

ABSTRACT
Mutated cancer antigens, or neoantigens, represent compelling immunological targets and appear to underlie the success of several forms of immunotherapy. While there are anecdotal reports of neoantigen-specific T cells being present in the peripheral blood and/or tumors of cancer patients, effective adoptive cell therapy (ACT) against neoantigens will require reliable methods to isolate and expand rare, neoantigen-specific T cells from clinically available biospecimens, ideally prior to clinical relapse. Here, we addressed this need using “mini-lines”, large libraries of parallel T cell cultures, each originating from only 2,000 T cells. Using small quantities of peripheral blood from multiple time points in an ovarian cancer patient, we screened over 3.3 × 106 CD8+ T cells by ELISPOT for recognition of peptides corresponding to the full complement of somatic mutations (n = 37) from the patient's tumor. We identified ten T cell lines which collectively recognized peptides encoding five distinct mutations. Six of the ten T cell lines recognized a previously described neoantigen from this patient (HSDL1L25V), whereas the remaining four lines recognized peptides corresponding to four other mutations. Only the HSDL1L25V-specific T cell lines recognized autologous tumor. HSDL1L25V-specific T cells comprised at least three distinct clonotypes and could be identified and expanded from peripheral blood 3–9 months prior to the first tumor recurrence. These T cells became undetectable at later time points, underscoring the dynamic nature of the response. Thus, neoantigen-specific T cells can be expanded from small volumes of blood during tumor remission, making pre-emptive ACT a plausible clinical strategy.
KEYWORDS: neoantigen, immunotherapy, adoptive T cell therapy, mutations, T cells, mini-line, ovarian cancer
Introduction
A growing body of evidence indicates that mutated antigens, or neoantigens, underlie the success of several cancer immunotherapies. For example, neoantigen-reactive T cells are often found in the tumor-infiltrating lymphocyte (TIL) product of patients who respond to TIL-based adoptive cell therapy (ACT).1-4 Furthermore, clinical responses to PD-1 and CTLA-4 blockade correlate with tumor mutation burden, and tumors that have escaped anti-PD-1 antibody treatment can lose neoantigens.5-7 Murine studies have shown that vaccines targeting neoantigens can eradicate tumors, and early clinical trial results suggest neoantigens can also be deliberately targeted with vaccines or ACT in humans.8-11 Theoretically, neoantigens have the advantages of being exquisitely tumor-specific and unable to induce central tolerance. For these reasons, they are widely considered one of the most promising classes of target antigen for cancer immunotherapy.12,13
Despite these advantages, neoantigens represent challenging targets in tumors with low-to-intermediate mutation burdens. We previously estimated that there may be only a 1–5% probability of any one mutation giving rise to a naturally processed and presented MHC class I epitope for which a spontaneous T cell response is present.14 For example, we calculated that a typical high-grade serous ovarian cancer (HGSC) with the median number of expressed mutations for that malignancy would have only a 39% likelihood of harbouring one or more bona fide neoantigens.14 In support of this, when we assessed TIL from three HGSC patients for recognition of autologous neoantigens, we detected only one positive response out of 79 mutations evaluated across the three patients.15 However, like many neoantigen studies, this analysis was designed to detect spontaneously induced (i.e. pre-existing) T cell responses. Other bona fide neoantigens might be present in tumors but fail to induce spontaneous immune responses. Therefore, it is unclear how efficiently naive, neoantigen-reactive T cells become activated and recruited into the anti-tumor immune response. Are all possible neoantigen-reactive T cells found within the TIL compartment, or does patient blood harbor additional neoantigen-reactive T cells that escape detection by conventional methods?
In theory, several mechanisms may make neoantigen-reactive T cells undetectable in the TIL compartment. First, neoantigens may be presented in a non-inflammatory context, resulting in failed T cell priming or T cell anergy.16 Second, inadequate expression of adhesion molecules on T cells or on the tumor vasculature may impair T cell infiltration or retention in tumor tissue.17,18 Third, the tumor microenvironment may harbor immunosuppressive cytokines and cell types (e.g. regulatory T cells) that functionally impair neoantigen-specific T cells.19,20 For these reasons, peripheral blood could theoretically be a more bountiful reservoir of neoantigen-reactive T cells compared to the TIL compartment.
In support of this notion, we recently showed that neoantigen-specific T cells, although present at exceedingly low frequencies, could nonetheless be expanded from the peripheral blood of lymphoma patients by priming with peptide-pulsed dendritic cells (DC).21 In another study, DC-based vaccines were shown to prime de novo neoantigen-specific T cell responses in melanoma patients.22 In two additional studies, neoantigen-specific T cells were successfully expanded from the peripheral blood of HLA-matched healthy donors.23,24 Although the sample sizes were small, these studies collectively suggest that a larger proportion of somatic mutations might give rise to bona fide MHC epitopes than previously suggested by studies in which only pre-existing TIL responses were evaluated.
Despite these encouraging results, there are several major challenges associated with the identification of neoantigen-reactive T cells in peripheral blood. This is especially true for naive T cells, which are present at exceptionally low frequencies (1/104 to 1/107) in blood and therefore require significant amplification to reach detectable levels.25-27 Moreover, the activation and expansion of naive T cells in vitro is usually performed using DC-based priming methods, creating a requirement for large quantities of peripheral blood from which to generate DCs. However, many cancer patients have co-morbidities that render them unable to donate sufficient volumes of blood for this purpose.
Library-based screening approaches represent an alternative means to identify naïve, neoantigen-reactive T cells using small volumes of blood. For example, Geiger et al. used polyclonal stimuli to activate and expand thousands of parallel, small-scale T cell cultures from the peripheral blood of healthy donors.28 Each “mini-line” was derived from approximately 2,000 T cells and underwent 1,000- to 5,000-fold polyclonal expansion. Thus, after expansion each T cell clone was theoretically still at a frequency of 1 in 2,000 yet amplified to an absolute number of 1,000-5,000 cells, which is sufficient for detection by conventional “recall” methods such as ELISPOT and flow cytometry.28 The mini-line method was used to efficiently activate, detect, and isolate MART1- and KLH-specific T cells without the need for DCs. More recently, a study using this technique identified and cloned T cells recognizing shared tumor-associated antigens from healthy donor PBMC.29 However, the mini-line method has not previously been used for the identification of neoantigen-specific naïve cells.
Here we used a similar mini-line approach to screen for neoantigen-specific T cells in the peripheral blood of a patient with HGSC. We studied a patient from whom a neoantigen-specific CD8+ T cell response had previously been identified among tumor-associated lymphocytes (TAL) from a recurrent ascites sample.15 From this patient, we had access to PBMC from four time points: the time of surgery and 3-, 6-, and 12-months post-surgery. We also had tumor cells, TAL, and whole exome sequencing data from three time points: primary (time of surgery), first recurrence (9 months post-surgery), and second recurrence (15 months post-surgery). We had previously identified a total of 37 missense mutations across the three time points,30 providing a list of potential neoantigens. Here, using the mini-line screening approach, we demonstrate the feasibility of identifying and expanding neoantigen-specific T cells from small volumes of autologous peripheral blood collected prior to clinical relapse, opening the possibility of creating neoantigen-specific T cell products for pre-emptive ACT.
Results
Mini-line screening to identify neoantigen-specific T cells in PBMC
To determine whether neoantigen-reactive T cells could be detected and isolated from small quantities of patient peripheral blood, we investigated a temporal series of blood and tumor samples donated by an HGSC patient who was described in prior studies15,30 (Patient 3; Fig. 1). As previously reported, the patient's primary tumor expressed MHC class I and II and showed clear evidence of spontaneous tumor immunity as indicated by the presence of intraepithelial TIL expressing CD3, CD8, CD4 and CD20.15 Previously, whole exome sequencing of tumor tissue from three time points identified a total of 37 non-synonymous somatic mutations, of which one was found to be recognized by TAL from the first recurrent tumor sample.15 The patient donated blood at four time points (pre-surgery and 3-, 6-, and 12-months post-surgery); however, only small quantities of PBMC (2–20 × 106 cells) were available from each time point. For mini-line screening, CD8+ T cells were purified by negative magnetic selection from each of four PBMC samples, and a total of 3.3 × 106 CD8+ T cells were seeded into 96-well plates to yield 1,632 wells at 2,000 CD8+ T cells/well. Each mini-line culture was subjected to the Rapid Expansion Protocol (REP),31 resulting in 1,000- to 5,000-fold expansion distributed across eight replicate plates.
Figure 1.

Clinical course for Patient 3. The dotted line shows blood CA-125 levels as a measure of tumor burden. Horizontal lines indicate the time of chemotherapy treatment (CATX = carboplatin and paclitaxel, CAG = carboplatin and gemcitabine, DX = pegylated liposomal doxorubicin). This figure was modified from previous articles 15,30.
We used a previously described set of peptides to identify potentially immunogenic mutated CD8+ T cell epitopes among the 37 somatic mutations.15 Mutant peptides with the best predicted binding score for each mutation were assessed, as well as all mutant epitopes predicted to bind to autologous HLA allele products with an IC50 < 50 nM (Supplementary Table 1). Additionally, for each mutation we used three overlapping 15-mer peptides (spanning the mutation) to provide an unbiased assessment of T cell reactivity, independent of epitope predictions.
Each mini-line was assessed by IFN-γ ELISPOT under the following stimulation conditions: (1) media as a negative control, (2) CEF11 peptide as a positive control for detecting memory CD8+ T cell responses, (3) MART1 peptide as a positive control for detecting naive CD8+ T cell responses,32 (4) the minimal mutant peptide pool (i.e. all predicted MHC class I epitopes), and (5) the 15-mer mutant peptide pool. For those mini-lines that were found to react to mutant peptides, wildtype versions of the peptides were later used to assess specificity (below). Fig. 2A shows an example of one mini-line plate stimulated with each peptide combination. The overall screen can be summarized as follows. All 1,632 mini-lines among four PBMC sample time points responded to the CEF11 peptide, indicating a high frequency of T cells specific for this antigen. Furthermore, 23 MART1-reactive wells were detected from among 400 mini-lines (8 × 105 CD8+ T cells), providing evidence that the approach could detect naive, antigen-specific T cells in patient PBMC. For the mutant peptide pools, 24 reactive T cell cultures were identified across the four time points (1,632 mini-lines) and were selected for further analysis (Fig. 2B). Since each mini-line was derived from only about 2,000 CD8+ T cells, and only 24 out of 1,632 mini-lines were reactive to the mutant peptide pools, we reasoned that all mutation-reactive T cells in a single mini-line were likely derived from a single T cell and represented a clonal response (according to the Poisson distribution, λ = 0.0148 and 99.3% of positive wells should contain only a single positive cell).
Figure 2.

Mini-line screen of peripheral blood T cells. CD8+ T cells were expanded using the mini-line protocol and assessed by IFN-γ ELISPOT. X-axes represent single mini-lines stimulated in individual ELISPOT wells with media alone (green), MART1 minimal peptide (red), a pool of all predicted minimal mutant peptides for this patient (blue), or an overlapping 15-mer pool encompassing each of 37 mutations (purple). Positivity was defined as > 13 spots and > 3-fold more than the average of the other stimulations for the well. A. Example of results from one mini-line plate (plate #1 from the 1 year blood draw) stimulated with the indicated peptides. Asterisks indicate positive wells. B. IFN-γ ELISPOT counts for the 24 positive mini-line wells (1,608 negative wells omitted) that were advanced to subsequent experiments. X-axis represents the plate number, row, and column of positive mini-line wells. Blood draw time points are noted below the X-axis.
Generation of highly enriched T cell lines that recognized tumor-specific mutations
To identify the mutations underlying the observed T cell responses, all mutation-reactive mini-lines were REP-expanded and screened by IFN-γ ELISPOT assay against each of 37 minimal epitope pools (one pool per mutation). Fig. 3A shows IFN-γ ELISPOT data from two representative experiments. Ten lines were unresponsive to any of the mutations, likely representing either false positives in the initial screen or failure of T cells to proliferate to detectable levels during REP expansion. In total, 14 T cell lines recognizing 7 mutations were confirmed reactive by ELISPOT. The responses involved the genes KIAA1467 (n = 1 T cell line), NOX4 (n = 2), CAPN7 (n = 1), ZNF41 (n = 1), MARK2 (n = 1), OR4C11 (n = 1), and HSDL1 (n = 7) (Table 1). T cell responses were not detected against any of the remaining 30 of 37 total mutations.
Figure 3.

Isolation of highly enriched, mutation-reactive T cell lines. For each panel, examples of T cell mini-lines 1F3 and 4C5 are shown. A. Mini-lines identified in the screen were REP expanded and stimulated in duplicate wells with peptides encompassing each tumor-specific mutation (N = 37 mutations) in ELISPOT assays. Bars represent the number of IFN-γ spot forming cells in each well. X-axis represents each individual mutation peptide pool with the reactive peptide indicated. B. FACS sorting plots for T cell line that proved reactive to a single mutation. CD8 (X-axis) and 4-1BB (Y-axis) double positive cells were FACS sorted. C. IFN-γ ELISPOT assay of T cell lines responding to individual minimal peptides contained within the mutation specific peptide pool. Bars represent the number of spot forming cells per well. Amino acids sequences of each peptide are shown on the X-axis. D. Flow cytometry plots of T cell lines stained with antibodies towards Vβ regions (X-axis) and 4-1BB (Y-axis). T cell lines were stained with the full complement of Vβ antibodies, and only the positive Vβ stains are shown (Vβ 13.1 for well 1F3; and Vβ 1 for well 4C5). E. IFN-γ ELISPOT assay of T cell cross-reactivity to WT versions of minimal epitopes. Lines represent the number of IFN-γ spot forming cells responding to 10-fold titrations of cognate mutated minimal epitope (squares) and the corresponding wild type peptide (triangles).
Table 1.
Work up of each putatively positive T cell line identified in mini-line screen of peripheral blood CD8+ T cells.
| Blood sample | Clone number | Gene(mutation) recognized | Enriched line derived? | Minimal Epitope | Vβ | Purity (%) | Tumor Reactive | Expressed in cDNA? | Avidity |
|---|---|---|---|---|---|---|---|---|---|
| Pre-surgery | 2F5 | KIAA1467(Q92E) | No | ||||||
| Pre-surgery | 3C2 | HSDL1(L25V) | Yes | CYMEAVAL | 1 | 99 | Yes | Yes | High |
| 3 month | 4D9 | HSDL1(L25V) | No | ||||||
| 3 month | 4H2 | NOX4(F197I) | Yes | FWYTHNLIF | 14 | 85 | No | Yes | Low |
| 3 month | 4A8 | CAPN7(Del) | Yes | FQETHLPI | 14 | 65 | No | Yes | Low |
| 3 month | 4D8 | ZNF41(G67E) | Yes | HLLSVEYQI | 13.6 | 88 | No | Yes | Low |
| 6 month | 4G9 | NOX4(F197I) | No | Yes | |||||
| 6 month | 1D9 | HSDL1(L25V) | Yes | CYMEAVAL | 13.6 | 88 | Yes | Yes | High |
| 6 month | 2D1 | HSDL1(L25V) | Yes | CYMEAVAL | 13.6 | 94 | Yes | Yes | High |
| 6 month | 2G2 | HSDL1(L25V) | Yes | CYMEAVAL | 5.2 | 80 | Yes | Yes | High |
| 6 month | 4C5 | HSDL1(L25V) | Yes | CYMEAVAL | 1 | 91 | Yes | Yes | High |
| 6 month | 5D8 | HSDL1(L25V) | Yes | CYMEAVAL | 5.2 | 80 | Yes | Yes | High |
| 1 year | 1D4 | MARK2(E13K) | No | ||||||
| 1 year | 1F3 | OR4C11(S154Y) | Yes | IH YTAQIIL | 13.1 | 80 | No | No | Low |
To generate highly-enriched mutation-reactive T cell cultures, reactive mini-lines were stimulated with pools of the relevant minimal peptides and FACS-sorted based on antigen-induced 4-1BB upregulation (Fig. 3B). The sorted T cells were REP-expanded and assessed by IFN-γ ELISPOT to confirm mutation reactivity and to identify the minimal epitope for each T cell line. At this stage, enriched T cell lines were derived for ten of 14 sorted T cell cultures and exhibited high reactivity to one or more mutant peptides from the relevant pool; we operationally defined the minimal epitope for each T cell line as the shortest mutant peptide that elicited a maximal response (Fig. 3C and Table 1). Four of the sorted T cell lines were non-reactive, suggesting the original responding T cell clone had been lost during expansion. Of the ten successfully expanded T cell lines reactive to mutant minimal peptides, six recognized HSDL1L25V peptides, and the four remaining lines recognized NOX4F197I, CAPN7(Del), ZNF41G67E, and OR4C11S154Y peptides (Table 1).
The clonality of each T cell line was assessed using flow cytometry-based Vβ antibody spectratyping, and in each case a dominant Vβ was identified that comprised between 40% and 95% of the T cell culture. Vβ spectratyping combined with 4-1BB staining performed on peptide-stimulated cultures confirmed that T cells expressing the dominant Vβ were peptide specific (Fig. 3D and Supplementary Fig. 1). Three different Vβ genes were identified among the six HSDL1-reactive T cell lines assessed, indicating that this mutant antigen was recognized by at least three distinct T cell clones (Table 1).
To assess the avidity and specificity of peptide recognition, T cell lines were stimulated in IFN-γ ELISPOT assays with titrated amounts of high-purity minimal peptides or the wild type counterpart. Each of the HSDL1L25V-reactive T cell lines responded to mutant peptide at concentrations ranging from 10 – 100 pM (Fig. 3E and Supplementary Fig. 2A). In contrast, T cell lines recognizing mutant peptides from NOX4, CAPN7, ZNF41, or OR4C11 required higher peptide concentrations ranging from 10 – 100 nM (Fig. 3E and Supplementary Figure 2B). None of the T cell lines demonstrated strong cross reactivity to the corresponding wild type peptides (Figure 3E and Supplementary Figures 2A, B).
T cell recognition of ascites tumor cells
We used IFN-γ ELISPOT assays to determine whether mutation-reactive T cell lines recognized purified tumor samples (i.e. CD45- cell populations) derived from ascites collected at three disease time points. For a positive control, we used an HSDL1L25V-specific CD8+ T cell clone (Clone 1) that was previously shown to be tumor-reactive.15 Similar to Clone 1, each of the six HSDL1L25V-specific T cell lines responded weakly to the primary ascites tumor cells, strongly to the first recurrence ascites tumor cells, and at intermediate levels to the second recurrence ascites tumor cells (Fig. 4). In contrast, the four T cell lines specific for peptides corresponding to NOX4F197I, CAPN7(Del), ZNF41G67E, or OR4C11S154Y failed to respond to ascites tumor cells from any time point, despite responding strongly to their respective minimal peptides (Fig. 4). A MART1-specific T cell line was used as a negative control, and as expected, failed to recognize ascites from any time point. Sanger sequencing of cDNA prepared from ascites tumor samples demonstrated that all mutations were transcribed at all time points, except mutant OR4C11 (an olfactory receptor) (Supplementary Figure 3). Thus, only the HSDL1L25V mutation gave rise to a bona fide MHCI epitope that was presented at detectable levels on the surface of tumor cells.
Figure 4.

Tumor recognition by mutation-specific T cell lines. Each highly purified T cell line was stimulated in IFN-γ ELISPOT wells in duplicate with media alone, MART1 peptide, autologous B cells, cognate minimal epitope, or purified tumor cells from three ascites collection time points. “Mutant epitope” for each T cell line is the minimal peptide containing the mutation within the gene written above each graph. A MART1-reactive T cell clone was used as a negative control T cell line. Bars represent the number of IFN-γ spots detected for each stimulus.
Clonal diversity of HSDL1L25V-reactive T cells in ascites
In our previous study, we identified and isolated a single HSDL1L25V-reactive CD8+ T cell clone (referred to as Clone 1), which was present in TAL at first recurrence and used the TCR-Vβ 13.6 gene.15 The HSDL1L25V-reactive CD8+ T cell lines identified in the mini-line screen used not only Vβ 13.6 but also Vβ 5.2 and Vβ 1. We assessed the first recurrence ascites sample to determine whether HSDL1L25V-reactive T cells infiltrating the ascites compartment also expressed these latter two Vβ genes. A CD8+ TAL line from the first recurrence ascites sample was generated by high-dose IL-2 expansion, stimulated with the minimal HSDL1L25V epitope, and stained with antibodies to 4-1BB and a panel of Vβ genes. This revealed that the HSDL1L25V-specific T cells from ascites used the same three Vβ genes as those from peripheral blood: Vβ13.6 (46.7% of 4-1BB+ cells), Vβ5.2 (17.9%), and Vβ1 (10.3%) (Fig. 5). Thus, using the mini-line approach, we were able to isolate from peripheral blood HSDL1L25V-reactive T cells that, at least at the level of Vβ usage, matched those found in the ascites compartment.
Figure 5.
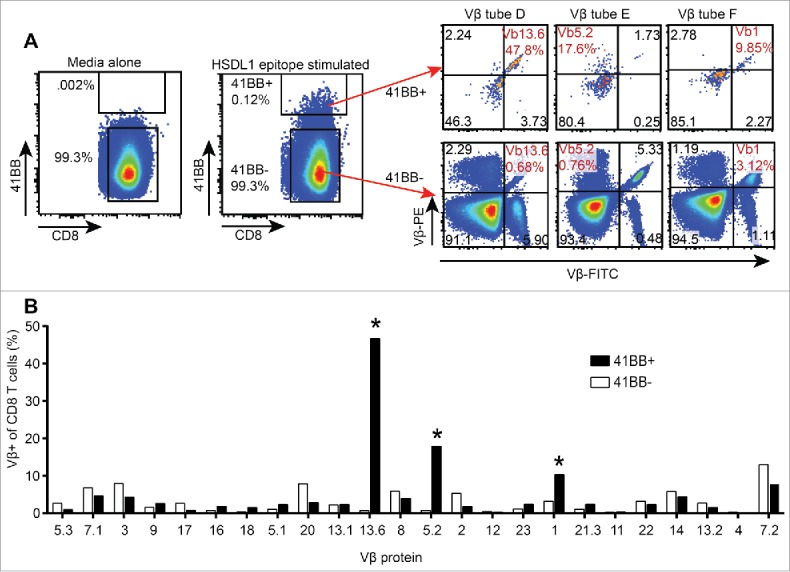
Vβ spectratyping of expanded TIL from first recurrence ascites. CD8+ TAL were stimulated with the HSDL1L25V peptide epitope or media alone and stained with antibodies to CD8, 4-1BB and each of 8 tubes of the Vβ spectratyping kit. A. HSDL1L25V epitope stimulated CD8+ TAL were gated as 4-1BB+ and 4-1BB−, and the Vβ spectratype plots of 4-1BB+ and 4-1BB− TAL are shown. The Vβ of HSDL1L25V-reactive T cells found in peripheral blood are highlighted in red. B. Bars represent the percentage of all 4-1BB+ (black) or 4-1BB− (white) T cells expressing each Vβ protein. Asterisks show the Vβ of HSDL1L25V-specific T cell lines isolated from peripheral blood.
Temporal dynamics of HSDL1L25V-specific T cell responses
The identification of multiple HSDL1L25V-reactive T cells in peripheral blood provided an opportunity to elucidate the dynamics of the T cell response to this neoantigen beyond our initial description of Clone 1 (Vβ 13.6).15 One HSDL1L25V-reactive T cell line (using Vβ 1) was isolated from the pre-surgery peripheral blood sample, one was isolated from the 3-month time point (with insufficient cells to assess Vβ usage), and five lines (using Vβ 1, Vβ 5.2 and Vβ 13.6) were isolated from the 6-month blood sample (Fig. 6). In contrast, no HSDL1L25V-reactive T cells were identified at the 12-month time point after screening 105 CD8+ T cells. In summary, using the mini-line approach and small volumes of patient blood, we were able to identify and expand at least three neoantigen-specific CD8+ T cell clones 3–9 months prior to first recurrence.
Figure 6.

Summary of sample collection, genomic data, and immunological findings for Patient 3. The timeline indicates the order of events from the time of primary surgery (Month 0) through to death (Month 17). Dark bars indicate chemotherapy treatments. HSDL1L25V VAF = Variant Allele Frequency from whole exome sequencing data 15,30, which is a measure of mutation abundance; TAL = Tumor Associated Lymphocytes (derived from ascites samples); PBMC = Peripheral Blood Mononuclear Cells; and T cells = HSDL1L25V-reactive T cell lines. The HSDL1 response at 3 months time point was not assessed for Vβ usage due to an insufficent number of cells for analysis.
Discussion
To interrogate both the naïve and activated neoantigen-specific T cell repertoire in HGSC, we adapted a published method for detecting antigen-specific T cells from small quantities of peripheral blood.28 In total, ten T cell lines were generated that recognized peptides corresponding to five distinct mutations: HSDL1L25V, NOX4F197I, CAPN7(Del), ZNF41G67E, and OR4C11S154Y. Of these, only the HSDL1L25V-reactive T cell lines recognized autologous tumor. We speculate that the other four mutations either failed to give rise to authentic neoantigens or were subject to prior immune editing that eliminated neoantigen expression. In either event, our data suggest that this patient's tumor expressed only a single neoantigen, HSDL1L25V, at detectable levels. Encouragingly, we identified at least three distinct CD8+ T cell clones (according to TCR Vβ usage) against this neoantigen, all of which were detectable three months prior to first recurrence. Thus, this study provides the first assessment of both the activated and naive T cell repertoire against the full complement of tumor-specific mutations in HGSC. Furthermore, it demonstrates a method to isolate neoantigen-reactive T cells from small quantities of peripheral blood prior to clinical relapse, opening the possibility of performing pre-emptive ACT to prevent relapse.
Our prior description of the CD8+ T cell response to HSDL1L25V (i.e. Clone 1) revealed important changes during progression from primary to recurrent to fatal disease.15 Consistent with the widespread genomic changes that occured over time with this patient's tumor,30 the HSDL1L25V mutation was rare in the primary ascites sample (approximately 4% of read counts in the whole exome sequencing data) but then became abundant at first recurrence (about 55% of read counts) and persisted at second recurrence (about 60% of read counts). The corresponding mutant epitope (CYMEAVAL) was presented by tumor cells with similar temporal dynamics. In contrast, Clone 1 was undetectable in the primary ascites sample, reached a peak at first recurrence (<0.4% of CD8+ TAL), and was undetectable at second recurrence.15 Thus, rather than undergoing immune editing, the HSDL1L25V mutation persisted at second recurrence, while Clone 1 disappeared from the TAL compartment. The current study further elucidates the temporal dynamics of the T cell response to this neoantigen (Fig. 6). HSDL1L25V-specific T cells were rare in PBMC from the primary and 3-month time points, in that only one HSDL1L25V-reactive T cell was found per 1.4 × 106 and 8 × 105 CD8+ T cells, respectively. However, at the 6-month blood draw, HSDL1L25V-reactive T cells may have been more prevalent, in that we identified five independent T cell lines (and at least 3 independent clonotypes) from 106 CD8+ T cells. By the 12-month time point, HSDL1L25V-reactive T cells were not detected upon screening to a depth of 105 CD8+ T cells. Thus, although the low numerators preclude an accurate estimate of precursor frequencies, these findings suggest that the temporal dynamics of the HSDL1L25V-specific T cell response were similar in the blood and ascites compartments. The results further suggest that the T cell response was primed or enhanced between 3 and 6 months and then declined between 6 and 12 months. Although speculative, the initial enhancement of the T cell response might have been due to the immunostimulatory effects of chemotherapy,33 in accord with our recent report that neoadjuvant chemotherapy is associated with increased TIL in HGSC.34 Another contributing factor could be the recovery of circulating lymphocytes that occurs following primary treatment of HGSC,35 which in theory could add new T cell clones to the peripheral repertoire.
Generating neoantigen-reactive T cells from small quantities of patient peripheral blood may be highly beneficial for ACT trials. Many patients are deemed ineligible for TIL-based ACT due to medical, logistical, or immunological barriers to producing a clinical TIL product. For example, surgical resection may be contraindicated due to the anatomical location of the tumor36 or potential morbidity associated with tumor resection.37 In contrast, peripheral blood is readily accessible from cancer patients. Indeed, one group demonstrated the ability to stimulate patient PBMCs in vitro with irradiated autologous tumor cells and generate T cell products for ACT.38 A subset of these patients experienced clinical responses, demonstrating the efficacy of using peripheral blood as a source of therapeutic T cells.38 Our work builds on this concept by enabling the generation of T cell products against defined neoantigens. The use of peripheral blood allows sampling of a more diverse neoantigen-reactive T cell repertoire than might be obtained from TIL. Furthermore, in the case of heterogeneous tumors with multiple metastases, the increased T cell diversity in peripheral blood may allow detection of metastasis-specific T cells absent from surgically resectable tumor nodules.
It is unclear why T cells recognizing NOX4F197I, CAPN7(Del), ZNF41G67E, or OR4C11S154Y peptides failed to respond to patient tumor. A recent study showed that immune pressure on mutated antigens can select for melanoma cells that either lose the mutated allele or express low quantities of the mutated gene.39 We found that one of the mutations (in OR4C11S154Y, an olfactory receptor gene) was not expressed at any time point, thereby explaining the lack of tumor recognition by the corresponding T cell line (Supplementary Figure 3). In contrast, NOX4F197I, CAPN7(Del), and ZNF41G67E were expressed at all time points, at least at the level of mRNA. One possibility is that these mutations failed to give rise to authentic neoantigens due to lack of processing and/or presentation of the mutant peptide on the MHC class I pathway. Alternatively, one or more of these mutations may have triggered a T cell response, which in turn selected for tumor cells that no longer expressed or presented the neoepitope (i.e. immune editing). Ideally, one would like to predict mutations giving rise to bona fide, expressed neoantigens. In this regard, we note that the HSDL1L25V-reactive T cells responded at an approximately 1,000-fold lower peptide concentration than the other mutation-reactive T cell lines, which might provide a criterion for predicting bona fide neoantigens.
Our work corroborates and expands on prior studies that evaluated neoantigen-reactive T cells in peripheral blood versus TIL. One study found that the activated, neoantigen-specific T cell repertoire from patient leukaphoresis product faithfully represented the neoantigen-specific T cell repertoire in TIL samples.40 Likewise, we found that T cells recognizing HSDL1L25V were present in both blood and ascites at first recurrence, and the Vβ repertoire of HSDL1L25V-specific T cells was the same in both compartments. In another study, healthy donor PBMC were used a source of neoantigen-reactive T cells.23 The authors identified several bona fide mutation-reactive T cell clones that were not detected in the patient's TIL compartment, suggesting that tumors can present neoantigens that are ignored by the patient's immune system. Accordingly, we showed that the primary tumor of Patient 3 expressed the HSDL1L25V neoantigen several months prior to the detection of a spontaneous T cell response in TAL, suggesting this neoantigen was ignored at early time points.
Despite the benefits of using mini-lines to generate neoantigen-reactive T cells, our approach has limitations. For example, low-frequency mutation-reactive T cells may have escaped detection due to the stochastic nature of sampling the peripheral blood T cell repertoire and subsequent in vitro T cell expansion and screening. Additionally, to generate enough clonal T cells for ACT, the method would require multiple rounds of REP expansion, and in our hands, some T cell lines failed to expand beyond four REP cycles. Furthermore, it remains to be determined whether the T cell activation protocol used here is as efficient as other methods such as in vivo priming or in vitro priming with DC, macrophages, genetically modified cells, or acellular materials.41,42 It is also unknown whether this technique can be used to expand antigen-specific T cells from TIL, which may be exhausted and refractory to ex vivo expansion. Nonetheless, when blood volume is limiting, the method described here provides a feasible means to generate autologous, antigen-specific T cells for ACT.
The mini-line method allowed us to isolate neoantigen-specific T cells from peripheral blood several months prior to their appearance in the TAL compartment. Indeed, during this patient's first remission period, one could have used 20 ml of blood to generate a tri-clonal T cell product that recognized a prominent mutation in the pending recurrent tumor. To translate this approach to the therapeutic setting, one could potentially collect serial blood samples from a patient, monitor the mutational profile of their cancer via circulating tumor DNA, and when indicated use the mini-line method to generate a T cell product for use in pre-emptive ACT. However, a remaining challenge highlighted by this and previous studies is the relatively low mutation load in HGSC, which severely limits the number of neoantigens available for therapeutic targeting. For such tumor types, the screening method described here may also prove helpful for identifying and validating additional classes of tumor antigens.
Methods
Biospecimens and clinical data
The participant (Patient 3) was enrolled with informed written consent in the BC Cancer Agency/Tumor Tissue Repository's “Immune Response to Ovarian Cancer” (IROC) study, which was approved by the Research Ethics Board of the BC Cancer Agency/University of British Columbia. Ascites samples were collected at primary surgery and during palliative paracentesis. Ascites cells were separated from supernatant by centrifugation (300 g × 10 min), and cryopreserved in 10% DMSO (Sigma), 50% FBS (Gibco), 40% complete media (RPMI + HEPES [Gibco] supplemented with 10% FBS), 50 µM β-mercaptoethanol (Sigma), 2 µM L-glutamine (Hyclone), and 1 mM sodium pyruvate (Hyclone). PBMC from Patient 3 and healthy donors were isolated using density centrifugation with Ficoll (GE Healthcare: 17-1440-03) as previously described.43 Patient HLA alleles (A*02:01, A*30:01, B*39:01, B*44:03, C*14:03, and C*07:02) were determined from exome sequencing data using published methods.15,44
Epitope prediction and peptide pools
NetMHCpan2.445 was used to predict the binding affinity of mutated peptides to Patient 3 HLA allele products. All possible mutated 8, 9, 10, and 11-mer sequences spanning each mutation were assessed. All peptides that were predicted to bind the patient's MHCI with an IC50 < 50 nM were purchased (crude, Genscript). In addition, for each mutation without a peptide predicted to bind with IC50 < 50 nM, the best predicted binding peptide (IC50 > 50 nM) was purchased. Thus, every mutation was represented by at least one predicted MHCI-binding mutant peptide (Supplementary Table 1). Peptides were solubilised in DMSO at 10 mg/ml. All predicted epitope peptides that encompassed the same Patient 3 mutation were pooled into a single mutation-specific “minimal” peptide pool, resulting in 37 minimal peptide pools. A separate master peptide pool contained every predicted mutant-epitope (76 peptides in total). Three 15mer peptides that overlapped by 11 amino acids were purchased and pooled for each mutation, ensuring that every mutant epitope 11 amino acids or smaller would be represented within at least one mutant 15mer peptide. Aliquots of each of the 15mer pools were combined to make a master pool of 111 mutant 15mer peptides. Additionally, the patient had been assessed for recognition of the CEF peptide pool (containing established minimal peptides from Cytomegalovirus, Epstein-Barr Virus, and Influenza Virus) and found to be highly reactive towards the CEF11 peptide, which was used as a positive control peptide in ELISPOT assays.
Generation of T cell lines from ascites
Tumor associated lymphocyte (TAL) lines were generated using a previously described protocol.31 Briefly, bulk ascites was thawed in complete RPMI, washed, and resuspended to 106 cells/ml in complete RPMI supplemented with 6,000 U/ml of human IL-2 (generously provided by the National Cancer Institute, USA). One ml of cell suspension was added to each well of a 24-well plate. Media was changed every two to three days, and cells were split as necessary to maintain concentration at approximately 106 cells/ml. After 2 weeks of T cell expansion in high dose IL-2, untouched CD8+ T cells were isolated using magnetic separation (Miltenyi: 130-096-495), and a modified rapid expansion protocol (REP)31 was performed using irradiated (50 Gy) PBMCs from healthy donors (PBMC: T cell ratio = 100:1), IL-2 (300 U/ml3), and CD3 agonist antibody (30 ng/ml, eBioscience: 16-0037-85). T cells were expanded for two weeks and split to maintain a concentration of approximately 106 cells/ml. After two weeks, T cells were rested for 48 hours in low dose IL-2 (1 U/ml) to eliminate background IFN-γ secretion, and IL-7 (10 ng/ml (Preprotech: 200–07) to prevent T cell apoptosis.
Generation of CD8+ T cell mini-lines from peripheral blood
Cryopreserved PBMC were thawed, washed, and resuspended in 3% FBS in PBS (Gibco). CD8+ T cells were enriched by magnetic negative depletion using the CD8+ T Cell Isolation Kit (Miltenyi: 130-096-495) according to the manufacturer's protocol. Enriched cells were stained with viability-dye-EF780 (0.5 μl dye/2 ml cell suspension) (eBioscience: 65-0865-18), CD8-FITC (1/100, BioLegend: 300906) and CD4-PE (1/100, BioLegend: 357404) and assessed on a FacsCalibur flow cytometer using FlowJo analysis software (VX.07). Cells were counted using Viacount (Cat#: 4000-0130) on a Guava cell cytometer, and 2,000 cells were added to each well of a round bottom 96 well plate. To serve as feeder cells, healthy donor PBMC were thawed, washed, and irradiated (50 Gy), and 2 × 105 cells were added to each well. Complete RPMI media was supplemented with human IL-2 (300 U/ml) and anti-CD3 antibody (30 ng/ml). Half of the media was removed and replaced with fresh media including additives on day 5 and every two days afterwards. Cells were split into replicate plates when cell pellets exceeded approximately 3 mm in diameter. After two weeks of expansion, one replicate plate was kept in reserve for future studies, and seven replicate plates were pooled using 96 well deep well tubes (Corning: 4408), keeping each original culture separate from all others. Pooled cultures were washed in complete RPMI, resuspended in resting media containing IL-2 (1 U/ml) and IL-7 (10 ng/ml), and distributed to 96 well flat bottom plates to rest. After 48 hours rest, replicate wells were pooled and washed in preparation for ELISPOT screens (described below). Wells from the reserve plate that corresponded to the mutation-reactive wells in the ELISPOT were harvested and subjected to a REP.31 All the T cells from each identified well were added to T25 flasks (Falcon: CA29185-300) and stimulated with IL-2 (300 U/ml), agonist CD3 antibody (30 ng/ml) and feeder cells (2 × 107 irradiated [50 Gy] PBMC from healthy donors) in 10 ml of complete RPMI. Cells were counted every two to three days and split as necessary to maintain a concentration of approximately 106/ml. After two weeks of expansion, cells were rested for two days in 1 U/ml of IL-2 and 10 ng/ml of IL-7.
IFN-γ ELISPOT assays
IFN-γ ELISPOT assays were performed according to the manufacturer's protocol (Mabtech) as previously described.14,15 Briefly, nitrocellulose plates (MSIP, Millipore: MSIPS4W10) were coated with 10 μg/ml of IFN-γ capture antibody (mAb 1-D1K) (Mabtech: 3420-3-1000) and blocked with complete media. T cells were added to each well and stimulated with peptides or tumor cells as indicated. For tumor recognition experiments, bulk ascites samples were depleted of CD45+ leukocytes by magnetic separation using CD45 MicroBeads (Miltenyi: 130-045-801) according to the manufacturer's protocol, and 2 × 104 purified tumor cells were added to each ELISPOT well. After 20 hours of incubation at 37°C and 5% CO2, plates were washed and 1 μg/ml of biotinylated IFN-γ antibody (Mabtech: 3420-6-1000) was added. Plates were developed with Vectastain Elite ABC kit (Vector Labs: PK-6100) and Vectastain AEC substrate reagent (Vector Labs: SK-4200). Plates were analyzed on an ELISPOT automatic plate reader (AID). For some experiments, autologous B cells were used as negative controls and generated as previously described.21 For each plate of mini-line expanded T cell cultures, five replicate ELISPOT plates were used to assess reactivity to peptides. Five random cultures from each mini-line plate were counted using Viacount on a Guava cytometer and showed that each culture contained between 2 × 106 and 5 × 106 T cells. Thus, 4 × 105 to 106 cells were added to each well in five replicate ELISPOT plates and were subjected to five conditions: (1) media alone, (2) CEF11 peptide (from EBV, GLCTLVAML) (5 µg/ml), (3) MART1 minimal peptide (ELAGIGILTV) (Genscript) (5 µg/ml), (4) minimal mutated peptide pool (50 µg/ml), and (5) 15mer mutated peptide pool (50 µg/ml). Post hoc analysis of the first ELISPOT screen indicated that a cutoff > 13 spots and > 3-fold over background eliminated almost all false positive calls (positive in media wells), and this cutoff was used for all mini-line screens.
Flow cytometry and FACS sorting
REP expanded T cell lines were incubated with cognate peptide (10 µg/ml) for 24 hours. Cells were then washed in complete RPMI and incubated with the surface staining antibodies CD8-FITC and 4-1BB-APC (1/100, BioLegend: 309810). Cells were assessed on an Influx cell sorter, and 4-1BB+CD8+ T cells were sorted into 200 µl of complete RPMI. FACS data were analyzed using FlowJo VX.07. For Vβ spectratyping, sorted T cell lines were stimulated with cognate peptide for 24 hours, washed, and stained with viability dye-EF780 (1 μl/0.5 ml suspension, eBioscience: 65-0863-18). After a 30-minute incubation, cells were washed and stained with surface antibodies for CD8-BV421 (1/200, BioLegend: 301036) and 4-1BB-APC. They were then divided into eight aliquots, each of which was stained with a different Vβ antibody cocktail (1/10, Beckman Coulter: IM3497) according to the manufacturer's protocol. Cells were detected on a FacsCalibur flow cytometer and analyzed using FlowJo VX.07.
PCR and Sanger sequencing of cDNA
RNA was extracted from purified tumor cells from each time point using RNeasy-plus mini kit (Qiagen: 74134), with two rounds through DNA eliminator columns. RNA was converted to cDNA using iScript cDNA synthesis kit (BIO-RAD: 1708890), and Platinum PCR supermix (Invitrogen: 11306-016) was used to PCR amplify cDNA surrounding each mutation loci for 35 cycles. All primers for transcribed genes spanned introns (to ensure the amplified products were derived from cDNA rather than genomic DNA), and the sequences were as follows: HSDL1L25V forward CTGCTGTTGACAGTTTCTACCT, reverse ACTTCTCCTCGTTCCGACTAA; ZNF41: forward GACTTCAGCAAGGAGGAGTG, reverse CCCAATAGCCTCACCTGAAC; CAPN7 forward AATCAGCATGCAGCTTTACTTT, reverse CTTGGTCCTCGTAGCTCAATC; NOX4 forward CTGGGCTCTTCCATACAAATC, reverse AGTTTCTAACTATGATATCTTCTGGTAT; OR4C11 forward TCAAATAGGGTCCACAGAAAGG, reverse CAGGTCTGCATCATCCTGATT. Primers were validated on healthy donor DNA. PCR amplicons were Sanger sequenced in both directions using GENEWIZ sequencing service.
Supplementary Material
Disclosure statement
The authors report no conflicts of interest.
Acknowledgments
We thank Drs. John Webb, David Kroeger, and Kwame Twumasi-Boateng for their expert input into experimental design.
Funding
This work was funded by the BC Cancer Foundation, Terry Fox Research Institute, Cancer Research Society, Canadian Breast Cancer Foundation, Canadian Institutes of Health Research (grants 341987 and 201110GSD-277623-204857), BioCanRx (Canada's Networks of Centres of Excellence Program), and Genome BC (Project B19BCH).
References
- 1.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747-52.doi: 10.1038/nm.3161. PMID:23644516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, Rosenberg SA. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183(3):1185-92.doi: 10.1084/jem.183.3.1185. PMID:8642260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clini. Invest. 2015;125(10):3981-91.doi: 10.1172/JCI82416. PMID:26389673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014;20(13):3401-10. doi: 10.1158/1078-0432.CCR-14-0433. PMID:24987109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207-11.doi: 10.1126/science.aad0095. PMID:26359337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017;7(3):264-76.doi: 10.1158/2159-8290.CD-16-0828. PMID:28031159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572-6.doi: 10.1038/nature14001. PMID:25428506 [DOI] [PubMed] [Google Scholar]
- 9.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577-81.doi: 10.1038/nature13988. PMID:25428507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641-5.doi: 10.1126/science.1251102. PMID:24812403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li F, Chen C, Ju T, Gao J, Yan J, Wang P, Xu Q, Hwu P, Du X, Lizée G. Rapid tumor regression in an Asian lung cancer patient following personalized neo-epitope peptide vaccination. Oncoimmunology. 2016;5(12):e1238539.doi: 10.1080/2162402X.2016.1238539. PMID:28123873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743-50.doi: 10.1101/gr.165985.113. PMID:24782321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69-74.doi: 10.1126/science.aaa4971. PMID:25838375 [DOI] [PubMed] [Google Scholar]
- 14.Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, Holt RA, Nelson BH. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PloS one. 2016;11(5):e0155189.doi: 10.1371/journal.pone.0155189. PMID:27192170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wick DA, Webb JR, Nielsen JS, Martin SD, Kroeger DR, Milne K, Castellarin M, Twumasi-Boateng K, Watson PH, Holt RA. Surveillance of the tumor mutanome by T cells during progression from primary to recurrent ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(5):1125-34.doi: 10.1158/1078-0432.CCR-13-2147. PMID:24323902 [DOI] [PubMed] [Google Scholar]
- 16.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437(7055):141-6.doi: 10.1038/nature03954. PMID:16136144 [DOI] [PubMed] [Google Scholar]
- 17.Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opi. Immuno. 2015;33:55-63.doi: 10.1016/j.coi.2015.01.011. PMID:25665467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014;20(6):607-15.doi: 10.1038/nm.3541. PMID:24793239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munn DH, Bronte V. Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2016;39:1-6.doi: 10.1016/j.coi.2015.10.009. PMID:26609943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta. 2014;1845(2):182-201. PMID:24440852 [DOI] [PubMed] [Google Scholar]
- 21.Nielsen JS, Sedgwick CG, Shahid A, Zong Z, Brumme ZL, Yu S, Liu L, Kroeger DR, Treon SP, Connors JM. Toward Personalized Lymphoma Immunotherapy: Identification of Common Driver Mutations Recognized by Patient CD8+ T Cells. Clin. Cancer Res. 2016;22(9):2226-36.doi: 10.1158/1078-0432.CCR-15-2023. PMID:26631611 [DOI] [PubMed] [Google Scholar]
- 22.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Böschen ML, Lund-Johansen F, Olweus J. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352(6291):1337-41.doi: 10.1126/science.aaf2288. PMID:27198675 [DOI] [PubMed] [Google Scholar]
- 24.Nielsen JS, Chang AR, Wick DA, Sedgwick CG, Zong Z, Mungall AJ, Martin SD, Kinloch NN, Ott-Langer S, Brumme ZL. Mapping the human T cell repertoire to recurrent driver mutations in MYD88 and EZH2 in lymphoma. Oncoimmunology. 2017;6(7):e1321184.doi: 10.1080/2162402X.2017.1321184. PMID:28811957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195(5):657-64.doi: 10.1084/jem.20001021. PMID:11877489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alanio C, Lemaitre F, Law HK, Hasan M, Albert ML. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved pr.doi:ecursor frequencies. Blood. 2010;115(18):3718-25. doi: 10.1182/blood-2009-10-251124. PMID:20200354 [DOI] [PubMed] [Google Scholar]
- 27.Hombrink P, Raz Y, Kester MG, de Boer R, Weissbrich B, von dem Borne PA, Busch DH, Schumacher TN, Falkenburg JH, Heemskerk MH. Mixed functional characteristics correlating with TCR-ligand koff -rate of MHC-tetramer reactive T cells within the naive T-cell repertoire. Eur. J. Immunol. 2013;43(11):3038-50.doi: 10.1002/eji.201343397. PMID:23893393 [DOI] [PubMed] [Google Scholar]
- 28.Geiger R, Duhen T, Lanzavecchia A, Sallusto F. Human naive and memory CD4+ T cell repertoires specific for naturally processed antigens analyzed using libraries of amplified T cells. J Exp Med. 2009;206(7):1525-34.doi: 10.1084/jem.20090504. PMID:19564353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Theaker SM, Rius C, Greenshields-Watson A, Lloyd A, Trimby A, Fuller A, Miles JJ, Cole DK, Peakman M, Sewell AK. T-cell libraries allow simple parallel generation of multiple peptide-specific human T-cell clones. J. Immunol. Methods. 2016;430:43-50.doi: 10.1016/j.jim.2016.01.014. PMID:26826277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castellarin M, Milne K, Zeng T, Tse K, Mayo M, Zhao Y, Webb JR, Watson PH, Nelson BH, Holt RA. Clonal evolution of high-grade serous ovarian carcinoma from primary to recurrent disease. J. Pathol. 2013;229(4):515-24.doi: 10.1002/path.4105. PMID:22996961 [DOI] [PubMed] [Google Scholar]
- 31.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003;26(4):332-42.doi: 10.1097/00002371-200307000-00005. PMID:12843795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pittet MJ, Valmori D, Dunbar PR, Speiser DE, Lienard D, Lejeune F, Fleischhauer K, Cerundolo V, Cerottini JC, Romero P. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 1999;190(5):705-15.doi: 10.1084/jem.190.5.705. PMID:10477554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell. 2015;28(6):690-714.doi: 10.1016/j.ccell.2015.10.012. PMID:26678337 [DOI] [PubMed] [Google Scholar]
- 34.Lo CS, Sanii S, Kroeger DR, Milne K, Talhouk A, Chiu DS, Rahimi K, Shaw PA, Clarke BA, Nelson BH. Neoadjuvant Chemotherapy of Ovarian Cancer Results in Three Patterns of Tumor-Infiltrating Lymphocyte Response with Distinct Implications for Immunotherapy. Clin. Cancer Res. 2017;23(4):925-34.doi: 10.1158/1078-0432.CCR-16-1433. PMID:27601594 [DOI] [PubMed] [Google Scholar]
- 35.Milne K, Alexander C, Webb JR, Sun W, Dillon K, Kalloger SE, Gilks CB, Clarke B, Köbel M, Nelson BH. Absolute lymphocyte count is associated with survival in ovarian cancer independent of tumor-infiltrating lymphocytes. J. Transl. Med. 2012;10:33. doi: 10.1186/1479-5876-10-33. PMID:22369276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svane IM, Verdegaal EM. Achievements and challenges of adoptive T cell therapy with tumor-infiltrating or blood-derived lymphocytes for metastatic melanoma: what is needed to achieve standard of care? Cancer Immunol Immunothe. 2014;63(10):1081-91.doi: 10.1007/s00262-014-1580-5. PMID:25099366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou JY, Kelly MG, Yu H, McAlpine JN, Azodi M, Rutherford TJ, Schwartz PE. Neoadjuvant chemotherapy lessens surgical morbidity in advanced ovarian cancer and leads to improved survival in stage IV disease. Gynecol Oncol. 2007;105(1):211-7.doi: 10.1016/j.ygyno.2006.11.025. PMID:17239941 [DOI] [PubMed] [Google Scholar]
- 38.Verdegaal EM, Visser M, Ramwadhdoebe TH, van der Minne CE, van Steijn JA, Kapiteijn E, Haanen JB, van der Burg SH, Nortier JW, Osanto S. Successful treatment of metastatic melanoma by adoptive transfer of blood-derived polyclonal tumor-specific CD4+ and CD8+ T cells in combination with low-dose interferon-alpha. Cancer Immunol Immunother. 2011;60(7):953-63.doi: 10.1007/s00262-011-1004-8. PMID:21431917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, Haanen JB. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536(7614):91-5.doi: 10.1038/nature18945. PMID:27350335 [DOI] [PubMed] [Google Scholar]
- 40.Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22(4):433-8.doi: 10.1038/nm.4051. PMID:26901407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leitner J, Kuschei W, Grabmeier-Pfistershammer K, Woitek R, Kriehuber E, Majdic O, Zlabinger G, Pickl WF, Steinberger P. T cell stimulator cells, an efficient and versatile cellular system to assess the role of costimulatory ligands in the activation of human T cells. J Immunol Methods. 2010;362(1-2):131-41.doi: 10.1016/j.jim.2010.09.020. PMID:20858499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eggermont LJ, Paulis LE, Tel J, Figdor CG. Towards efficient cancer immunotherapy: advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014;32(9):456-65.doi: 10.1016/j.tibtech.2014.06.007. PMID:24998519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.English D, Andersen BR. Single-step separation of red blood cells. Granulocytes and mononuclear leukocytes on discontinuous density gradients of Ficoll-Hypaque. J Immunol Methods. 1974;5(3):249-52.doi: 10.1016/0022-1759(74)90109-4. [DOI] [PubMed] [Google Scholar]
- 44.Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, Holt RA. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012;4(12):95.doi: 10.1186/gm396. PMID:23228053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1-13.doi: 10.1007/s00251-008-0341-z. PMID:19002680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.