

Abstract
Induction of the detoxifying enzyme heme oxygenase-1 (HO-1) is a critical protective host response to cellular injury associated with inflammation and oxidative stress. We previously found that HO-1 protein expression is reduced in brains of HIV-infected individuals with HIV-associated neurocognitive disorders (HAND) and in HIV-infected macrophages, where this reduction associates with enhanced glutamate release and neurotoxicity. Because HIV-infected macrophages are a small component of the cellular content of the brain, the reduction of macrophage HO-1 expression likely accounts for a small portion of brain HO-1 loss in HIV infection. We therefore investigated the contribution of astrocytes, the major pool of brain HO-1. We identified immunoproteasome-mediated HO-1 degradation in astrocytes as a second possible mechanism of brain HO-1 loss in HIV infection. We demonstrate that prolonged exposure of human fetal astrocytes to interferon-gamma (IFNγ), an HIV-associated CNS immune activator, selectively reduces expression of HO-1 protein without a concomitant reduction in HO-1 RNA, increases expression of immunoproteasome subunits, and decreases expression of constitutive proteasome subunits, consistent with a shift towards increased immunoproteasome activity. In HIV-infected brain HO-1 protein reduction also associates with increased HO-1 RNA expression and increased immunoproteasome expression. Finally, we show that IFNγ treatment of astrocytic cells reduces HO-1 protein half-life in a proteasome-dependent manner. Our data thus suggest unique causal links among HIV infection, IFNγ-mediated immunoproteasome induction, and enhanced HO-1 degradation, which likely contribute to neurocognitive impairment in HAND. Such IFNγ-mediated HO-1 degradation should be further investigated for a role in neurodegeneration in inflammatory brain conditions.
Keywords: interferon gamma, immunoproteasome, HIV-associated neurocognitive disorders, HAND
Brief Summary
Kovacsics et al. identify immunoproteasome degradation of heme oxygenase-1 (HO-1) in interferon gamma-stimulated astrocytes as a plausible mechanism for the observed loss of HO-1 protein expression in the brains of HIV-infected individuals, which likely contributes to the neurocognitive impairment in HIV-associated neurocognitive disorders.
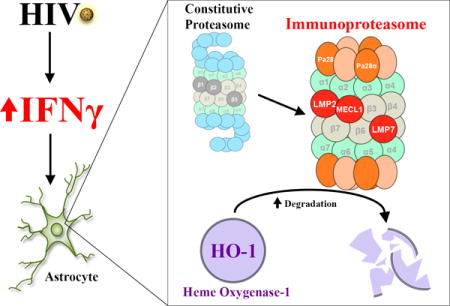
Introduction
We recently identified decreased expression of the phase II detoxifying enzyme heme oxygenase-1 (HO-1) in the brain prefrontal cortex of individuals with HIV-associated neurocognitive disorders (HAND), a spectrum of neurocognitive impairment that is diagnosed using neuropsychological testing and functional status assessments, and we further showed that HO-1 loss is linked to glutamate dysregulation in cultured HIV-infected macrophages (Gill et al. 2014; Gill et al. 2015). The heme oxygenase family consists of two expressed isoforms, HO-1 and HO-2, each of which catalyzes the degradation of free heme and generates equimolar amounts of carbon monoxide, bilirubin, and iron (Bauer et al. 2008; Morse and Choi 2002; Morse and Choi 2005; Ryter et al. 2000; Ryter et al. 2002). The HO-1 isoform is highly inducible with limited basal expression in most cell types among which neurons may express the lowest levels (Ambegaokar and Kolson 2014), while the HO-2 isoform is constitutively expressed in all cell types, including neurons (Munoz-Sanchez and Chanez-Cardenas 2014). Induced expression of HO-1 serves a critical function in the host’s response to acute cellular injury, including that resulting from inflammation and oxidative stress. (Ambegaokar and Kolson 2014; Gozzelino et al. 2010; Morse and Choi 2002; Morse and Choi 2005). The cytoprotective effects of HO-1 have been linked to its enzymatic degradation of the pro-oxidant heme and subsequent generation of carbon monoxide and bilirubin, which have anti-inflammatory and antioxidant properties; nonenzymatic cytoprotective functions have also been proposed (Gozzelino et al. 2010; Hori et al. 2002; Lin et al. 2007).
HO-1 expression is tightly controlled through transcriptional regulation by both repressive and activating factors associated with oxidative stress, including the antioxidant response activator, nuclear factor erythroid 2-related factor 2 (Nrf2) (Alam and Cook 2007; Salinas et al. 2004). Nrf2 binds to the antioxidant response element (ARE) present within the promoter of HO-1 (HMOX1) and other antioxidant genes, and induces expression of more than 200 cytoprotective and detoxifying effector genes, including NAD(P)H dehydrogenase [quinone] 1 (NQO1), and glutathione peroxidase 1 (GPX1) (Ishii et al. 2000). The net effect of Nrf2-driven transcriptional responses is the promotion of cell survival through restoration of cellular homeostasis, redox stability, and removal of injurious products.
Our previous in vivo studies showed reduced HO-1 protein expression in brain specimens from patients with a clinical diagnosis of HAND as well as in patients with a pathological diagnosis of HIV-encephalitis (HIVE) (Gill et al. 2015). HIVE is characterized by the presence of HIV-infected cells in the brain parenchyma, accompanied by formation of multi-nucleated giant cells, microglial nodules, microgliosis, astrogliosis, and perivascular inflammation (Gelman 2015). This loss of HO-1 protein expression in the brain of HIV-infected individuals is not accompanied by a lower protein expression of other ARE proteins, including NQO1 and GPX1, which suggests that the loss of HO-1 protein in HIV infection is relatively selective among the ARE proteins. This further suggests that transcriptional down-regulation of the ARE promoter element is not likely to be the driving force for the selective lowering of brain HO-1 in HAND (Cross et al. 2011; Gill et al. 2014). In contrast, our in vitro studies of HIV infection of macrophages (a major reservoir for HIV replication in the brain) showed that HIV replication results in selective reduction of both HO-1 RNA and protein expression; the parallel decrease in HO-1 RNA and protein suggests a transcriptional mechanism of HO-1 suppression that is relevant specifically to HIV infection of brain macrophages (Gill et al. 2015). However, macrophages represent a relatively small fraction of the total cellular content of the brain and neurons express very little to no basal HO-1. Astrocytes, which have detectable basal and rapidly inducible HO-1 expression, represent a large cellular pool of brain HO-1. We therefore sought to identify possible mechanisms of HIV-associated dysregulation of HO-1 expression in astrocytes, which are not a source of productive HIV replication (Boutet et al. 2001), but which could be modulated by inflammatory pathways triggered by HIV replication in brain macrophages.
Given the associations between HIV infection, brain prefrontal cortex HO-1 protein loss and markers of immune activation, we hypothesized that HIV-driven neuroinflammation can dysregulate astrocyte HO-1 expression in HIV infected brain (Gill et al. 2014). We determined the effects of several HIV-relevant immune mediators, including TNFα, IFNγ, and lipopolysaccharide (LPS) on heme oxygenase expression in primary human fetal astrocytes under conditions of both short- and long-term exposure, to mimic conditions of acute and chronic HIV brain infection relevant to HAND pathogenesis (Schrier et al. 2015). We considered both transcriptional and post-transcriptional mechanisms for the selective HO-1 loss. Through additional analyses of human brain tissue specimens and examination of cultured astrocytes, we have demonstrated the following: i) low HO-1 protein expression in HIV-infected brain tissue is accompanied by increased HO-1 RNA expression and induction of the immunoproteasome; ii) prolonged exposure to IFNγ in cultured human fetal astrocytes reduces HO-1 protein expression and increases HO-1 RNA and immunoproteasome expression; and iii) IFNγ decreases HO-1 protein half-life in cultured astrocytic cells and this effect is blocked by proteasome inhibitors. In fetal astrocytes the effects of IFNγ are biphasic, with an initial induction of HO-1 expression and a later (~15 days) sustained reduction of HO-1 expression, accompanied by a sustained induction of immunoproteasome subunit expression and reduction in constitutive proteasome subunit expression. This suggests that the reduction of HO-1 expression in the brain can be mediated by chronic effects of IFNγ exposure in astrocytes. Clincopathological effects of IFNγ expression in the CNS in HIV-infected individuals are supported by the demonstration of a strong correlation between the presence of IFNγ-expressing CD8+ T lymphocytes in cerebrospinal fluid and the severity of neurocognitive impairment in such individuals (Schrier et al. 2015). Thus, our studies have identified a plausible, novel mechanism of HO-1 loss in HIV-infected brain that is modulated by chronic IFNγ-induction of immunoproteasome-mediated degradation in astrocytes.
Materials and Methods
Supplemental Materials and Methods
Detailed descriptions of human cohort characteristics and additional protocols used can be found in Supplemental Methods and Supplemental Tables 1 and 2.
National NeuroAIDS Tissue Consortium (NNTC) brain autopsy cohort
A cohort consisting of 90 HIV-positive individuals and 66 HIV-negative individuals was selected from the NNTC autopsy cohort and was assembled by the Texas NeuroAIDS Research Center (Morgello et al. 2001; Nguyen et al. 2010). Among the 90 HIV-positive cases, 14 subjects had pathologically confirmed encephalitis (HIVE) and 76 subjects did not have HIVE (HIV+/HIVE−). NNTC site neuropathologists assigned the diagnosis of HIVE by the criteria of Budka et al. (Budka et al. 1991). The neurocognitive diagnosis of HAND was assigned by the supervising neuropsychologist according to the Frascati Criteria as reviewed by an experienced neurologist (Antinori et al. 2007; Woods et al. 2004). Sixty-three percent of HIV-positiveindividuals were antiretroviral therapy (ART)-experienced. HIV-negative subjects were significantly older than HIV-positive subjects (6.6 years), but did not significantly differ from HIV-positive subjects in gender, ethnicity, race, or post-mortem interval. Further details on demographic and clinical data of this cohort were described previously (Gill et al. 2014; Nguyen et al. 2010).
Cell culture and treatment of human primary fetal astrocytes and U251 cells
Cultures of primary human fetal astrocytes were generously provided by the Temple University Comprehensive NeuroAIDS Center (Supplemental Methods). Human primary fetal astrocyte cultures were prepared by the Comprehensive NeuroAIDS Center at Temple University School of Medicine. Astrocyte cultures were fed with astrocyte growth media (DMEM/F-12 supplemented with 15% FBS, 50ug/mL Gentamicin, 5ug/mL Fungizone, 2 mM L-glutamine, and 10ug/mL insulin) and maintained at 37°C and 5% CO2. The human U251 MG cell line, derived from human glioblstoma astrocytoma, was cultured in growth medium (DMEM supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/mL penicillin, and 100 μg/mL streptomycin) and maintained at 37°C and 5% CO2. Stock solutions of purified LPS (Sigma-Aldrich), recombinant human TNFα (Peprotech), and recombinant human IFNγ (Peprotech) were prepared in sterile phosphate buffered saline (PBS). Cells were exposed to vehicle (PBS), LPS (1 μg/mL), TNFα (10 ng/mL), and IFNγ (10 ng/mL) alone or in combination for indicated lengths of time and medium and treatments were replaced every 3 days. Stock solutions of the cell-permeable proteasome inhibitor MG132 (Sigma-Aldrich) were prepared in sterile DMSO and cells were treated with MG132 (5 μM) 2 hours prior to and throughout pulse-chase assays to inhibit proteasome activity.
[35S] Pulse-chase assay
U251 cells transiently expressing FLAG-HO-1 and continuously exposed to IFNγ (or vehicle) for 72 hours were used in pulse-chase assays. Detailed transfection methods are available in Supplemental Methods. Cells were treated with 5 μM of the cell permeable proteasome inhibitor MG132 (or dimethyl sulfoxide vehicle) for 2 hours, washed in HBSS, and starved for 25 minutes in labeling media (DMEM without L-glutamine/L-methionine/L-cystine (Corning) supplemented with 3% dialyzed FBS (HyClone), 2 mM L-glutamine, 1 mM sodium pyruvate, and 10 mM HEPES). Cells were then pulsed for 25 minutes with ~85 μCi [35S]L-methionine/L-cysteine (Perkin Elmer) diluted into 1 mL fresh labeling media, washed once with chase medium containing excess unlabeled L-methionine/L-cysteine (DMEM supplemented with 10% FBS, 2 mM L-Glutamine, 10 mM HEPES, and 5 mM each of L-methionine and L-cysteine (Calbiochem), and either immediately lysed (0 hour) or incubated in fresh chase medium at 37°C, 5% CO2 for indicated times (2, 4, 8, 12 hours). Cells were continuously exposed to IFNγ, MG132, and appropriate vehicles throughout each pre-treatment, starve, pulse, and chase period.
At indicated time points, cells were lysed for 30 minutes on ice in immunoprecipitation buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, and 1% Triton X-100) supplemented with Complete Protease Inhibitor Cocktail (Roche Applied Science), PhosSTOP Phosphatase Inhibitor Cocktail (Roche Applied Science), and 20 mM iodoacetmide (Sigma-Aldrich) and lysates were spun at 12,000 × g for 15 minutes at 4°C. An aliquot of the resulting supernatant fractions was used to determine total cellular protein degradation using [35S] liquid scintillation counting (see Supplemental Methods). Equivalent amounts of protein from supernatant fractions were incubated with anti-FLAG M2 affinity gel (Sigma-Aldrich) overnight at 4°C to isolate FLAG-HO-1 and eluted proteins were separated by SDS-PAGE. Dried gels were exposed overnight to a storage phosphor screen (Molecular Dynamics) and [35S]-labeled FLAG-HO-1 was detected by phosphorimaging with Typhoon FLA 7000 (GE Healthcare). Background-corrected signal intensity of bands was determined using Image Studio Lite (LI-COR Biosciences). Signal intensity across time was calculated relative to 0 hour within each treatment condition and data were combined from three independent biological replicates.
Statistical Analysis
All quantifications are expressed as mean ± standard error mean (SEM) and protein and RNA expression data were log transformed. In the NNTC brain autopsy cohort, HIV subgroups were compared by one-way ANOVA followed by Holm-Sidak post-test and linear trends were analyzed by Pearson’s correlation with line of best fit determined by linear regression. In cell culture experiments, statistical comparisons of two groups were made by paired t-test and comparisons of three or more groups were made by repeated measures one-way ANOVA with Holm-Sidak post test. Data from pulse-chase experiments were fit with one-phase exponential decay and extra sum of squares F test was used to compare the rate of decay (k). Effects of MG-132 were analyzed by two-way RM-ANOVA with Holm-Sidak post test. All statistical analyses were performed using GraphPad Prism 7 software (GraphPad Software). Significance was defined as P <0.05.
Study Approvals
Handling of NNTC-derived human autopsy specimens was in accordance with human subject protection protocols at participating institutions. Written consent was obtained for subjects at four collection sites in the USA: (a) The University of Texas Medical Branch, Galveston, Texas; (b) Mount Sinai Medical Center, New York, New York; (c) University of California, San Diego, California; and (d) University of California, Los Angeles, Los Angeles, California. Human fetal astrocyte cultures provided by the Temple University Comprehensive NeuroAIDS Center were prepared from fetal brain tissue obtained in full compliance with National Institutes of Health and Temple University ethical guidelines and approved by the IRB of Temple University.
Results
Reduced HO-1 protein in the prefrontal cortex is accompanied by increased HO-1 RNA in HIV-infected individuals
To determine whether transcriptional suppression of brain HO-1 in HIV infection was likely, HO-1 RNA expression was quantified in the dorsolateral prefrontal cortex (DLPFC) from the same HIV autopsy cohort that was studied previously (HIV-negative, HIV−, n=66; HIV-infected without encephalitis, HIV+/HIVE−, n=76; HIV-infected with encephalitis, HIVE+, n=14). HO-1 RNA was significantly increased in HIV+/HIVE− individuals as compared to HIV− individuals, with a further increase in HIVE+ individuals (Fig. 1A). In contrast, as we previously reported, HO-1 protein expression was significantly reduced in both HIV+/HIVE− and HIVE+ individuals in comparison with HIV− individuals (Fig. 1B) (Gill et al. 2014). HO-1 protein expression negatively correlated with HO-1 RNA expression in HIV+ individuals (HIV+/HIVE− and HIVE+), but there was no correlation in HIV− individuals (Fig. 1C). The discordance between HO-1 protein and RNA expression in HIV+ individuals suggested that CNS HIV infection might reduce HO-1 protein expression through a post-transcriptional mechanism.
Figure 1. HO-1 RNA is elevated and HO-1 protein is reduced in the dorsolateral prefrontal cortex of HIV-infected individuals.
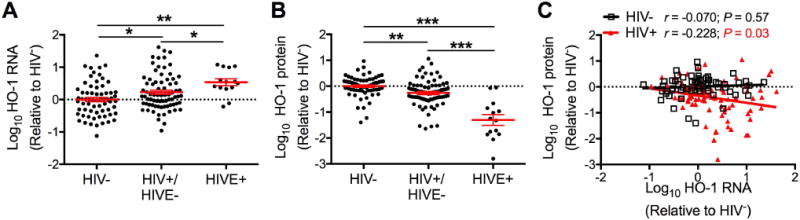
(A) HO-1 RNA expression in the DLPFC determined by real-time quantitative PCR. HIV− = (HIV-negative, n=66); HIV+/HIVE− = (HIV-positive without HIV encephalitis, n = 76); and HIVE = (HIV-positive with HIV encephalitis, n = 14). (B) HO-1 protein expression determined by Western blot in 66 HIV−, 75 HIV+/HIVE−, and 14 HIVE brain tissue samples. Fold-change in expression was calculated relative to the average of HIV− groups after normalization to GAPDH (RNA) or β-tubulin (protein). Data were log transformed and mean HIV− group value set to 0 (dotted line). Solid black lines indicate mean ± SEM. Groups were analyzed by ANOVA with post hoc Holm-Sidak test. *P < 0.05; **P < 0.01; ***P < 0.001. (C) Correlation between HO-1 RNA and protein expression in 65 HIV− and 89 HIV+ (75 HIV+/HIV− and 14 HIVE) was determined by Pearson’s correlation with line of best fit determined by linear regression.
Induction of immunoproteasome subunits is associated with reduced expression of HO-1 protein in the brains of HIV-infected individuals
The observed discordance between HO-1 protein and RNA expression could result from decreased HO-1 RNA translation or enhanced HO-1 protein degradation. Prior work showed that there is higher expression of immunoproteasome subunits within the DLPFC of HIV-positive individuals compared with HIV-negative individuals (Nguyen et al. 2010), so we hypothesized that HIV-driven induction of immunoproteasomes might enhance HO-1 degradation and ultimately reduce HO-1 protein expression. We correlated HO-1 RNA and protein levels with expression of immunoproteasome protein subunits LMP7 and PA28α, quantified previously by Western blot. HO-1 RNA correlated positively and significantly with LMP7 (Fig. 2A) and PA28α (Fig. 2D) protein in both HIV-positive and HIV-negative individuals. In contrast, in HIV-positive individuals, but not HIV-negative individuals, HO-1 protein negatively correlated with LMP7 (Fig. 2B) and PA28α (Fig. 2E) protein, which supported the hypothesis that HO-1 protein reduction and immunoproteasome induction may be functionally related and mediated by an HIV-driven process within the brain. HO-2 protein was not significantly correlated with LMP7 (Fig. 2C) or PA28α (Fig. 2F) protein in either HIV-positive or HIV-negative individuals, which suggested a relatively specific relationship between HO-1 isoform expression and immunoproteasome induction.
Figure 2. HO-1 protein loss is associated with immunoproteasome induction in HIV-infected brain.
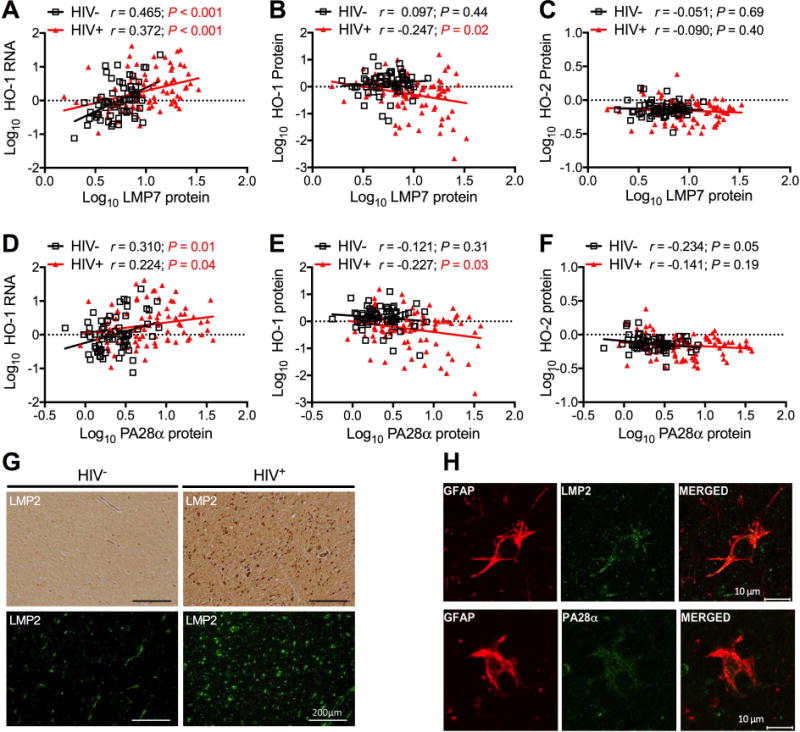
Expression of immunoproteasome subunits LMP7 (A, B, C) and PA28α (D, E, F) was determined by Western blot in DLPFC samples. Immunoproteasome expression was correlated with (A, D) HO-1 RNA in 64 HIV− and 87 HIV+ samples and with (B, E) HO-1 and (C, F) HO-2 protein in 65 HIV− and 88 HIV+ DLPFC samples. Correlations were determined by Pearson’s correlation with line of best fit determined by linear regression. (G) Immunoproteasome subunit LMP2 was visualized in subcortical white matter by immunohistochemistry (top panels) and immunofluorescence (bottom panels). Scale bar = 200 μm. (H) Immunoproteasome subunits were localized to astrocytes by dual indirect immunofluorescence staining of LMP2 (top panels) or PA28α (bottom panels) with the astrocyte marker GFAP in subcortical white matter of HIVE cases. Scale bar =10 μm.
To further define the association between immunoproteasome expression and HIV infection we attempted to localize subunit expression using immunohistochemistry and indirect immunofluorescence labeling. LMP2 immunoreactivity in frontal white matter was increased in subjects with HIVE compared with HIV-negative subjects (Fig. 2G) and in tissue from individuals with HIVE, LMP2 and PA28α colocalized with the astrocyte marker GFAP (glial fibrillary acidic protein) (Fig. 2H). LMP2 and PA28α expression was also detected in other glial cell types in frontal white matter in subjects with HIVE, as demonstrated by colocalization with the oligodendrocyte marker OMG (oligodendrocyte myelin glycoprotein) and the macrophage marker CD68 (Cluster of Differentiantion 68) (data not shown). These experiments suggested that in HIV-infected brain tissue immunoproteasome subunits are increased not only in those lineages that support HIV infection (macrophages and microglia), but also in lineages incapable of harboring productive HIV infection (astrocytes, oligodendrocytes). We therefore wanted to determine the effects of HIV-associated immune activators on HO-1 and proteasome subunit expression in cultured cells and focused specifically on astrocytes, as they are the major cellular source of HO-1 in the brain.
Prolonged IFNγ exposure reduces HO-1 protein, but not RNA, in human primary fetal astrocytes
Astrocytes are known to respond to various inflammatory stimuli associated with HIV infection, including TNFα, LPS, and IFNγ, and we hypothesized that such HIV-relevant immune stimuli might reduce HO-1 protein expression within astrocytes. Human primary fetal astrocytes were exposed to TNFα, LPS, and IFNγ alone or in combination for increasing periods of time (up to 15 days) to mimic the chronic inflammatory state of HIV-infected individuals. First, we determined effects of acute (24 hours) exposure. Expression of HO-1 (Fig. 3A) and HO-2 (Fig. 3B) RNA was not significantly altered by any of the inflammatory stimuli. HO-1 protein (Fig. 3C,D) was increased by TNFα exposure, alone or in combination with LPS. Notably, the increase in HO-1 protein expression with TNFα exposure was not observed when IFNγ was also present (Fig. 3C,D). HO-2 protein levels did not change under any treatment condition (Fig. 3E) and a modest, but significant increase in the ARE protein NQO1 was observed under several treatment conditions (Fig. 3F). We thus concluded that 24-hour exposure of astrocytes to TNFα, a known inducer of ARE gene expression, increases HO-1 and NQO1 expression, with no effect on HO-2 expression, indicating expected functional responses in our human fetal astrocyte cultures.
Figure 3. Acute (24 hours) exposure to IFNγ does not significantly reduce HO-1 expression in astrocytes.
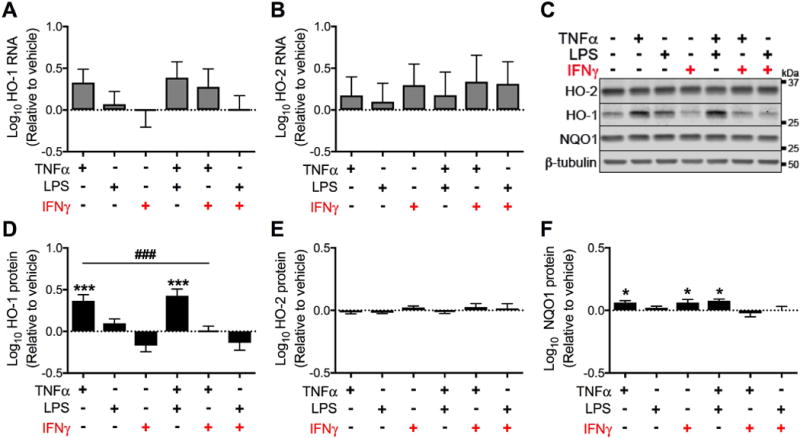
Primary human fetal astrocytes were exposed to TNFα, LPS, and IFNγ (alone or in combination) for 24 hours. (A) HO-1 and (B) HO-2 RNA expression was determined by real-time quantitative PCR. Fold change in expression was calculated relative to vehicle after normalization to ACTB (β-Actin). (C) Representative Western blot from a single biological replicate. Quantification of (D) HO-1, (E) HO-2 and (F) NQO1 protein expression relative to vehicle after normalization to β-tubulin. Data were log transformed and values represent mean ± SEM (n = 4 biological replicates) of the fold change in expression from vehicle (dotted line). Statistical comparisons were made by RM-ANOVA with post hoc Holm-Sidak test. *P < 0.05; ***P < 0.001; ****P < 0.0001 vs. vehicle. ###P < 0.001 for indicated comparison.
Given the ability of IFNγ to block TNFα-mediated HO-1 induction (Fig. 3D), we further investigated effects of immune activators over longer exposure times. Prolonged exposure to IFNγ (15 days) produced markedly discordant effects on HO-1 RNA and protein expression (Fig. 4). Expression of HO-1 RNA was not significantly altered by prolonged exposure to any of the inflammatory stimuli (Fig. 4A), but HO-1 protein was significantly reduced by IFNγ alone or in combination with TNFα or LPS (Fig. 4C,D). Less stringent statistical testing revealed a trend for increased HO-1 RNA following exposure to IFNγ alone or in combination with TNFα or LPS (Fig. 4A) (paired t-test vs. vehicle: IFNγ, P = 0.03; IFNγ + TNFα, P = 0.08; IFNγ + LPS, P = 0.03). In contrast, HO-2 RNA (Fig. 4B) and protein (Fig. 4C,E) were significantly increased after exposure to IFNγ, suggesting that IFNγ differentially regulates expression of these heme oxygenase isoforms. Finally, IFNγ significantly increased expression of NQO1 (Fig. 4F), an ARE effector protein, further supporting a relatively specific effect of IFNγ on HO-1 protein.
Figure 4. Prolonged (15 days) exposure to IFNγ significantly reduces HO-1 protein expression, but not RNA expression in astrocytes.
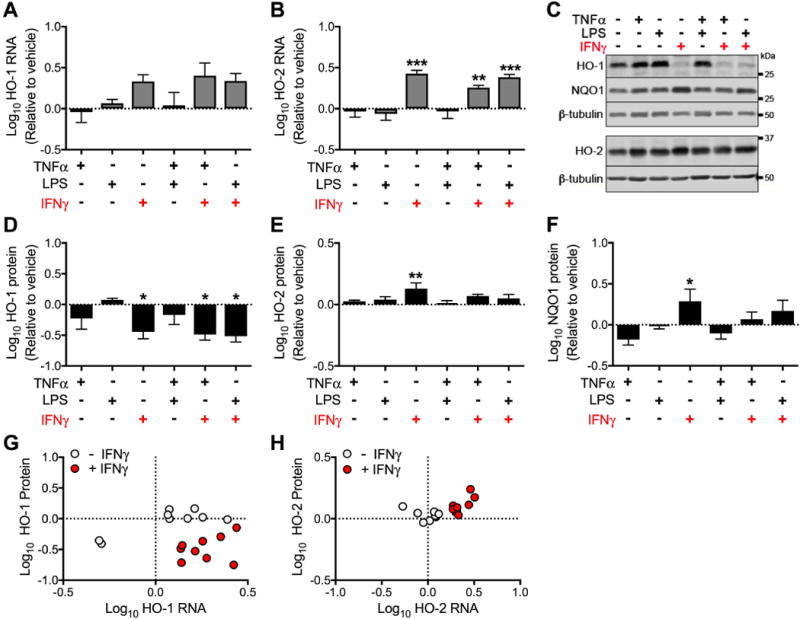
Primary human fetal astrocytes were exposed to TNFα, LPS, and IFNγ (alone or in combination) for 15 days. Media and treatments were replaced every 3 days. (A) HO-1 and (B) HO-2 RNA expression was determined by real-time quantitative PCR. Fold change in expression was calculated relative to vehicle after normalization. (C) Representative Western blot from a single biological replicate. Quantification of (D) HO-1, (E) HO-2 and (F) NQO1 protein expression relative to vehicle after normalization. Data were log transformed and values represent mean ± SEM (n = 4 biological replicates) of the fold change in expression from vehicle (dotted line). Statistical comparisons to vehicle were made by RM-ANOVA with post hoc Holm-Sidak test. *P < 0.05; **P < 0.01; ***P < 0.001. Fold change in RNA and protein expression of (G) HO-1 and (H) HO-2 were individually plotted for each treatment condition from (n = 3 biological replicates, treatments and protein and RNA extraction occured in parallel wells). Solid black circles represent TNFα, LPS, and TNFα + LPS; solid red circles represent IFNγ, IFNγ + TNFα, and IFNγ + LPS.
To represent the degree of concordance between protein and RNA expression, we plotted data from each biological replicate grouped according to the absence or presence of IFNγ. Exposure to IFNγ, alone or in combination with TNFα and LPS, was significantly associated with reduced HO-1 protein expression and increased RNA expression (Fig. 4G). These treatments were also significantly associated with increased HO-2 protein and RNA expression (Fig. 4H). These data demonstrate that prolonged exposure of primary human fetal astrocytes to IFNγ consistently reduces HO-1 protein expression, but not HO-1 RNA. These results suggested that the effects of IFNγ on astrocytes could account for the discordant pattern of HO-1 protein and RNA expression observed within the brain of HIV-infected individuals.
IFNγ induces expression of immunoproteasome subunits and reduces expression of constitutive proteasome subunits in astrocytes
HO-1 protein is an established target of proteasome-mediated degradation (Boname et al. 2014; Lin et al. 2008; Lin et al. 2013), and proteasome function and composition is markedly influenced by IFNγ (Stohwasser et al. 2000). We therefore asked whether the loss of HO-1 protein in IFNγ exposed astrocytes was associated with induction of the immunoproteasome. Accordingly, we determined the expression of the immunoproteasome regulatory subunit PA28α, the immunoproteasome catalytic subunits LMP2 and LMP7, and the constitutive catalytic subunits β1, β2, and β5 under the conditions of prolonged (15 days) exposure that consistently reduced HO-1 protein expression. PA28α protein was significantly increased after 15 days of exposure to TNFα alone and IFNγ alone or in combination with TNFα or LPS (Fig. 5A,B). Contrastingly, LMP7 (Fig. 5C) and LMP2 (Fig. 5E) proteins were significantly increased by each of the immune activators (and combinations thereof). Induction was strongest in the presence of IFNγ. In contrast, constitutive proteasome subunits β5 (Fig. 5C), β2 (Fig. 5D), and β1 (Fig. 5E) were significantly decreased by IFNγ alone or in combination with TNFα or LPS. TNFα or LPS alone did not alter expression of β5, β2, or β1. Interestingly, induction of immunoproteasome subunits without concurrent loss of constitutive proteasome subunits, as observed following prolonged TNFα exposure, is not associated with reduced HO-1 protein expression (Fig. 4D). Reduced HO-1 protein was observed following prolonged exposure to IFNγ (Fig. 4D), in association with a near complete loss of constitutive proteasome subunits. The latter finding suggested that replacement constitutive proteasomes with immunoproteasomes might be necessary for this reduction in HO-1 protein.
Figure 5. Prolonged (15 days) exposure to IFNγ significantly induces immunoproteasome subunit expression and decreases constitutive proteasome subunit expression in astrocytes.
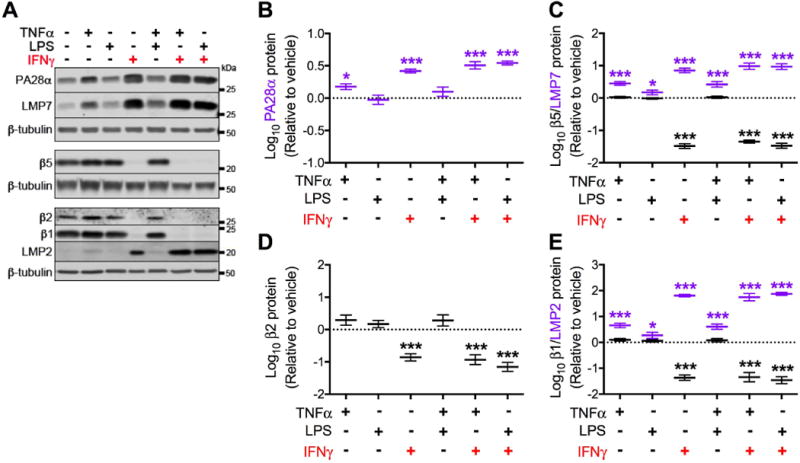
Primary human fetal astrocytes were exposed to TNFα, LPS, and IFNγ (alone or in combination) for 15 days. Media and treatments were replaced every 3 days. (A) Representative Western blot of immunoproteasome (PA28α, LMP7, LMP2) and constitutive proteasome (β5, β2, β1) subunits from a single biological replicate. Quantification of (B) PA28α, (C) LMP7 and β5, (D) β2 and (E) LMP2 and β1 protein expression relative to vehicle after normalization. Data were log transformed and values represent mean ± SEM (n = 4 biological replicates) of the fold change from vehicle (dotted line). Statistical comparisons to vehicle were made by RM-ANOVA with post hoc Holm-Sidak test. *P < 0.05; ***P < 0.001; **** P < 0.0001.
IFNγ-driven modulation of proteasome subunit expression and HO-1 protein reduction is dose-dependent
Given that we identified prolonged IFNγ exposure as a unique cause of reduced HO-1 protein expression in astrocytes, we determined the effects of IFNγ over a concentration range that includes those reported in the CSF of HIV-infected individuals (1–10 pg/mL) (Kamat et al. 2012). The response of human fetal astrocytes to a prolonged exposure to IFNγ appeared to be biphasic, with increased HO-1 protein expression observed up to 100 pg/ml, an apparent inflection point between 100 pg/mL and 1 ng/ml, and then a significant reduction in HO-1 protein at ≥ 10 ng/mL (Fig. 6A and Supp. Info. Fig. 1A). In contrast, IFNγ consistently increased expression of both HO-2 and NQO1 proteins (Supp. Info. Fig. 1B,C). At doses between 100 pg/mL and 1 ng/ml IFNγ, the observed reduction in HO-1 protein was accompanied by a significant reduction in the expression of constitutive proteasome subunits β2, β5, and β1 (Fig 6B and Supp. Info. Fig 1D,E). In contrast, immunoproteasome subunits PA28α, LMP7, and LMP2 were significantly increased starting at 10 pg/mL IFNγ, with significantly enhanced induction observed at 1 ng/mL IFNγ (Fig 6B,C and Supp. Info. Fig. 1E). As reduced HO-1 protein was associated with induction of immunoproteasome subunits and concurrent loss of constitutive proteasome subunits, these data gave added support to the hypothesis that the replacement of constitutive proteasomes by immunoproteasomes is critical for IFNγ-driven HO-1 protein loss in astrocytes.
Figure 6. IFNγ effects on expression of proteasome subunits and HO-1 are dose-dependent.
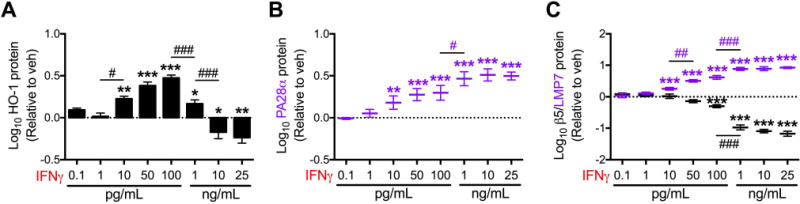
Primary human fetal astrocytes were exposed to IFNγ (0.1 pg/mL – 25 ng/mL) for 15 days. Media and treatments were replaced every 3 days. Western blot densitometry quantification of (A) HO-1, (B) PA28α, (C) LMP7, and β5 protein expression relative to vehicle after normalization to β-tubulin. Data were log transformed and values represent mean ± SEM (n = 4 biological replicates) of the fold change from vehicle (dotted line). Statistical comparisons to vehicle were made by RM-ANOVA with post hoc Holm-Sidak test. *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001. Statistical comparisons between consecutive IFNγ doses were made by RM-ANOVA with post hoc Holm-Sidak test. #P < 0.05; ##P < 0.01; ###P < 0.001; ####P < 0.0001.
IFNγ-driven modulation of proteasome subunit expression and HO-1 reduction is time-dependent
To assess the kinetics of immunoproteasome induction and constitutive proteasome loss associated with IFNγ exposure, astrocytes were exposed to IFNγ (10 ng/mL) or vehicle continuously for up to 15 days. We hypothesized that the loss of constitutive proteasome subunits would be slow given the relatively long half-life of constitutive proteasome complexes (~5 days) and HO-1 protein expression would be decreased only at later time points associated with this progressive loss of constitutive proteasomes (Heink et al. 2005; Khan et al. 2001; Tanaka and Ichihara 1989). HO-1 RNA expression was increased by day 9 and sustained through day 15 (Supp. Info. Fig. 2A), however no significant increase in HO-1 protein was seen at any time point and HO-1 protein was significantly reduced at day 15 (Supp. Info. Fig. 2C,D). In contrast, HO-2 RNA and protein were significantly increased by day 3 and this persisted through day 15 (Supp. Info. Fig. 2B,C,E). Furthermore, the ARE protein NQO1 was significantly increased by IFNγ at day 9 and this persisted through day 15 (Supp. Info. Fig. 2C,F). We thus concluded that the discordant effect of prolonged IFNγ on HO-1 RNA and protein expression is relatively specific to HO-1.
The expression of both immunoproteasome and constitutive proteasome subunits also consistently changed with duration of IFNγ exposure. Immunoproteasome subunits PA28α, LMP7, and LMP2 were increased by day 3 and remained elevated at each subsequent time point (Supp. Info. Fig. 2G,I,J). PA28α expression remained at the same elevation achieved at day 3, while expression of LMP7 and LMP2 increased through day 6 and then remained at the same elevated level. In contrast, the constitutive proteasome subunits β2, β5, and β1were progressively reduced through day 15 (Supp. Info. Fig. 2H–J). The progressive loss of β5 and β1 at each time point suggests that the loss of these constitutive subunits combined with the increased expression of the immunoproteasome subunits may account for the reduction of HO-1 protein expression. This is consistent with previously reported IFNγ-driven replacement of constitutive proteasomes by immunoproteasomes (Groettrup et al. 2001; Heink et al. 2005; Meiners et al. 2014; Rivett et al. 2001; Tanaka 2013) and it is further consistent with a role for the immunoproteasome in mediating HO-1 protein loss with IFNγ treatment.
IFNγ increases the rate of HO-1 protein degradation mediated by proteasome-dependent mechanisms
Given the coordinated reduction in HO-1 protein expression and induction of immunoproteasomes with reciprocal loss of constitutive proteasomes in IFNγ-exposed astrocytes, we hypothesized that HO-1 protein is degraded more rapidly by the immunoproteasome. To test this, we measured HO-1 protein half-life in astrocytic cells exposed to IFNγ. We used the U-251 MG astrocytic cell line, derived from a human glioblastoma, as the use of human primary fetal in pulse-chase experiments is technically impractical. We first examined effects of IFNγ on expression of HO-1, HO-2, and proteasome subunits to ensure that this cell line appropriately reproduced the effects observed in primary human astrocytes. After 3 days of exposure to IFNγ, HO-1 protein was significantly reduced while HO-1 RNA and HO-2 RNA and protein expression was significantly increased, similar to our observations in primary fetal astrocytes (Supp. Info. Fig. 3). Additionally, IFNγ increased the expression of immunoproteasome subunits LMP7 and LMP2 and decreased expression of the corresponding constitutive β5 and β2 subunits (Supp. Info. Fig. 3). Thus, the discordance between RNA and protein levels seen for HO-1, but not HO-2, and the shift to expression of immunoproteasomes after IFNγ exposure in this cell line recapitulates the effects of prolonged IFNγ seen in human primary fetal astrocytes.
We next determined the rate of FLAG-HO-1 degradation in transfected U-251 cells exposed to IFNγ for 3 days using [35S] liquid scintillation following FLAG-HO-1 immunoprecipitation. As measured by [35S] liquid scintillation counting of TCA-precipitable lysates, IFNγ did not alter the rate of total cellular protein degradation (Fig. 7A). Pulse chase experiments demonstrated that IFNγ significantly increased the rate of FLAG-HO-1 protein degradation, reducing the half-life of HO-1 protein from approximately 14 hours to 7.7 hours (Fig. 7B,C). We next determined the contribution of proteasome activity to HO-1 degradation by pre-treating cells with MG-132, a potent inhibitor of proteasome activity. Treatment with MG-132 significantly increased the amount of [35S]-labeled FLAG-HO1 protein remaining at each time point in both vehicle and IFNγ treatment conditions (Fig. 7B,C), suggesting that the observed loss of HO-1 protein is mediated, at least in part, by activity of proteasome complexes. These data demonstrate that HO-1 protein is degraded more rapidly in cells exposed to IFNγ and strongly implicate immunoproteasomes as the source of enhanced HO-1 protein degradation.
Figure 7. IFNγ increases the rate of proteasome-dependent HO-1 degradation.
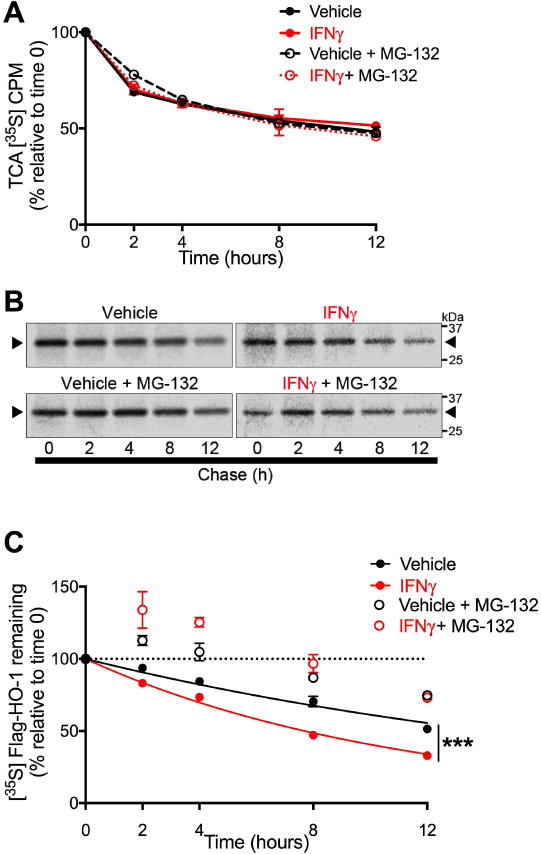
U251 cells transiently expressing FLAG-HO-1 and exposed to IFNγ (10 ng/mL) for 3 days were used in pulse chase assays to measure HO-1 degradation. Cells were pulse labeled with [35S]methionine/cysteine and chased for indicated time points. Degradation mediated by proteasome activity was assessed by treating cells 2 hours prior to labeling with the proteasome inhibitor MG-132 (5 μM) or DMSO (vehicle). (A) Intracellular protein degradation was determined by [35S] liquid scintillation counting of TCA-precipitable lysate fractions. Radioactivity was measured as counts per minute (CPM) and are expressed as a percentage relative to time 0 within each independent experiment (n=3). Data represent mean ± SEM. Statistical analysis of treatment conditions at each time point was performed by two-way RM-ANOVA with post-hoc Holm-Sidak test. (B) Representative phosphorimage used for densitometric quantification of radiolabeled FLAG-HO-1. Black arrows indicate radiolabeled FLAG-HO-1. (C) Degradation of HO-1 protein was determined by densitometric quantification of radiolabeled FLAG-HO-1 and are expressed as a percentage relative to time 0 within each independent experiment (n=3). Data represent mean ± SEM. Protein half-life in vehicle and IFNγ conditions (closed circles) was determined by one-phase exponential decay and rate of decay (k) was compared by Extra sum of squares F test. ***P < 0.001. The effects of MG-132 at each time point was analyzed by two-way RM-ANOVA with post hoc Holm-Sidak test (significance not shown in figure). IFNγ vs. IFNγ+MG-132: P < 0.001 at all time points. PBS vs. PBS + MG-132: P < 0.01 at 8hr; P < 0.001 at 2, 4, and 12 hr.
Discussion
The introduction of antiretroviral therapy (ART) has significantly transformed HIV infection from a uniformly lethal disease into a manageable chronic condition in which neurocognitive dysfunction persists in a significant proportion of ART-treated individuals (Broder 2010; Heaton et al. 2011; Saylor et al. 2016; Simioni et al. 2010). Despite abundant evidence implicating persistent inflammation and oxidative stress in the pathogenesis of HIV-associated neurocognitive disorders, the mechanisms by which these processes drive neurocognitive impairment in HIV-infected individuals remain incompletely understood (Babbe et al. 2000; Bandaru et al. 2007; Eden et al. 2007). We previously identified reduced HO-1 protein in HIV-infected brain as a host factor potentially contributing to HAND pathogenesis and we further suggested targeting HO-1 brain loss as an adjunctive neuroprotective strategy in ART-treated individuals (Gill et al. 2014; Gill et al. 2015).
Our new data demonstrate that the decreased HO-1 protein expression in HIV-infected brain is accompanied by increased HO-1 RNA expression, suggesting a post-transcriptional mechanism of HIV-mediated HO-1 protein loss, and we have identified enhanced immunoproteasome-mediated degradation of HO-1 in IFNγ-exposed astrocytes as a potential mechanism of HO-1 loss in the HIV-infected brain. In both HIV-infected brain and in human astrocytes exposed to IFNγ, we observed reduced HO-1 protein expression but did not detect reduced expression of other members of the ARE/Nrf2-driven family nor the ARE-independent heme oxygenase isoform HO-2 (Gill et al. 2014), which suggests that HO-1 is selectively affected. Furthermore, this decrease in HO-1 protein was not accompanied by decreased HO-1 RNA expression in either HIV-infected brain or astrocytes exposed to IFNγ in vitro. Additionally, we have previously demonstrated that HIV-infected subjects have increased brain expression of immunoproteasomes subunits (PA28α and LMP7) and that this increased expression associates with neurocognitive impairment, particularly with neuropsychological subtests associated with executive functioning and speed of processing (Nguyen et al. 2010). We now show that IFNγ treatment of primary human astrocytes increases expression of regulatory and catalytic immunoproteasome subunits (PA28α, LMP2, LMP7) while progressively decreasing expression of constitutive catalytic proteasome subunits (β1, β2, β5) and that the loss of constitutive proteasome subunits continues (15 days) beyond the time at which immunoproteasome induction is maximal (9 days). This continued, progressive loss of constitutive proteasome subunits in IFNγ-exposed astrocytes appears necessary for the reduced expression of HO-1 protein, which suggests a progressive replacement of constitutive proteasomes by immunoproteasomes. Finally, treatment of astrocytic cells with IFNγ significantly reduced HO-1 protein half-life by approximately 2-fold, and this was blocked by a broad-spectrum proteasome inhibitor. This implicates proteasome activity in HO-1 degradation, and we believe the immunoproteasome is responsible for the enhanced degradation of HO-1 protein as the reduced HO-1 protein half-life observed after IFNγ exposure is associated with increased expression of immunoproteasome subunits and reduced expression of constitutive subunits. However, a role for constitutive proteasomes cannot be absolutely ruled out because MG-132 not only inhibits activity of immunoproteasomes but also activity of constitutive proteasomes. Nonetheless, these observations strongly suggest a plausible link between IFNγ-induced accelerated immunoproteasome-mediated HO-1 degradation in astrocytes and brain HO-1 loss in HIV-infected individuals with HAND. This potential mechanism is particularly relevant in light of recent reports of a correlation between the presence of IFNγ-secreting CD8+ T lymphocytes and increased IFNγ in CSF and the diagnosis of HAND in HIV-infected individuals (Cassol et al. 2014; Schrier et al. 2015).
We examined effects of both acute (24 hours) and prolonged (15 days) exposure to immune activators associated with HIV infection of the brain because neurodegeneration associated with HIV infection is believed to progress over years, during which time persistent immune activation within the CNS is observed. The immune activators TNFα, LPS, and IFNγ are elevated in HIV-infected individuals and associated with HAND, and each has been shown to modulate the expression of HO-1 after acute exposure in various cell types, although effects on HO-1 after prolonged exposure have not been clearly defined (Ambegaokar and Kolson 2014; Ancuta et al. 2008; Brenchley et al. 2006; Griffin 1997; Wagener et al. 2003; Wesselingh et al. 1997). Increased expression of IFNγ and neopterin, an indirect measure of IFNγ activity produced by macrophages and T cells, is observed in serum, CSF, and brain parenchyma in HIV-infected individuals, including those receiving ART (Fuchs et al. 1989a; Fuchs et al. 1989b; Griffin et al. 1991; Kamat et al. 2012; Shapshak et al. 2004). Our observation that prolonged IFNγ exposure reduces HO-1 protein expression in human primary astrocytes and is associated with induction of immunoproteasome subunits and concomitant loss of constitutive proteasome subunits is novel and likely relevant to HIV neuropathogenesis, as we have observed a significant negative correlation between expression of HO-1 protein and immunoproteasome subunits in HIV-infected brain autopsy specimens. Furthermore, we detected immunoproteasome subunit expression by indirect immunofluorescence labeling in astrocytes, oligodendrocytes and macrophages in prefrontal brain tissue sections from individuals with HIVE. We thus speculate that chronic CNS inflammation associated with HIV infection drive immunoproteasome expression and associated HO-1 degradation.
The concomitant increase in immunoproteasome subunit expression and progressive decrease in constitutive proteasome subunit expression likely reflects a process of gradual replacement of constitutive proteasome complexes with functional immunoproteasome complexes that are responsible for enhanced degradation of HO-1 protein. This apparent requirement for replacement of constitutive proteasomes by immunoproteasomes implies that the functional balance between activities of constitutive and immunoproteasome complexes influences HO-1 protein expression. Newly synthesized immunoproteasome subunits do not replace corresponding constitutive subunits within existing proteasome complexes; instead, they are preferentially incorporated over constitutive subunits into de novo complexes (Fruh et al. 1994; Heink et al. 2005; Nandi et al. 1997). Thus, modest induction of immunoproteasome subunits without concurrent loss of constitutive proteasomes is unlikely to dramatically shift the balance between constitutive proteasomes and immunoproteasomes (Aki et al. 1994). Although IFNγ induces expression of immunoproteasome subunits, it does not suppress the expression of constitutive subunits, and thus it does not directly drive the loss of constitutive proteasome complexes (Akiyama et al. 1994; Nandi et al. 1996). Increased incorporation of immunoproteasome subunits into nascent complexes effectively excludes constitutive subunits and leaves them vulnerable to rapid degradation in this “free” form (Heink et al. 2005; Murata et al. 2009). As more immunoproteasome complexes are formed, the pool of pre-existing constitutive proteasome complexes is reduced through degradation, with a half-life of approximately 5 days that is unaffected by IFNγ (Nandi et al. 1997). The extent of constitutive proteasome replacement by immunoproteasomes within the HIV-infected brain is not yet clear, but the increased expression of immunoproteasome subunits that we observed likely represents a functionally important shift favoring immunoproteasome complexes that results in loss of HO-1 protein.
In the human brain specimens that we examined, we identified discordant correlations between expression of immunoproteasome subunits and expression of HO-1 RNA and protein. In HIV-positive individuals, but not in HIV-negative individuals, there was a significant negative correlation between expression of immunoproteasome subunits and expression of HO-1 protein. Conversely, there was a significant positive correlation between expression of HO-1 RNA and expression of immunoproteasome subunits in both HIV-infected and non-infected individuals. This suggests that HIV infection does indeed drive the discordance between expression of HO-1 RNA and protein through a post-translational mechanism, and our in vitro astrocyte experiments suggest that this mechanism is immunoproteasome degradation of HO-1. While in this study we specifically identify and define IFNγ-induced immunoproteasome degradation of HO-1, other proinflammatory cytokines as well as oxidative stress induce the immunoproteasome (Ferrington and Gregerson 2012). Thus, HIV-associated CNS inflammation and oxidative stress in addition to elevated IFNγ is likely also contributing to immunproteasome induction and subsequent degradation of HO-1.
The mechanism responsible for the proposed IFNγ-induced degradation of HO-1 protein by immunoproteasome complexes is not clear. Several mechanisms of post-transcriptional regulation of HO-1 expression have indeed been identified and include regulation by miRNA, de-adenylation and altered turnover of HO-1 RNA, HO-1 RNA interactions with RNA-binding proteins, and HO-1 protein degradation by the ubiquitin proteasome system; whether any of these processes are directly modulated by IFNγ is not known (Amadio et al. 2014; Boname et al. 2014; Cheng et al. 2013; Leautaud and Demple 2007; Lin et al. 2008; Lin et al. 2013; Ma et al. 2013). Intrinsic properties of the catalytic subunits of immunoproteasomes might directly promote HO-1 degradation, as immunoproteasomes show enhanced cleavage rates and increased preference for specific cleavage sites compared to constitutive proteasomes (Basler et al. 2004; Dahlmann et al. 2000; Mishto et al. 2014; Raule et al. 2014; Zanker et al. 2013). Alternatively, IFNγ may regulate upstream components of the ubiquitin proteasome system to enhance immunoproteasome access to HO-1 protein. Recent work has shown that intramembrane cleavage of HO-1 by signal peptide peptidase (SPP) and ubiquitination by the E3 ubiquitin ligase TRC8 are critical for removal of the HO-1 tail anchor from the ER and subsequent proteasome-dependent degradation (Boname et al. 2014; Hsu et al. 2015; Lin et al. 2013). Modulating effects of IFNγ on the activity of SPP and TRC8 are not known, but these are potential mechanisms that may enhance degradation of HO-1 protein.
Our studies provide compelling evidence that enhanced degradation of HO-1 by immunoproteasomes within astrocytes may drive reduced expression of HO-1 protein in HIV-infected brain and they suggest that persistently elevated IFNγ is a critical modulator linking chronic immune activation to the pathogenesis of HAND. When present at moderate levels and/or for brief periods, IFNγ may exert beneficial effects during HIV infection through induction of HO-1 expression, which would be expected to limit inflammation and oxidative stress in various tissues. IFNγ-mediated induction of immunoproteasomes has been suggested to be a beneficial host response that protects cells from proteotoxic stress and enhances the cellular capacity to degrade oxidized proteins accumulating as a result of inflammation and oxidative stress (Seifert et al. 2010). Immunoproteasomes are thought to subserve a rapid and transient response to stress, exhibiting a faster rate of assembly and significantly shorter half-life than constitutive proteasomes (Heink et al. 2005). Prolonged induction of immunoproteasomes may dysregulate the expression and/or degradation of cellular proteins other than HO-1, which could contribute to pathological processes. For example, studies suggest that immunoproteasomes enhance the production and presentation of antigenic peptides derived from myelin basic protein, suggesting similar effects on other endogenous proteins could contribute to autoimmune disorders (Belogurov et al. 2014; Belogurov et al. 2015; Kuzina et al. 2013).
Our previous studies suggest that HO-1-inducing therapies could offer a therapeutic approach for neuroprotection in HIV-infected individuals and perhaps other neurodegenerative diseases. We previously showed that HIV infection of monocyte-derived macrophages drives HO-1 protein and RNA loss in a time-dependent manner and this is associated with neurotoxic levels of extracellular glutamate (Gill et al. 2015). Induction of HO-1 expression in HIV-infected macrophages reduces extracellular levels of glutamate and associated neurotoxicity (Gill et al. 2014); a role for HO-1 in modulation of glutamate homeostasis and neuroprotection within the brain seems likely. Induction of HO-1 may thus be therapeutically beneficial not only in HIV infection where HO-1 expression is reduced but also other neurodegenerative diseases associated with neuroinflammation, immunoproteasome activation, and/or glutamate-mediated excitotoxicity.
Supplementary Material
Main Points.
In HIV infection, decreased brain HO-1 protein expression accompanies increased HO-1 RNA and immunoproteasome expression
In astrocytes IFNγ increases immunoproteasome subunit expression and proteasome-dependent HO-1 protein degradation
Acknowledgments
This study utilized primary human fetal astrocytes provided by core facilities of the Comprehensive NeuroAIDS Center at Temple University School of Medicine. The National NeuroAIDS Tissue Consortium provided autopsy tissue specimens and clinical and paraclinical data.
Funding
This work was supported in part by NIH R01 grants MH095671 (D.L.K.), MH104134 (D.L.K.), and NS072005 (B.B.G.) and F30 grant MH102120 (A.J.G.). The National NeuroAIDS Tissue Consortium received support from MH083507, MH083501, MH083500, MH083506, and MH083545. The Comprehensive NeuroAIDS Center at Temple University received support from NIMH P30MH092177.
Dr. Dennis Kolson serves on the Steering Committee for the National Institutes of Health/National Institute of Mental Health National NeuroAIDS Tissue Consortium. Dr. Benjamin Gelman has served as the Chair of the National NeuroAIDS Tissue Consortium Steering Committee.
Abbreviations
- ARE
antioxidant response element
- ART
antiretroviral therapy
- DLPFC
dorsolateral prefrontal cortex
- GFAP
glial fibrillary acidic protein
- GPX1
glutathione peroxidase 1
- HAND
HIV-associated neurocognitive disorders
- HIVE
HIVE-encephalitis
- HO-1
heme oxygenase-1
- HO-2
heme oxygenase-2
- IFNγ
interferon gamma
- LPS
lipopolysaccharide
- NNTC
National NeuroAIDS Tissue Consortium
- NQO1
NAD(P)H dehydrogenase [quinone] 1
- NRF2
nuclear factor erythroid 2-related factor 2
- OMG
oligodendrocyte myelin glycoprotein
- SEM
standard error of the mean
- TNFα
tumor necrosis factor alpha
Footnotes
Conflict of Interest Statement
The other authors have no conflicts of interest.
References
- Aki M, Shimbara N, Takashina M, Akiyama K, Kagawa S, Tamura T, Tanahashi N, Yoshimura T, Tanaka K, Ichihara A. Interferon-gamma induces different subunit organizations and functional diversity of proteasomes. J Biochem. 1994;115:257–69. doi: 10.1093/oxfordjournals.jbchem.a124327. [DOI] [PubMed] [Google Scholar]
- Akiyama K, Yokota K, Kagawa S, Shimbara N, Tamura T, Akioka H, Nothwang HG, Noda C, Tanaka K, Ichihara A. cDNA cloning and interferon gamma down-regulation of proteasomal subunits X and Y. Science. 1994;265:1231–4. doi: 10.1126/science.8066462. [DOI] [PubMed] [Google Scholar]
- Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36:166–74. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- Amadio M, Scapagnini G, Davinelli S, Calabrese V, Govoni S, Pascale A. Involvement of ELAV RNA-binding proteins in the post-transcriptional regulation of HO-1. Front Cell Neurosci. 2014;8:459. doi: 10.3389/fncel.2014.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambegaokar SS, Kolson DL. Heme oxygenase-1 dysregulation in the brain: implications for HIV-associated neurocognitive disorders. Curr HIV Res. 2014;12:174–88. doi: 10.2174/1570162X12666140526122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008;3:e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–99. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbe H, Roers A, Waisman A, Lassmann H, Goebels N, Hohlfeld R, Friese M, Schroder R, Deckert M, Schmidt S, et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med. 2000;192:393–404. doi: 10.1084/jem.192.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaru VV, McArthur JC, Sacktor N, Cutler RG, Knapp EL, Mattson MP, Haughey NJ. Associative and predictive biomarkers of dementia in HIV-1-infected patients. Neurology. 2007;68:1481–7. doi: 10.1212/01.wnl.0000260610.79853.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M, Youhnovski N, Van Den Broek M, Przybylski M, Groettrup M. Immunoproteasomes down-regulate presentation of a subdominant T cell epitope from lymphocytic choriomeningitis virus. J Immunol. 2004;173:3925–34. doi: 10.4049/jimmunol.173.6.3925. [DOI] [PubMed] [Google Scholar]
- Bauer M, Huse K, Settmacher U, Claus RA. The heme oxygenase-carbon monoxide system: regulation and role in stress response and organ failure. Intensive Care Med. 2008;34:640–8. doi: 10.1007/s00134-008-1010-2. [DOI] [PubMed] [Google Scholar]
- Belogurov A, Jr, Kudriaeva A, Kuzina E, Smirnov I, Bobik T, Ponomarenko N, Kravtsova-Ivantsiv Y, Ciechanover A, Gabibov A. Multiple sclerosis autoantigen myelin basic protein escapes control by ubiquitination during proteasomal degradation. J Biol Chem. 2014;289:17758–66. doi: 10.1074/jbc.M113.544247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belogurov A, Jr, Kuzina E, Kudriaeva A, Kononikhin A, Kovalchuk S, Surina Y, Smirnov I, Lomakin Y, Bacheva A, Stepanov A, et al. Ubiquitin-independent proteosomal degradation of myelin basic protein contributes to development of neurodegenerative autoimmunity. FASEB J. 2015;29:1901–13. doi: 10.1096/fj.14-259333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boname JM, Bloor S, Wandel MP, Nathan JA, Antrobus R, Dingwell KS, Thurston TL, Smith DL, Smith JC, Randow F, et al. Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J Cell Biol. 2014;205:847–62. doi: 10.1083/jcb.201312009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet A, Salim H, Taoufik Y, Lledo PM, Vincent JD, Delfraissy JF, Tardieu M. Isolated human astrocytes are not susceptible to infection by M- and T-tropic HIV-1 strains despite functional expression of the chemokine receptors CCR5 and CXCR4. Glia. 2001;34:165–77. [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Broder S. The development of antiretroviral therapy and its impact on the HIV-1/AIDS pandemic. Antiviral Res. 2010;85:1–18. doi: 10.1016/j.antiviral.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budka H, Wiley CA, Kleihues P, Artigas J, Asbury AK, Cho ES, Cornblath DR, Dal Canto MC, DeGirolami U, Dickson D, et al. HIV-associated disease of the nervous system: review of nomenclature and proposal for neuropathology-based terminology. Brain Path. 1991;1:143–52. doi: 10.1111/j.1750-3639.1991.tb00653.x. [DOI] [PubMed] [Google Scholar]
- Cassol E, Misra V, Dutta A, Morgello S, Gabuzda D. Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS. 2014;28:1579–91. doi: 10.1097/QAD.0000000000000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: New players in micromanaging redox homeostasis. Free Radic Biol Med. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- Cross SA, Cook DR, Chi AW, Vance PJ, Kolson LL, Wong BJ, Jordan-Sciutto KL, Kolson DL. Dimethyl fumarate, an immune modulator and inducer of the antioxidant response, suppresses HIV replication and macrophage-mediated neurotoxicity: a novel candidate for HIV neuroprotection. J Immunol. 2011;187:5015–25. doi: 10.4049/jimmunol.1101868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlmann B, Ruppert T, Kuehn L, Merforth S, Kloetzel PM. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J Mol Biol. 2000;303:643–53. doi: 10.1006/jmbi.2000.4185. [DOI] [PubMed] [Google Scholar]
- Eden A, Price RW, Spudich S, Fuchs D, Hagberg L, Gisslen M. Immune activation of the central nervous system is still present after >4 years of effective highly active antiretroviral therapy. J Infect Dis. 2007;196:1779–83. doi: 10.1086/523648. [DOI] [PubMed] [Google Scholar]
- Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci. 2012;109:75–112. doi: 10.1016/B978-0-12-397863-9.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruh K, Gossen M, Wang K, Bujard H, Peterson PA, Yang Y. Displacement of housekeeping proteasome subunits by MHC-encoded LMPs: a newly discovered mechanism for modulating the multicatalytic proteinase complex. EMBO J. 1994;13:3236–44. doi: 10.1002/j.1460-2075.1994.tb06625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs D, Chiodi F, Albert J, Asjo B, Hagberg L, Hausen A, Norkrans G, Reibnegger G, Werner ER, Wachter H. Neopterin concentrations in cerebrospinal fluid and serum of individuals infected with HIV-1. AIDS. 1989a;3:285–8. doi: 10.1097/00002030-198905000-00006. [DOI] [PubMed] [Google Scholar]
- Fuchs D, Hausen A, Reibnegger G, Werner ER, Werner-Felmayer G, Dierich MP, Wachter H. Interferon-gamma concentrations are increased in sera from individuals infected with human immunodeficiency virus type 1. J Acquir Immune Defic Syndr. 1989b;2:158–62. [PubMed] [Google Scholar]
- Gelman BB. Neuropathology of HAND With Suppressive Antiretroviral Therapy: Encephalitis and Neurodegeneration Reconsidered. Curr HIV/AIDS Rep. 2015;12:272–9. doi: 10.1007/s11904-015-0266-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill AJ, Kovacsics CE, Cross SA, Vance PJ, Kolson LL, Jordan-Sciutto KL, Gelman BB, Kolson DL. Heme oxygenase-1 deficiency accompanies neuropathogenesis of HIV-associated neurocognitive disorders. J Clin Invest. 2014;124:4459–72. doi: 10.1172/JCI72279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill AJ, Kovacsics CE, Vance PJ, Collman RG, Kolson DL. Induction of Heme Oxygenase-1 Deficiency and Associated Glutamate-Mediated Neurotoxicity Is a Highly Conserved HIV Phenotype of Chronic Macrophage Infection That Is Resistant to Antiretroviral Therapy. J Virol. 2015;89:10656–67. doi: 10.1128/JVI.01495-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- Griffin DE. Cytokines in the brain during viral infection: clues to HIV-associated dementia. J Clin Invest. 1997;100:2948–51. doi: 10.1172/JCI119847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE, McArthur JC, Cornblath DR. Neopterin and interferon-gamma in serum and cerebrospinal fluid of patients with HIV-associated neurologic disease. Neurology. 1991;41:69–74. doi: 10.1212/wnl.41.1.69. [DOI] [PubMed] [Google Scholar]
- Groettrup M, Khan S, Schwarz K, Schmidtke G. Interferon-gamma inducible exchanges of 20S proteasome active site subunits: why? Biochimie. 2001;83:367–72. doi: 10.1016/s0300-9084(01)01251-2. [DOI] [PubMed] [Google Scholar]
- Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17:3–16. doi: 10.1007/s13365-010-0006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heink S, Ludwig D, Kloetzel PM, Kruger E. IFN-gamma-induced immune adaptation of the proteasome system is an accelerated and transient response. Proc Natl Acad Sci U S A. 2005;102:9241–6. doi: 10.1073/pnas.0501711102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori R, Kashiba M, Toma T, Yachie A, Goda N, Makino N, Soejima A, Nagasawa T, Nakabayashi K, Suematsu M. Gene transfection of H25A mutant heme oxygenase-1 protects cells against hydroperoxide-induced cytotoxicity. J Biol Chem. 2002;277:10712–8. doi: 10.1074/jbc.M107749200. [DOI] [PubMed] [Google Scholar]
- Hsu FF, Yeh CT, Sun YJ, Chiang MT, Lan WM, Li FA, Lee WH, Chau LY. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene. 2015;34:2360–70. doi: 10.1038/onc.2014.166. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–9. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- Kamat A, Lyons JL, Misra V, Uno H, Morgello S, Singer EJ, Gabuzda D. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr. 2012;60:234–43. doi: 10.1097/QAI.0b013e318256f3bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, van den Broek M, Schwarz K, de Giuli R, Diener PA, Groettrup M. Immunoproteasomes largely replace constitutive proteasomes during an antiviral and antibacterial immune response in the liver. J Immunol. 2001;167:6859–68. doi: 10.4049/jimmunol.167.12.6859. [DOI] [PubMed] [Google Scholar]
- Kuzina ES, Chernolovskaya EL, Kudriaeva AA, Zenkova MA, Knorre VD, Surina EA, Ponomarenko NA, Bobik TV, Smirnov IV, Bacheva AV, et al. Immunoproteasome enhances intracellular proteolysis of myelin basic protein. Dokl Biochem Biophys. 2013;453:300–3. doi: 10.1134/S1607672913060070. [DOI] [PubMed] [Google Scholar]
- Leautaud V, Demple B. Regulation of heme oxygenase-1 mRNA deadenylation and turnover in NIH3T3 cells by nitrosative or alkylation stress. BMC Mol Biol. 2007;8:116. doi: 10.1186/1471-2199-8-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PH, Chiang MT, Chau LY. Ubiquitin-proteasome system mediates heme oxygenase-1 degradation through endoplasmic reticulum-associated degradation pathway. Biochim Biophys Acta. 2008;1783:1826–34. doi: 10.1016/j.bbamcr.2008.05.008. [DOI] [PubMed] [Google Scholar]
- Lin PH, Lan WM, Chau LY. TRC8 suppresses tumorigenesis through targeting heme oxygenase-1 for ubiquitination and degradation. Oncogene. 2013;32:2325–34. doi: 10.1038/onc.2012.244. [DOI] [PubMed] [Google Scholar]
- Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem. 2007;282:20621–33. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- Ma N, Xiang Y, Zhang Y, Zhao X, Zhou L, Gao X. The balance mediated by miRNAs and the heme oxygenase 1 feedback loop contributes to biological effects. J Cell Biochem. 2013;114:2637–42. doi: 10.1002/jcb.24631. [DOI] [PubMed] [Google Scholar]
- Meiners S, Keller IE, Semren N, Caniard A. Regulation of the proteasome: evaluating the lung proteasome as a new therapeutic target. Antioxid Redox Signal. 2014;21:2364–82. doi: 10.1089/ars.2013.5798. [DOI] [PubMed] [Google Scholar]
- Mishto M, Liepe J, Textoris-Taube K, Keller C, Henklein P, Weberruss M, Dahlmann B, Enenkel C, Voigt A, Kuckelkorn U, et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur J Immunol. 2014;44:3508–21. doi: 10.1002/eji.201444902. [DOI] [PubMed] [Google Scholar]
- Morgello S, Gelman BB, Kozlowski PB, Vinters HV, Masliah E, Cornford M, Cavert W, Marra C, Grant I, Singer EJ. The National NeuroAIDS Tissue Consortium: a new paradigm in brain banking with an emphasis on infectious disease. Neuropathol Appl Neurobiol. 2001;27:326–35. doi: 10.1046/j.0305-1846.2001.00334.x. [DOI] [PubMed] [Google Scholar]
- Morse D, Choi AM. Heme oxygenase-1: the “emerging molecule” has arrived. Am J Respir Cell Mol Biol. 2002;27:8–16. doi: 10.1165/ajrcmb.27.1.4862. [DOI] [PubMed] [Google Scholar]
- Morse D, Choi AM. Heme oxygenase-1: from bench to bedside. Am J Respir Crit Care Med. 2005;172:660–70. doi: 10.1164/rccm.200404-465SO. [DOI] [PubMed] [Google Scholar]
- Munoz-Sanchez J, Chanez-Cardenas ME. A review on hemeoxygenase-2: focus on cellular protection and oxygen response. Oxid Med Cell Longev. 2014;2014:604981. doi: 10.1155/2014/604981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Yashiroda H, Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol. 2009;10:104–15. doi: 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- Nandi D, Jiang H, Monaco JJ. Identification of MECL-1 (LMP-10) as the third IFN-gamma-inducible proteasome subunit. J Immunol. 1996;156:2361–4. [PubMed] [Google Scholar]
- Nandi D, Woodward E, Ginsburg DB, Monaco JJ. Intermediates in the formation of mouse 20S proteasomes: implications for the assembly of precursor beta subunits. EMBO J. 1997;16:5363–75. doi: 10.1093/emboj/16.17.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TP, Soukup VM, Gelman BB. Persistent hijacking of brain proteasomes in HIV-associated dementia. Am J Pathol. 2010;176:893–902. doi: 10.2353/ajpath.2010.090390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raule M, Cerruti F, Cascio P. Enhanced rate of degradation of basic proteins by 26S immunoproteasomes. Biochim Biophys Acta. 2014;1843:1942–7. doi: 10.1016/j.bbamcr.2014.05.005. [DOI] [PubMed] [Google Scholar]
- Rivett AJ, Bose S, Brooks P, Broadfoot KI. Regulation of proteasome complexes by gamma-interferon and phosphorylation. Biochimie. 2001;83:363–6. doi: 10.1016/s0300-9084(01)01249-4. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Kvam E, Tyrrell RM. Heme oxygenase activity. Current methods and applications. Methods Mol Biol. 2000;99:369–91. doi: 10.1385/1-59259-054-3:369. [DOI] [PubMed] [Google Scholar]
- Ryter SW, Otterbein LE, Morse D, Choi AM. Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance. Mol Cell Biochem. 2002:234–235. 249–63. doi: 10.1023/A:1015957026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas M, Wang J, Rosa de Sagarra M, Martin D, Rojo AI, Martin-Perez J, Ortiz de Montellano PR, Cuadrado A. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004;578:90–4. doi: 10.1016/j.febslet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC. HIV-associated neurocognitive disorder - pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12:309. doi: 10.1038/nrneurol.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrier RD, Hong S, Crescini M, Ellis R, Perez-Santiago J, Spina C, Letendre S, Group H Cerebrospinal fluid (CSF) CD8+ T-cells that express interferon-gamma contribute to HIV associated neurocognitive disorders (HAND) PLoS One. 2015;10:e0116526. doi: 10.1371/journal.pone.0116526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, Prozorovski T, Lange N, Steffen J, Rieger M, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142:613–24. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- Shapshak P, Duncan R, Minagar A, Rodriguez de la Vega P, Stewart RV, Goodkin K. Elevated expression of IFN-gamma in the HIV-1 infected brain. Front Biosci. 2004;9:1073–81. doi: 10.2741/1271. [DOI] [PubMed] [Google Scholar]
- Simioni S, Cavassini M, Annoni JM, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave JP, Giacobini E, Hirschel B, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24:1243–50. doi: 10.1097/QAD.0b013e3283354a7b. [DOI] [PubMed] [Google Scholar]
- Stohwasser R, Giesebrecht J, Kraft R, Muller EC, Hausler KG, Kettenmann H, Hanisch UK, Kloetzel PM. Biochemical analysis of proteasomes from mouse microglia: induction of immunoproteasomes by interferon-gamma and lipopolysaccharide. Glia. 2000;29:355–65. [PubMed] [Google Scholar]
- Tanaka K. The proteasome: from basic mechanisms to emerging roles. Keio J Med. 2013;62:1–12. doi: 10.2302/kjm.2012-0006-re. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Ichihara A. Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem Biophys Res Commun. 1989;159:1309–15. doi: 10.1016/0006-291x(89)92253-5. [DOI] [PubMed] [Google Scholar]
- Wagener FA, Volk HD, Willis D, Abraham NG, Soares MP, Adema GJ, Figdor CG. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol Rev. 2003;55:551–71. doi: 10.1124/pr.55.3.5. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Takahashi K, Glass JD, McArthur JC, Griffin JW, Griffin DE. Cellular localization of tumor necrosis factor mRNA in neurological tissue from HIV-infected patients by combined reverse transcriptase/polymerase chain reaction in situ hybridization and immunohistochemistry. J Neuroimmunol. 1997;74:1–8. doi: 10.1016/s0165-5728(96)00160-9. [DOI] [PubMed] [Google Scholar]
- Woods SP, Rippeth JD, Frol AB, Levy JK, Ryan E, Soukup VM, Hinkin CH, Lazzaretto D, Cherner M, Marcotte TD, et al. Interrater reliability of clinical ratings and neurocognitive diagnoses in HIV. J Clin Exp Neuropsychol. 2004;26:759–78. doi: 10.1080/13803390490509565. [DOI] [PubMed] [Google Scholar]
- Zanker D, Waithman J, Yewdell JW, Chen W. Mixed proteasomes function to increase viral peptide diversity and broaden antiviral CD8+ T cell responses. J Immunol. 2013;191:52–9. doi: 10.4049/jimmunol.1300802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.