

ABSTRACT
Adoptive therapy with engineered T cells shows promising results in treating patients with malignant disease, but is challenged by incomplete responses and tumor recurrences. Here, we aimed to direct the tumor microenvironment in favor of a successful immune response by local secretion of interleukin (IL-) 12 and IL-18 by sadministered T cells. To this end, we engineered T cells with a melanoma-specific T cell receptor (TCR) and murine IL-12 and/or IL-18 under the control of a nuclear-factor of activated T-cell (NFAT)-sensitive promoter. These T cells produced IL-12 or IL-18, and consequently enhanced levels of IFNγ, following exposure to antigen-positive but not negative tumor cells. Adoptive transfer of T cells with a TCR and inducible (i)IL-12 to melanoma-bearing mice resulted in severe, edema-like toxicity that was accompanied by enhanced levels of IFNγ and TNFα in blood, and reduced numbers of peripheral TCR transgene-positive T cells. In contrast, transfer of T cells expressing a TCR and iIL-18 was without side effects, enhanced the presence of therapeutic CD8+ T cells within tumors, reduced tumor burden and prolonged survival. Notably, treatment with TCR+iIL-12 but not iIL-18 T cells resulted in enhanced intra-tumoral accumulation of macrophages, which was accompanied by a decreased frequency of therapeutic T cells, in particular of the CD8 subset. In addition, when administered to mice, iIL-18 but not iIL-12 demonstrated a favorable profile of T cell co-stimulatory and inhibitory receptors. In conclusion, we observed that treatment with T cells engineered with a TCR and iIL18 T cells is safe and able to skew the tumor microenvironment in favor of an improved anti-tumor T cell response.
KEYWORDS: cytokines, solid tumors, T cells, TCR gene therapy, tumor microenvironment
Introduction
Up to now, dozens of clinical trials have demonstrated the effectiveness of adoptive therapy with T cells that are gene-engineered with T cell receptors (TCRs) or chimeric antigen receptors (CARs). Therapeutic successes are most prominent for the treatment of hematological malignancies,1-6 whereas in the majority of patients with solid tumors, initial regressions are challenged by rapid tumor recurrences.
Patient responsiveness to immune therapies appears to be related to numbers and location of tumor infiltrating lymphocytes (TILs).7,8 Tumors that have low or negligible numbers of TILs may demonstrate intrinsic changes that result in limited T cell infiltration, movement and activation, and as such pose barriers to T cell treatments when compared to tumors with high numbers of TILs.9,10 In fact, predictive value towards response to clinical T cell therapies has recently been attributed to determinants of immunogenicity, such as extent of antigen presentation, composition of immune infiltrates, and presence of inhibitors of T cell activity.11 Expectedly, intervention with such determinants has the ability to enhance the sensitivity of tumors for T cell treatment.12
Here, we investigated to what extent local depositions of IL-12 and IL-18 in the tumor tissue would enhance the efficacy of adoptive T cell therapy. IL-12 is a heterodimer that consists of the subunits p35 and p40 and, although primarily produced by antigen-presenting cells, it is also a product of T cells.13,14 Initial studies showed IL-12's ability to enhance the cytolytic activity of T cells,15 aid in the recruitment of macrophages,16 and induce a TNFα-mediated shift towards M1-type macrophages.17,18 IL-18 is a pro-inflammatory IL-1-like cytokine that is primarily produced by macrophages and is able to drive T and NK cell maturation (reviewed in19). Both cytokines enable an immunological shift towards Th1-type T cell responses. Notably, IL-12 increases the proportion of T cells expressing the IL-18 receptor 1 subunit (IL-18R1), resulting in further enhancement of IFNγ production by IL-12 plus IL-18-stimulated T cells.20,21 Despite IL-12's beneficial impact on anti-tumor immune responses in preclinical models, which also holds in early-phase trials, systemic administration of recombinant IL-12 has been reported to result in hepatotoxicity and high systemic levels of IFNγ.22,23 In contrast, administration of IL-18 revealed no such toxicities in studies with melanoma and renal cell cancer patients, however, did not reveal clinical responses either.24-26 We argue that delivery of these two cytokines directly at the site of the tumor, either alone or together, will minimize toxic effects and at the same time maximize immune-stimulatory effects. In earlier studies, we have shown that the use of CAR-engineered T cells with inducible IL-12 (iIL-12) enhanced tumor killing without toxicity by increasing the numbers and activation of tumor infiltrating macrophages.18 In the current study, we have extended these findings and exploited TCR-engineered T cells to provide antigen-specific and intra-tumoral release of IL-12 and IL-18, and investigated the impact of these enhanced T cells on tumor growth, toxicity, T cell phenotype and function.
Materials and methods
Cell culture and reagents
The human embryonic kidney 293 T and Phoenix-Amp cell lines, both used to package retroviruses carrying RNA encoding TCRαβ and/or iIL-12 or iIL-18, were grown in DMEM with 10% (v/v) Fetal Bovine Serum (FBS; Greiner Bio-one Alphen a/d Rijn, The Netherlands), 200 nM L-glutamine, 1% (v/v) MEM non-essential amino acids and antibiotics (DMEM complete). Clones derived from mouse melanoma cell lines B16BL6 (B16WT) and B16:A2-YLEP (gene-engineered to express a fusion protein between the human glycoprotein (gp)100280-288 epitope (YLEPGPVTA) and the HHD molecule27) were cultured using the same medium, which in case of B16:A2-YLEP cells contained neomycin (1 mg/ml) (G418, Calbiochem, La Jolla, CA). In order to enhance HLA-A2 transgene expression, B16 cells were pre-treated with 100 IU/ml murine IFNγ (Sanquin, Amsterdam, The Netherlands) for 24 hours prior to in vitro assays. Mouse splenocytes were cultured in mouse T cell medium consisting of RPMI 1640 medium supplemented with 25 mM HEPES, 200 nM L-glutamine, 10% FBS, 1% MEM non-essential amino acids, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol, antibiotics and 50 IU/ml human recombinant IL-2 (Proleukin; Chiron, Amsterdam, The Netherlands). For flow cytometry the following antibodies and pMHC multimer were used: anti-TCR-Vβ14-FITC (Beckman Coulter, Fullerton, CA); CD3-PE, CD3-BV510, Ly6G-PE-Cy7, CD8-APC-Cy7, F4/80-APC-Cy7, CD4-BV650, 4-1BB-BV421, LAG3-BV421 (all BD Biosciences, San Jose, CA); TIM3-APC, BTLA-APC, PD-1-PE-Cy7, OX40-APC (all Biolegend, San Diego, CA); CD45-FITC, OX40-PE, CTLA4-PE, 4-1BB-PE, CD40 L-PE, CD4-Qdot605, LAG3-APC, ICOS-APC, ICOS-PE-Cy7, CD14-PerCP, CD335-eF660 (all eBiosciences, San Diego, CA); and gp100/A2Kb pMHC tetramer-PE (a kind gift by Prof. Andrew Sewell, Cardiff University, UK). For immunofluorescent stainings, the following primary and secondary antibodies were used: rat anti-mouse CD3 (clone 17A2), rat anti-mouse CD335 (clone 29A1.4), rat anti-mouse Ly6G (clone RB6-8C5) (all three from eBiosciences) and rat anti-mouse CD68 (clone FA-11; BioLegend); and donkey anti-rat IgG Alexa Fluor 488 (Life Technologies, Carlsbad, California, USA).
TCR and inducible IL-12 and IL-18 constructs
TCRα and β genes specific for the human gp100280-288 epitope presented by HLA-A2 (gp100/A2) were derived from CTL-296 clone,28 murinized and codon-optimized as described earlier.29 Subsequently, TCR genes were cloned into the pMP71 vector (kindly provided by prof. Wolfgang Uckert, Max-Delbrück Center, Berlin, Germany) with TCRα and β genes separated by an optimized T2 A ribosome skipping sequence. Sequences of murine IL-12 (a linked dimer consisting of p35 (NM_000882) and p40 (NM_002187)) and the processed form of IL-18 (NM_001562) were ordered from Geneart (not codon-optimized) and cloned into a pSIN vector, which contains 6 repeats of the NFAT response element and a minimal mIL-2 promotor, as described in.18 The plasmids with iIL-12 and iIL-18 contain a neomycin resistance gene to allow for selection of transduced T cells.
T cell transduction
Total mouse splenocytes were isolated, activated with Concanavalin A (0.5 µg/ml) and rhIL-2 in mouse T cell medium, and transduced with retroviral supernatant from a co-culture of single construct (either TCR, iIL-12 or iIL-18) transfected 293 T and Phoenix-Amp cell lines as described by Pouw and colleagues.29 To generate T cells that express TCR and either iIL-12 or iIL-18, T cells were simultaneously incubated with two retroviral supernatants in a 1:1 ratio (yielding TCR+iIL-12 or TCR+iIL-18 T cells). To obtain T cell populations that provide an inducible source for both cytokines, used in some experiments, TCR+iIL-12 and TCR+iIL-18 T cells were mixed in a 1:1 ratio (yielding TCR+iIL-12+iIL-18 T cells). Control (mock) T cells were transduced with empty retroviral vector. TCR and mock T cells were cultured as described above, whereas T cells harboring (in addition to the TCR) iIL-12 or iIL-18 underwent selection with 1 mg/ml of G418 starting at 24 h after transduction and lasting for 72 h (for a detailed flowchart of the T cell activation, transduction and selection procedure, please see Fig. 1A and supplementary figure 1 A, B).
Figure 1.

Protocol to generate T cells equipped with a TCR and an inducible cytokine construct. (A) Timeline for transduction procedure that yields TCR- and induced (i) cytokine-positive T cells Optimization of individual aspects of the procedure are described in materials and methods and supplementary figure 1. (B) At the end of T cell selection (day 7 after T cell activation) T cells that were transduced with TCR and iIL-12 or TCR+iIL-18 were labeled with CD8-APC antibody and gp100/HLA-A2 tetramer-PE. Mock and TCR-only transduced T cells were stained as controls. T cells were gated for live cells and dotplots are representative of three different experiments. Percentages in upper right quadrants represent fractions of CD8+ T cells binding to tetramer.
Adoptive T cell therapy
Experiments with mice were approved by the Experimental Animal Committee of the Erasmus MC Cancer Institute and carried out in accordance with institutional and national guidelines. For adoptive T cell therapy studies we used HLA-A2 transgenic (tg) mice that express a chimeric HLA-A*0201 transgene (HHD, referred to as HLA-A2).30 Adoptive transfer of T cells was done as described before.27 In short, at day -15, HLA-A2 tg mice were injected subcutaneously with 0.5 × 106 of a B16:A2-YLEP clone and at days -4 and -3 mice received a total of two Busulfan injections intraperitoneally (16.5 μg/kg ea.), followed a day later by a single Cyclophosphamide injection intraperitoneally (200 mg/kg). In some experiments, the mice were not transplanted with tumor cells, but followed the same conditioning and T cell treatment protocol. Mice were treated with T cells at day 0 and grouped according to one of the following treatments: mock T cells (number of T cells equal to TCR T cell group); TCR T cells (total number corrected for 7.5 × 106 transduced T cells); TCR+iIL-12 T cells; TCR+iIL-18 T cells; and TCR+iIL12+18 T cells). Tumor growth was measured by caliper 3 times a week and tumor volumes were estimated with the formula 0.4 x (AxB2) where A represents the largest diameter and B the diameter perpendicular to A. Tumor regression or response to treatment was defined as a >30% reduction in size compared to day 0, while relapse was defined as a size-increase of the tumor over the course of at least 3 days preceded by response to treatment. Survival was monitored daily for up to 45 days after administration of T cells. Body weight and edema like symptoms were recorded starting at the day of tumor inoculation and every third day thereafter. Peripheral blood was collected at day 6 after T cell transfer and at weekly intervals thereafter and used to determine numbers of administered T cells, whereas plasma was collected at days 16 and day 21 and used to measure cytokine levels. Tumor tissues of regressing tumors were collected at day 5 after T cell transfer and snap-frozen in liquid nitrogen and stored at -80°C for downstream assessment of cytokines in tumor lysates and performance of in situ immune fluorescence. Another part of tumor tissue was directly processed into TIL suspensions. To this end, tissue was cut into smaller fragments, incubated with collagenase (1 mg/ml; Sigma, St. Louis, MI) for 45 minutes at 37°C, checked microscopically and washed prior to flow cytometry.
Flow cytometry
T cells or TILs (5 × 105) were washed with PBS and incubated with antibodies at 4°C or with pMHC multimers at 37°C for 30 min. Following staining, T cells were washed again and fixed with 1% paraformaldehyde. Absolute T cell counts in mouse blood samples were determined using Flow-Count Fluorospheres. Events were acquired on a FACS Canto flow cytometer and analysed using FCS Express 4 software (BD Biosciences).
Measurement of cytokines
T cells (6 × 104/well of 96-wellsplate) were co-cultured with either antigen-positive or negative B16 cells (2 × 104/well) in a total volume of 200 µl of T cell assay medium (RPMI 1640, L-glutamine, 10% FBS, and antibiotics) for 24 h at 37°C and 5% CO2. Stimulation with medium was used as a control. Subsequently, supernatants were harvested and used to determine cytokine levels by standard ELISAs (IL-2, IL-10, IL-12, IFNγ, TNFα: eBioscience, San Diego, CA; IL-18: MBL, Nagoya, Japan). Plasma levels of selected cytokines were measured using the ProcartaPlex Mouse Th1/Th2/Th9/Th17/Th22/Treg Cytokine Panel (eBioscience) according to manufacturer's instructions. Tumor lysates were generated following sonication (3 cycles of 10 s using SoniPrep 150, MSE, London, UK) of frozen tissue suspended in PBS with protease inhibitors (Protease inhibitor cocktail tablets, Roche), and subsequently analyzed for concentrations of IL-12 and IL-18 via ELISA.
In situ immune fluorescence
Tissue sections were cut at 5 µm and fixed with acetone for 10 min, dried, washed, and blocked with PBS/10% donkey serum/0.3% Triton for 30 min prior to immune staining. Next, sections were incubated with the primary antibody overnight at 4°C, washed and incubated with the donkey anti-rat IgG Alexa Fluor 488 for 2 h at room temperature in the dark. Sections were covered with Vectashield, and kept subsequently at room temperature for 2 h and at 4°C overnight. Sections were examined microscopically (Leica, DM IL, 200x magnification) and photographed (Leica DFC 3000G camera and LAS4 software). Recorded photographs were analyzed using Fiji software.31 The number of pictures from each tumor ranged between 12 and 16, and using an in-house developed algorithm, the mean number of positively stained cells was determined, and normalized for percentages of nucleated cells (DAPI staining).
Statistical analyses
The different treatment groups were compared with the Student's t-tests, or the Mantel-Cox test in case of survival data, using GraphPad Prism5 (GraphPad Software, La Jolla, CA). P values < 0.05 were considered statistically significant.
Results
TCR+iIL-12 T cells, and to a lesser extent TCR+iIL-18 T cells, produce enhanced levels of IFNγ
We developed a protocol that yielded high numbers of T cells transduced with both the gp100/HLA-A2-specific TCR and iIL-12 or IL-18. Such inducible T cells showed maximal responsiveness towards antigen-positive tumor cells (see Fig. 1A and supplementary figure 1 A+B for details). Surface expression of TCR genes and binding to cognate pMHC ranged between 65–75%, irrespective of the presence of iIL-12 or iIL-18 (Fig. 1B). TCR+iIL-12 T cells produced >5.5 ng/106 cells of IL-12, and TCR+iIL18 T cells produced >75 pg/106 cells of IL-18 upon co-culture with antigen-positive cells (see Fig. 2A). Co-culturing TCR+iIL-12 T cells and TCR+iIL-18 T cells at a 1:1 ratio (TCR+iIL-12+iIL-18) demonstrated an expected drop in the antigen-specific production of IL-12 and IL-18. Notably, T cells that harbor iIL-12 produced minor amounts of IL-12 independent of TCR engagement, which was not the case for T cells harbouring iIL-18 (see Fig. 2A). When testing antigen-specific production of IFNγ by these T cell populations, we observed a significant increase in case T cells harbored iIL-12, iIL-18 or both (see Fig. 2B) when compared to TCR T cells. TCR+iIL-12 and TCR+iIL-12+iIL-18 T cells produced more than twice as much IFNγ than TCR+iIL-18 T cells. Furthermore, testing for antigen-specific production of IL-10 revealed that both TCR+iIL-12 and TCR+iIL-12+iIL-18 T cells produced significantly increased levels of this cytokine. While the various T cell populations also produced IL-2 as well as TNFα upon co-culture with antigen-positive B16 cells, their levels of were not affected by either iIL-12, IL-18 or both (see Fig. 2B).
Figure 2.
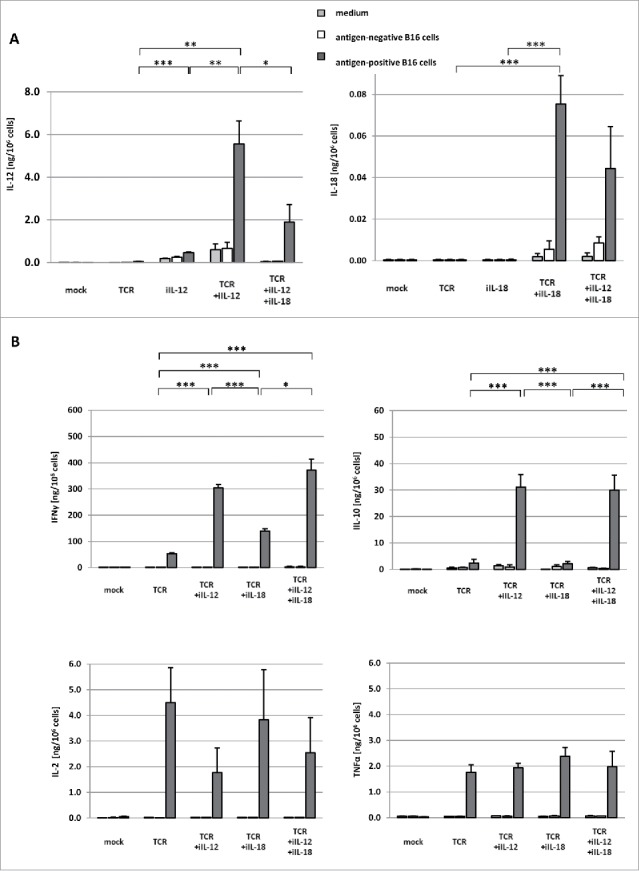
Inducible T cells produce IL-12 and IL-18 upon recognition of cognate antigen, which results in enhanced production of IFNγ. T cells were transduced with one or a combination of the following constructs: empty retroviral vector (mock); gp100 TCR (TCR); gp100 TCR and iIL-12 (TCR+iIL-12); gp100 TCR and iIL-18 (TCR+iIL-18). In an additional set of experiments, TCR+iIL-12 and TCR+iIL-18 T cells were mixed at a 1:1 ratio (TCR+iIL-12+iIL-18). These T cell populations were co-cultured with B16 melanoma cells that were either positive or negative for the gp100 target antigen at an effector:target (E:T) ratio of 3:1 for 24 h. As a control, T cells were cultured in medium only. (A) Levels of IL-12 and IL-18 in culture supernatants were measured via ELISA, and displayed as mean cytokine production per 106 T cells ±SEM (n = 5). (B) Levels of IFNγ, IL-10, IL-2 and TNFα in culture supernatants were measured via ELISA, and displayed as mean cytokine production per 106 T cells ±SEM (n = 5). Statistically significant differences between T cell populations that were co-cultured with antigen-positive B16 cells were calculated with Student's t-test: *p<0.05; **p<0.01; ***p<0.005.
Treatment of melanoma-bearing mice with TCR+iIL-18 T cells results in more cures, yet treatment with TCR+iIL-12 T cells adversely affects survival
We treated HLA-A2 transgenic mice with palpable B16:A2-YLEP tumor (∼400 mm3) either with 7.5 × 106 TCR, TCR+iIL-12, TCR+iIL-18, TCR+iIL-12+iIL-18 or mock T cells (see Materials and Methods for details). In the TCR T cell group, 66% of mice showed tumor regression, whereas in the TCR+iIL-12, TCR+iIL-18 and TCR+iIL-12+iIL-18 T cell groups these percentages were 66, 100 and 78, respectively (see Fig. 3A; for individual tumor growth curves, see supplementary Fig. 2). Moreover, treatment with TCR T cells resulted in complete responses (i.e., no detectable tumor) until the end of the experiment (day 45 after T cell transfer) in 11% of mice, a result that was not improved by treatment with TCR+iIL-12 T cells (see Fig. 3B). In contrast, treatment with TCR+iIL-18 or TCR+iIL-12+iIL-18 T cells resulted in complete responses in 33% of mice. In addition, in the group treated with TCR+iIL-12 T cells, mice suffered from tumor recurrence at earlier time points than mice in the group treated with TCR+iIL-18 T cells (see supplementary Fig. 2). Fig. 3B provides an overview of the fractions of complete, partial and no responses among the different treatment groups. When assessing survival, 29% and 14% of mice treated with TCR or TCR+iIL-12 T cells were alive at the end of experiment, respectively, whereas 57% and 43% of mice treated with TCR+iIL-18 or TCR+iIL-12+iIL-18 T cells were alive at the end of experiment (see Fig. 3C). Treatment-related mortality, defined as death of mice after T cell treatment not coinciding with tumor outgrowth, occurred in TCR+iIL-12 and TCR+iIL-12+iIL-18 T cell-treated mice, with percentages of mice that died within 14 days after T cell transfer being 33 and 22, respectively. Treatment-related mortality was absent in the TCR and TCR+iIL-18 T cell-treated mice (see Fig. 3B). Along these lines, we observed enhanced loss of weight in 56–78% (see supplementary Fig. 3) as well as edema-like toxicities in 29% of the treatment groups with iIL-12. Table I provides details on observed therapy-related effects.
Figure 3.

Treatment with TCR+iIL-18 T cells reduces tumor growth and prolongs survival. HLA-A2 transgenic mice bearing established tumors derived from inoculated B16:A2-YLEP cells were conditioned with Busulfan and Cyclophosphamide and treated with 7.5 × 106 T cells that were mock transduced or transduced with TCR, TCR+iIL-12, or TCR+iIL-18; or mice were treated with a mixture of 3.75 × 106 T cells transduced with TCR+iIL-12 and 3.75 × 106 T cells transduced with TCR+iIL-18 (TCR+iIL-12+iIL-18). (A) Waterfall graph of percentage change in tumor size between days 3 and 14 after T cell transfer, with each bar representing a single mouse. Tumor sizes were measured three times a week with a caliper. Dashed lines indicate average tumor size per treatment group. ‘†’ indicates death of mouse, in which case latest record of tumor size has been used. (B) Stacked bars representing percentages of mice with different responses to therapy at day 45 after T cell transfer. Complete response is defined as absence of a palpable tumor at the end of the experiment (day 45); partial response is defined as ≥ 30% tumor regression followed by relapse; no-response is defined as < 30% tumor regression; and therapy-related death is defined as death not coinciding with tumor progression (n = 9 per group). (C) Survival curves of treatment groups. Two mice of each group were sacrificed at day 5 after T cell transfer to collect tumors and are omitted from this figure (n = 7 per group).
Table 1.
Therapy-related side-effects of T cells gene-engineered with TCR and iIL-12 and/or iIL-18.
| Treatment | Maximum weight lossa | Edemab | Therapy-related deathc | Overall survivald |
|---|---|---|---|---|
| mock | 57% | 0% | 0% | 0% |
| TCR only | 11% | 0% | 0% | 29% |
| TCR+iIL-12 | 78% | 29% | 43% | 14% |
| TCR+iIL-18 | 0% | 0% | 0% | 57% |
| TCR+iIL-12+iIL-18 | 56% | 29% | 29% | 43% |
percentage of mice showing a loss of weight exceeding 10% of starting weight (day -15).
percentage of mice developing edema; mice were scored positive for edema according to both, visual evaluation by a pathologist and increase in body weight of >3.0% per day over a period of 7 or more days.
percentage of mice dying within the first 10 days after T cell administration while no tumor growth was detected.
percentage of mice alive at day 45.
Administration of TCR+iIL-12 T cells results in compromised T cell persistence and enhanced plasma levels of inflammatory cytokines
As peripheral T cell persistence has been directly linked to clinical anti-tumor efficacy,32 blood samples were taken at various time points after T cell transfer and analyzed by flow cytometry for the presence of TCR-engineered T cells. We observed increased numbers of pMHC-binding T cells in blood of TCR+iIL-18 and the TCR+iIL-IL-12+iIL-18 T cell-treated mice at day 6 after T cell transfer when compared to TCR T cell-treated mice (see Fig. 4A). Numbers of pMHC-binding T cells were decreased in the blood at day 6 after treatment with TCR+iIL-12 T cells. It is noteworthy, that the ratio of CD8 to CD4 positive T cells was significantly enhanced in the treatment groups that received treatment with iIL-18 when compared to the TCR+iIL-12 group (see Fig. 4B). These findings are in line with our in vitro observation that cultures of TCR+iIL-12 T cells yielded lower cell numbers when compared to cultures of TCR+iIL-18 or TCR T cells (see supplementary figure 1 A, lower panel). In addition to whole blood, we also collected plasma samples on day 16 after T cell transfer, which were analyzed via multiplex assay for the presence of cytokines (see Fig. 5). While plasma levels of IL-12 were low in all treatment groups (<7 pg/ml), plasma levels of IL-18 were considerably high in all groups (>700 pg/ml) and highest in mice treated with TCR+iIL-12+iIL-18 T cells (>1300 pg/ml). Of interest, in particular in relation to the above-mentioned toxicities, is the observation that plasma levels of IFNγ and TNFα were significantly higher in the TCR+iIL-12 and TCR+iIL-12+iIL-18 T cell groups (and negligible in the TCR and TCR+iIL-18 T cell groups). IL-2 plasma levels showed no significant difference among the tested groups. Plasma levels of IL-10 were increased in mice treated with TCR+iIL-12 T cells compared with those mice treated with TCR+iIL-18 and TCR+iIL-12+iIL-18 T cells.
Figure 4.

Treatment with TCR+iIL-12 T cells results in lowered numbers of CD8 and TCR transgene-positive T cells in blood. HLA-A2 transgenic mice bearing established B16:A2-YLEP tumors were conditioned and treated with T cells as described in legend to Figure 3. (A) Peripheral blood was collected from mice at the indicated time points after T cell transfer and absolute numbers of gp100/HLA-A2 pMHC-binding CD8 T cells were determined by flow cytometry. Data are presented as mean numbers per μl blood±SEM (n = 4–6). (B) depicts the mean ratio of CD8/CD4 positive therapeutic T cells at day 6 after T cell transfer ±SEM (n = 4–6). Statistically significant differences between treatment groups were calculated with Student's t-test: *p < 0.05; ** p< 0.01.
Figure 5.

Treatment with TCR+iIL-12 T cells results in increased plasma levels of inflammatory cytokines. Plasma samples were collected from mice on days 16 and 21 after T cell transfer and were screened for the presence of multiple cytokines via multiplex assay (“ProcartaPlex Mouse Th1/ Th2/ Th9/ Th17/ Th22/ Treg Cytokine Panel (17 plex)” by eBioscience). Concentrations of IL-12, IL-18, IFNγ, IL-2, TNFα and IL-10 on day 16 are presented as mean±SEM (n = 3). Statistically significant differences between treatment groups were calculated with Student's t-test: *p < 0.05; **p < 0.01.
Treatment with TCR+iIL-18 T cells results in enhanced accumulation of CD8 and TCR-transgene-positive T cells within the tumor
To better understand the in vivo behavior of TCR+iIL-12 and TCR+iIL-18 T cells, we assessed how the different treatments affect T cell infiltration into solid tumors and the phenotype of intra-tumoral T cells. To this end, we have performed flow cytometry analyses of TILs and observed an enhanced frequency of CD8+TCR+ T cells amongst CD3+ TILs upon treatment with TCR+iIL-18 T cells when compared to TCR+iIL-12 T cells (39 vs 9%) (see Fig. 6A, left graph). Looking into the ratio of CD8+ and CD4+ T cells, it became apparent that treatment with TCR+iIL-18 but not with iIL-12 T cells results in an enrichment of CD8+ TILs (∼ 7 vs 0.5, respectively; see Fig. 6A, right graph). Next, we studied the phenotype of TILs, in particular with respect to co-stimulatory as well as co-inhibitory receptor expression. The analysis of percentages of individual co-stimulatory or co-inhibitory receptors by CD8+ TILs, nor analysis of their co-expression by these cells, revealed no significant difference between treatment groups (see Fig. 6B and C). Also, phenotypic differences were not observed when analyzing CD4+ TILs or analyzing mean fluorescence intensities (data not shown).
Figure 6.
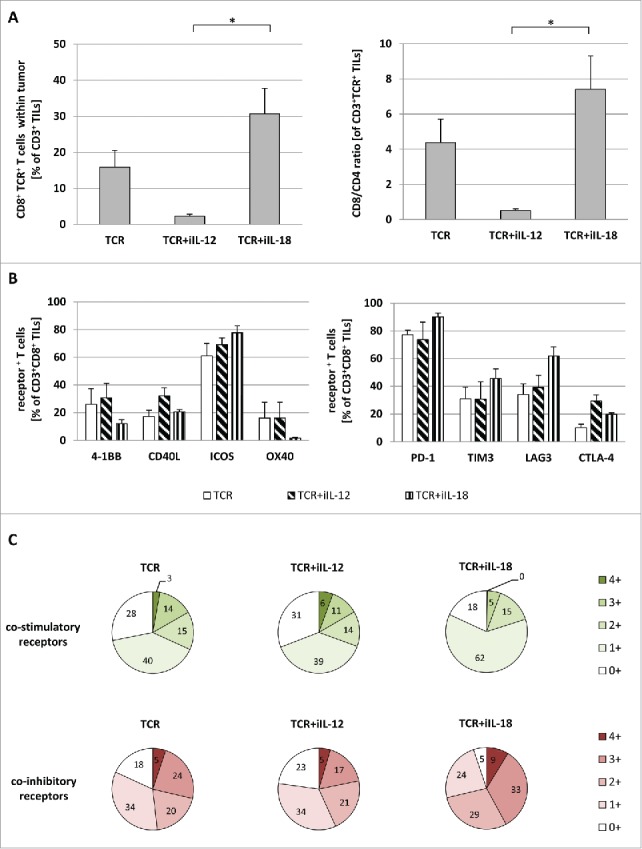
Upon treatment with TCR+iIL-18 T cells, numbers of CD8-positive TILs are enhanced without a change in expression of co-signaling receptors. Mice with regressing tumors (treated with TCR T cells with or without inducible cytokines) were sacrificed (n = 4 per group). TILs were isolated as described in Materials and Methods and analyzed via flow cytometry for the expression of CD3, CD4, CD8, TCR, 4–1BB, CD40 L, ICOS, OX40, PD-1, TIM3, LAG3 and CTLA-4. (A) depicts the percentage of CD8+TCR+ T cells amongst CD3+ TILs (left panel) and the CD8/CD4 ratio of TCR+ T cells within CD3+ TILs. Bars in (B) show the mean expression of individual co-stimulatory (left panel) and co-inhibitory (right panel) receptors of CD3+CD8+ TILs. (C) provides an overview of the degree of co-expression of co-stimulatory receptors (upper charts) as well as co-inhibitory receptors (lower charts). Statistically significant differences between treatment groups were calculated with Student's t-test: *p < 0.05.
Mice treated with TCR+iIL-12 T cells demonstrate extensive intra-tumoral infiltration of macrophages
In addition to numbers and phenotype of intra-tumoral T cells, we also assessed the cellular composition of the immune infiltrate in more detail. TILs from the different treatment groups were assessed by flow cytometry for the presence of T cells (CD3+), macrophages (CD68+), NK cells (CD335+) and neutrophils (Ly6G+). Strikingly, we observed that frequencies of macrophages were increased in mice receiving TCR+iIL-12 T cells, an observation that went hand in hand with a decrease in frequencies of T cells (see Fig. 7). Neither NK cell nor neutrophil numbers showed significant differences between treatment groups. To substantiate these findings we assessed the presence of IL-12 and IL-18 in lysates from the same tumor, and showed that the concentrations of IL-12 were higher in tumors from the groups treated with TCR+iIL-12 or TCR+iIL-12+iIL-18 T cells, whereas the concentrations of IL-18 were higher in tumors from the groups treated with TCR+iIL-18 when compared to treatment with TCR T cells. Endogenous levels of IL-18 ranged between 400 and 1000 pg/ml, while those of IL-12 were below 100 pg/ml (see supplementary Figure 4 A). Furthermore, we also assessed the composition of the immune infiltrate using in situ immune fluorescence, and quantified the various immune cell types per area tumor tissue. The numbers of T cells (CD3+), macrophages (F4/80+), NK cells (CD335+) and neutrophils (Ly6G+) that were found in the tumors from the different treatment groups confirmed the findings of the flow cytometric analysis (see supplementary Figure 4B and C). It is noteworthy that NK cells and neutrophils were readily detectable by flow cytometry, while staining for the same markers in tissues slices yielded no to negligible detection of either cell type. This discrepancy between both techniques most likely relates to the use of different antibodies in different applications.
Figure 7.
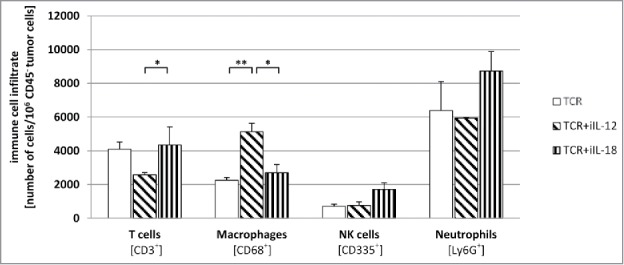
Mice treated with TCR+iIL-12 T cells show an enhanced number of tumor-infiltrating macrophages. Following T cell treatment, regressing tumors were collected at day 5 after T cell transfer. Tumors were dissociated into a single-cell suspension, stained with antibodies directed against CD3 (T cells), CD68 (macrophages), CD335 (NK cells) or Ly6G (neutrophils), and analyzed by flow cytometry. Figure depicts the mean number for each cell type ±SEM (n = 4). Statistically significant differences between treatment groups were calculated with Student's t-test: *p < 0.05; **p < 0.01.
Discussion
In this study, we compared the therapeutic impact of IL-12 and/or IL-18 secretion at the tumor site by antigen-triggered T cells. To this end, we equipped murine T cells both with gp100-specific TCRα/β genes and an inducible cytokine construct. T cells were antigen-responsive and, besides being producers of IL-12 or IL-18, produced enhanced levels of IFNγ. The production of IFNγ was prominent in case of TCR+iIL-12 T cells and highest in TCR+iIL-12+iIL-18 T cells, which is in line with previous reports where T cells were exposed to a combination of recombinant IL-12 and IL-18.33 TCR T cells harboring iIL-18, when transferred to mice bearing melanoma positive for the gp100 target antigen, resulted in enhanced and more durable anti-tumor responses when compared to T cells engineered with a TCR only or TCR and iIL-12. Furthermore, treatment with TCR+iIL-18 T cells, but not TCR+iIL-12 T cells, prolonged survival when compared to treatment with TCR T cells.
The low survival in the TCR+iIL-12 T cell treatment group is most likely a result of IL-12-mediated toxicities. Mice receiving TCR+iIL-12 or TCR+ iIL-12+iIL-18 T cells demonstrated elevated plasma levels of IFNγ and TNFα, clear weight loss, edema-like symptoms and in some cases death. The detection of IFNγ and TNFα in plasma is considered a consequence of IL-12-mediated productions of these cytokines by tumor-infiltrating immune cells.34 IFNγ is most likely derived from administered T cells following infiltration and activation in the tumor tissue. This notion is strengthened by our observation that CD335-positive NK cells, as an alternative source of IFNγ, were only present at low frequency within the tumors of treated mice. TNFα is most likely derived from macrophages since we detected an enhanced frequency of CD68 as well as F4/80-positive macrophages in tumors isolated from mice treated with TCR+iIL-12 T cells, and in vitro assays revealed no enhanced TNFα production by these T cells. Collectively, our findings extend previous clinical studies, where systemic administration of recombinant protein or cDNA encoding IL-12 was reported to result in enhanced production of IFNγ and TNFα, changes in blood vessel wall permeability, edema and even sepsis-like symptoms.34,35 In fact, a recent study demonstrated that treatment of melanoma patients with iIL-12-transduced TILs was accompanied by liver dysfunction, high fever, and hemodynamic instability.36 To further assess the observed toxicities, we also treated mice without tumor-implant with TCR+iIL-12 T cells and still observed enhanced weight loss when compared to mice treated with TCR T cells (see supplementary figure 3). These findings extend our in vitro observations showing target-independent release of IL-12 by these T cells (Fig. 2A) as well as those by Zhang and colleagues, who also reported non-specific release of IL-12 and reduced fitness when culturing iIL-12-expressing TILs.36 It is noteworthy that none of the mice without a tumor implant developed edema or showed therapy-related mortality, arguing that although toxicities are related to antigen-independent release of IL-12, they may exacerbate in the presence of antigen-positive tumor cells. Non-specific release of IL-12, in particular in the setting of TCR-engineered T cells, may be explained by the size of the inserted construct. IL-12 is a relatively large cytokine with 550 aa, while IL-18 encompasses 192 aa and, like other smaller constructs we tested (data not shown), shows no antigen non-specific release. Finally, we cannot exclude the contribution of host, co-treatment and tumor to the observed toxicities. In one of our earlier studies, where we tested iIL-12 in mouse models utilizing CAR-transduced T cells,18 we did not observe the toxicities as reported in this study, which may suggest that mouse strain (C57 BL/6 vs NIH-III), pre-conditioning protocol (Busulfan/Cyclophosphamide vs no pre-treatment), and type of tumor (melanoma cells vs colon carcinoma cells) affect the extent of IL-12-mediated toxicities.
Treatment with TCR+iIL-18 T cells, not revealing any toxicity, resulted in clearly enhanced anti-tumor responses. To better understand the immune-enhancing effect of this treatment, we have assessed numbers and phenotype of intra-tumoral T cells as well as the presence of other immune cell types within tumors. Notably, we observed an enhanced density of TCR-transduced TILs, in particular of the CD8 subset, when mice were treated with TCR+iIL-18 but not TCR+iIL-12 T cells. Enhanced numbers of CD8+ T cells within tumors are generally considered a beneficial factor with respect to tumor evolution and therapy response.7,37,38 The differential treatment effect with respect to numbers of TILs can be explained by two lines of arguments. First, our in vivo analysis of tumors showed that treatment with TCR+iIL-12 when compared to TCR+iIL-18 T cells results in an enhanced presence of macrophages (see Fig. 7 and supplementary Fig. 4). These results, together with enhanced TNFα levels in sera, argue that macrophages and their product TNFα may limit the infiltration of CD8+ T cells as was observed previously using various mouse models.39,40 Second, in vitro analysis of T cells demonstrated that iIL-18 when compared to iIL-12 provides T cells with high expression of co-stimulatory receptors and a low expression of co-inhibitory receptors (see supplementary figure 5). These traits may be representative of a preferred T cell phenotype at the time of adoptive transfer for an effective tumor response. Interestingly, when studying the phenotype of TILs, we observed no significant differences in the expression of co-stimulatory or inhibitory receptors by CD8+ TILs. When zooming in on TCR transgene-expressing TILs, however, we did observe that treatment with TCR+iIL-18 T cells resulted in enhanced expression of the co-stimulatory receptor ICOS and the co-inhibitory receptors PD-1 and CTLA-4, whereas treatment with TCR+iIL-12 T cells resulted in enhanced expression of the co-stimulatory receptors 4–1BB and CD40 L and the co-inhibitory receptors LAG3 and CTLA-4 (see supplementary figure 6). These latter data are difficult to interpret given the fact that numbers of TCR+ TILs are extremely low in case of treatment with TCR+iIL-12 T cells. With respect to treatment with TCR+iIL-18 T cells, ICOS may contribute to enhanced tumor infiltration and prolonged T cell persistence as was demonstrated in adoptive T cell therapy studies (41; Kunert, manuscript in preparation). PD-1, on the other hand, may mark successful antigen-specific TCR triggering and T cell activation.42 Notably, the fact that TCR+iIL-12 T cell treatment, when compared to TCR+iIL-18 T cells, results in the highest CTLA-4 and LAG3 expressions may provide an additional explanation for their lowered ability to mediate tumor clearance.
The above findings argue that IL-12 has potent immune-stimulatory effects, which may rapidly initiate the expression of immune checkpoints. In an effort to downscale negative feedback on T cell activation, we titrated down TCR+iIL-12 T cells and observed that lowering the numbers of these cells did not change the effects this cytokine has on the production of IFNγ nor the expression of PD-1 (supplementary Figure 7; same results were observed for production of IL-10 and expression of FOXP3, data not shown), arguing that it is difficult to maintain beneficial effects of IL-12 towards anti-tumor T cell activity while preventing the induction of a negative feedback. These data, together with the toxic effects we noted when using TCR+iIL-12 T cells, do not favor further therapeutic studies with T cells carrying iIL-12.
In conclusion, our findings demonstrate that the therapeutic use of TCR-engineered T cells equipped with inducible IL-12 leads to limited anti-tumor effects and severe toxicities in vivo, in part due to antigen non-specific release, whereas equipment of TCR T cells with inducible IL-18 results in enhanced anti-tumor responses without toxicities, most clearly related to increased accumulation of CD8 and TCR-transgene-positive T cells within tumors. Taken together, these findings advocate further studies towards the use of iIL-18 T cells to address therapy resistance in the setting of adoptive T cell therapy.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgement
The authors would like to thank the team of the Erasmus Optical Imaging Center, in particular Adriaan Houtsmuller, Gert van Capellen and Gert-Jan Kremers, for their support in the quantification of in situ stainings. Part of research was funded by the EU 7th framework ITN Grant ATTRACT.
References
- 1.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al.. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233-9. doi: 10.1200/JCO.2008.16.5449. PMID:18809613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, Levy D, Kubi A, Hovav E, Chermoshniuk N, et al.. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2010;16(9):2646-55. doi: 10.1158/1078-0432.CCR-10-0041. PMID:20406835 [DOI] [PubMed] [Google Scholar]
- 3.Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al.. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540-9. doi: 10.1200/JCO.2014.56.2025. PMID:25154820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al.. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917-24. doi: 10.1200/JCO.2010.32.2537. PMID:21282551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al.. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-17. doi: 10.1056/NEJMoa1407222. PMID:25317870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J, et al.. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914-21. doi: 10.1038/nm.3910. PMID:26193344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568-71. doi: 10.1038/nature13954. PMID:25428505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al.. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280 A in cancer patients. Nature. 2014;515(7528):563-7. doi: 10.1038/nature14011. PMID:25428504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegde PS, Karanikas V, Evers S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the Era of checkpoint inhibition. Clin Cancer Res. 2016;22(8):1865-74. doi: 10.1158/1078-0432.CCR-15-1507. PMID:27084740 [DOI] [PubMed] [Google Scholar]
- 10.Debets R, Donnadieu E, Chouaib S, Coukos G. TCR-engineered T cells to treat tumors: Seeing but not touching? Semin Immunol. 2016;28(1):10-21. doi: 10.1016/j.smim.2016.03.002. PMID:26997556 [DOI] [PubMed] [Google Scholar]
- 11.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18(1):248-62. doi: 10.1016/j.celrep.2016.12.019. PMID:28052254 [DOI] [PubMed] [Google Scholar]
- 12.Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S, Debets R. TCR-Engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of Tumor Milieu. Front Immunol. 2013;4:363. doi: 10.3389/fimmu.2013.00363. PMID:24265631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman RM, Romani N, Schuler G. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur J Immunol 1996;26(3):659-68. doi: 10.1002/eji.1830260323. PMID:8605935 [DOI] [PubMed] [Google Scholar]
- 14.Michelin MA, Abdalla DR, Aleixo AA, Murta EF. Peripheral helper lymphocytes produce interleukin 12 in cancer patients. Clin Med Insights Oncol. 2013;7:75-81. PMID:23515751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z, Palmer DC, Reger RN, Borman ZA, Zhang L, et al.. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70(17):6725-34. doi: 10.1158/0008-5472.CAN-10-0735. PMID:20647327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133-41. doi: 10.1182/blood-2011-12-400044. PMID:22354001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johansson A, Hamzah J, Ganss R. Intratumoral TNFalpha improves immunotherapy. Oncoimmunology. 2012;1(8):1395-97. doi: 10.4161/onci.20981. PMID:23243605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71(17):5697-706. doi: 10.1158/0008-5472.CAN-11-0103. PMID:21742772 [DOI] [PubMed] [Google Scholar]
- 19.Reddy P. Interleukin-18: recent advances. Curr Opin Hematol. 2004;11(6):405-10. doi: 10.1097/01.moh.0000141926.95319.42. PMID:15548995 [DOI] [PubMed] [Google Scholar]
- 20.Tominaga K, Yoshimoto T, Torigoe K, Kurimoto M, Matsui K, Hada T, Okamura H, Nakanishi K. IL-12 synergizes with IL-18 or IL-1beta for IFN-gamma production from human T cells. Int Immunol. 2000;12(2):151-60. doi: 10.1093/intimm/12.2.151. PMID:10653850 [DOI] [PubMed] [Google Scholar]
- 21.Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351-65. doi: 10.1084/jem.20120944. PMID:23209317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Portielje JE, Kruit WH, Schuler M, Beck J, Lamers CH, Stoter G, Huber C, de Boer-Dennert M, Rakhit A, Bolhuis RL, et al.. Phase I study of subcutaneously administered recombinant human interleukin 12 in patients with advanced renal cell cancer. Clin Cancer Res 1999;5(12):3983-9. PMID:10632329 [PubMed] [Google Scholar]
- 23.Ansell SM, Witzig TE, Kurtin PJ, Sloan JA, Jelinek DF, Howell KG, Markovic SN, Habermann TM, Klee GG, Atherton PJ, et al.. Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood. 2002;99(1):67-74. doi: 10.1182/blood.V99.1.67. PMID:11756154 [DOI] [PubMed] [Google Scholar]
- 24.Robertson MJ, Chang HC, Pelloso D, Kaplan MH. Impaired interferon-gamma production as a consequence of STAT4 deficiency after autologous hematopoietic stem cell transplantation for lymphoma. Blood. 2005;106(3):963-70. doi: 10.1182/blood-2005-01-0201. PMID:15817683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson MJ, Kirkwood JM, Logan TF, Koch KM, Kathman S, Kirby LC, Bell WN, Thurmond LM, Weisenbach J, Dar MM. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008;14(11):3462-9. doi: 10.1158/1078-0432.CCR-07-4740. PMID:18519778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, Kathman S, Pandite LN, Oei C, Kirby LC, et al.. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin Cancer Res. 2006;12(14 Pt 1):4265-73. doi: 10.1158/1078-0432.CCR-06-0121. PMID:16857801 [DOI] [PubMed] [Google Scholar]
- 27.Straetemans T, Berrevoets C, Coccoris M, Treffers-Westerlaken E, Wijers R, Cole DK, Dardalhon V, Sewell AK, Taylor N, Verweij J, et al.. Recurrence of melanoma following T cell treatment: continued antigen expression in a tumor that evades T cell recruitment. Mol Ther. 2015;23(2):396-406. doi: 10.1038/mt.2014.215. PMID:25363716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaft N, Willemsen RA, de Vries J, Lankiewicz B, Essers BW, Gratama JW, Figdor CG, Bolhuis RL, Debets R, Adema GJ, et al.. Peptide fine specificity of anti-glycoprotein 100 CTL is preserved following transfer of engineered TCR alpha beta genes into primary human T lymphocytes. J Immunol. 2003;170(4):2186-94. doi: 10.4049/jimmunol.170.4.2186. PMID:12574392 [DOI] [PubMed] [Google Scholar]
- 29.Pouw NM, Westerlaken EJ, Willemsen RA, Debets R. Gene transfer of human TCR in primary murine T cells is improved by pseudo-typing with amphotropic and ecotropic envelopes. J Gene Med. 2007;9(7):561-70. doi: 10.1002/jgm.1047. PMID:17471588 [DOI] [PubMed] [Google Scholar]
- 30.Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Perarnau B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2 m double knockout mice. J Exp Med. 1997;185(12):2043-51. doi: 10.1084/jem.185.12.2043. PMID:9182675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al.. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676-82. doi: 10.1038/nmeth.2019. PMID:22743772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173(12):7125-30. doi: 10.4049/jimmunol.173.12.7125. PMID:15585832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamura H, Kashiwamura S, Tsutsui H, Yoshimoto T, Nakanishi K. Regulation of interferon-gamma production by IL-12 and IL-18. Curr Opin Immunol. 1998;10(3):259-64. doi: 10.1016/S0952-7915(98)80163-5. PMID:9638361 [DOI] [PubMed] [Google Scholar]
- 34.Barrios B, Baez NS, Reynolds D, Iribarren P, Cejas H, Young HA, Rodriguez-Galan MC. Abrogation of TNFalpha production during cancer immunotherapy is crucial for suppressing side effects due to the systemic expression of IL-12. PLos One. 2014;9(2):e90116. doi: 10.1371/journal.pone.0090116. PMID:24587231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541-8. PMID:9326219 [PubMed] [Google Scholar]
- 36.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, et al.. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21(10):2278-88. doi: 10.1158/1078-0432.CCR-14-2085. PMID:25695689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, et al.. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203-13. doi: 10.1056/NEJMoa020177. PMID:12529460 [DOI] [PubMed] [Google Scholar]
- 38.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al.. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960-4. doi: 10.1126/science.1129139. PMID:17008531 [DOI] [PubMed] [Google Scholar]
- 39.Bertrand F, Rochotte J, Colacios C, Montfort A, Tilkin-Mariame AF, Touriol C, Rochaix P, Lajoie-Mazenc I, Andrieu-Abadie N, Levade T, et al.. Blocking tumor necrosis factor alpha enhances CD8 T-cell-dependent immunity in experimental melanoma. Cancer Res 2015;75(13):2619-28. doi: 10.1158/0008-5472.CAN-14-2524. PMID:25977337 [DOI] [PubMed] [Google Scholar]
- 40.Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, Clendenin C, Gladney WL, Knoblock DM, Guirnalda PD, et al.. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6 C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology. 2015;149(1):201-10. doi: 10.1053/j.gastro.2015.04.010. PMID:25888329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, Lee J, Posey AD Jr, Scholler J, Scholler N, et al.. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070-80. doi: 10.1182/blood-2013-10-535245. PMID:24986688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME, et al.. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246-59. doi: 10.1172/JCI73639. PMID:24667641 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.