacRae SL, Lin M, Zheng D, Vijg J. Comprehensive transcriptional landscape of aging mouse liver. BMC Genomics. 2015;16:899. eng. doi: 10.1186/s12864-015-2061-8. PMID:26541291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otranto M, Sarrazy V, Bonte F, Hinz B, Gabbiani G, Desmouliere A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh Migr. 2012;6:203–219. eng. doi: 10.4161/cam.20377. PMID:22568985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schumacher JD, Guo GL. Regulation of Hepatic Stellate Cells and Fibrogenesis by Fibroblast Growth Factors. Biomed Res Int. 2016;2016:8323747. eng. doi: 10.1155/2016/8323747. PMID:27699175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148:362–375. eng. doi: 10.1016/j.cell.2011.11.060. PMID:22265421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gallmeier E, Hernaez R, Gress TM. Controversy on the time to progression of pancreatic ductal adenocarcinoma. Gut. 2015;64:1676–1677. eng. doi: 10.1136/gutjnl-2014-309066. PMID:25731873. [DOI] [PubMed] [Google Scholar]
- 32.Yu J, Blackford AL, Dal Molin M, Wolfgang CL, Goggins M. Time to progression of pancreatic ductal adenocarcinoma from low-to-high tumour stages. Gut. 2015;64:1783–1789. eng. doi: 10.1136/gutjnl-2014-308653. PMID:25636698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuveson DA, Neoptolemos JP. Understanding metastasis in pancreatic cancer: A call for new clinical approaches. Cell. 2012;148:21–23. eng. doi: 10.1016/j.cell.2011.12.021. PMID:22265397. [DOI] [PubMed] [Google Scholar]
- 34.Collado M, Serrano M. Senescence in tumours: Evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. eng. doi: 10.1038/nrc2772. PMID:20029423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. eng. doi: 10.1016/j.cell.2008.03.039. PMID:18555778. [DOI] [PubMed] [Google Scholar]
- 36.Li A, King J, Moro A, Sugi MD, Dawson DW, Kaplan J, Li G, Lu X, Strieter RM, Burdick M, et al. . Overexpression of CXCL5 is associated with poor survival in patients with pancreatic cancer. Am J Pathol. 2011;178:1340–1349. eng. doi: 10.1016/j.ajpath.2010.11.058. PMID:21356384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, et al. . CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2016;29:832–845. eng. doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasan MR, Ho SHY, Owen DA, Tai IT. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int J Cancer. 2011;129:2115–2123. eng. doi: 10.1002/ijc.26179. PMID:21618508. [DOI] [PubMed] [Google Scholar]
- 39.Egberts J-H, Cloosters V, Noack A, Schniewind B, Thon L, Klose S, Kettler B, von Forstner C, Kneitz C, Tepel J, et al. . Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008;68:1443–1450. eng. doi: 10.1158/0008-5472.CAN-07-5704. PMID:18316608. [DOI] [PubMed] [Google Scholar]
- 40.van Cutsem E, Rivera F, Berry S, Kretzschmar A, Michael M, DiBartolomeo M, Mazier M-A, Canon J-L, Georgoulias V, Peeters M, et al. . Safety and efficacy of first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in metastatic colorectal cancer: The BEAT study. Ann Oncol. 2009;20:1842–1847. eng. doi: 10.1093/annonc/mdp233. PMID:19406901. [DOI] [PubMed] [Google Scholar]
- 41.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O'Reilly E, Wozniak TF, et al. . Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28:3617–3622. eng. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rougier P, Riess H, Manges R, Karasek P, Humblet Y, Barone C, Santoro A, Assadourian S, Hatteville L, Philip PA. Randomised, placebocontrolled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. Eur J Cancer. 2013;49:2633–2642. eng. doi: 10.1016/j.ejca.2013.04.002. PMID:23642329. [DOI] [PubMed] [Google Scholar]
- 43.Aiello NM, Bajor DL, Norgard RJ, Sahmoud A, Bhagwat N, Pham MN, Cornish TC, Iacobuzio-Donahue CA, Vonderheide RH, Stanger BZ. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat Commun. 2016;7:12819. eng. doi: 10.1038/ncomms12819. PMID:27628423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172 H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. eng. doi: 10.1016/j.ccr.2005.04.023. PMID:15894267. [DOI] [PubMed] [Google Scholar]
- 45.von Burstin J, Eser S, Paul MC, Seidler B, Brandl M, Messer M, von Werder A, Schmidt A, Mages J, Pagel P, et al. . E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–71, 371.e1-5. eng. doi: 10.1053/j.gastro.2009.04.004. PMID:19362090. [DOI] [PubMed] [Google Scholar]
- 46.Helm O, Mennrich R, Petrick D, Goebel L, Freitag-Wolf S, Roder C, Kalthoff H, Rocken C, Sipos B, Kabelitz D, et al. . Comparative characterization of stroma cells and ductal epithelium in chronic pancreatitis and pancreatic ductal adenocarcinoma. PLoS One. 2014;9:e94357.eng. doi: 10.1371/journal.pone.0094357. PMID:24797069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian J, Niu J, Li M, Chiao PJ, Tsao M-S. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–5053. eng. doi: 10.1158/0008-5472.CAN-04-3208. PMID:15958547. [DOI] [PubMed] [Google Scholar]
- 48.Proell V, Mikula M, Fuchs E, Mikulits W. The plasticity of p19 ARF null hepatic stellate cells and the dynamics of activation. Biochim Biophys Acta. 2005;1744:76–87. eng. doi: 10.1016/j.bbamcr.2004.12.009. PMID:15878400. [DOI] [PubMed] [Google Scholar]
- 49.Sebens Muerkoster S, Werbing V, Sipos B, Debus MA, Witt M, Grossmann M, Leisner D, Kotteritzsch J, Kappes H, Kloppel G, et al. . Druginduced expression of the cellular adhesion molecule L1CAM confers anti-apoptotic protection and chemoresistance in pancreatic ductal adenocarcinoma cells. Oncogene. 2007;26:2759–2768. eng. doi: 10.1038/sj.onc.1210076. PMID:17086212. [DOI] [PubMed] [Google Scholar]
- 50.Sebens Muerkoster S, Rausch AV, Isberner A, Minkenberg J, Blaszczuk E, Witt M, Folsch UR, Schmitz F, Schafer H, Arlt A. The apoptosis-inducing effect of gastrin on colorectal cancer cells relates to an increased IEX-1 expression mediating NF-kappa B inhibition. Onco-gene. 2008;27:1122–1134. eng. doi: 10.1038/sj.onc.1210728. PMID:17704804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.