

Abstract
Human respiratory syncytial virus (hRSV) is a major cause of morbidity and mortality in the pediatric, elderly, and immune compromised populations1,2. A gap in our understanding of hRSV disease pathology is the interplay between virally encoded immune antagonists and host components that limit hRSV replication. hRSV encodes for non-structural (NS) proteins that are important immune antagonists3–6; however, the role of these proteins in viral pathogenesis is incompletely understood. Here we report the crystal structure of hRSV NS1 protein, which suggests that NS1 is a structural paralog of hRSV matrix (M) protein. Comparative analysis of the shared structural fold with M revealed regions unique to NS1. Studies on NS1 WT or mutant alone or in recombinant RSVs demonstrate that structural regions unique to NS1 contribute to modulation of host responses, including inhibition of type I IFN responses, suppression of dendritic cell maturation, and promotion of inflammatory responses. Transcriptional profiles of A549 cells infected with recombinant RSVs show significant differences in multiple host pathways, suggesting that NS1 may have a greater role in regulating host responses than previously appreciated. These results provide a framework to target NS1 for therapeutic development to limit hRSV associated morbidity and mortality.
Human respiratory syncytial virus (hRSV) is a major cause of severe respiratory infections in children and a significant contributor to morbidity and mortality for the elderly and immune-compromised individuals. The virus causes 130,000 cases of clinical infection in children less than 5 years of age in the United States alone1,2. Currently we lack sufficient knowledge and understanding of several outcomes that are observed during hRSV infections, including complications arising from partial immunity to hRSV infections, reduced immune responses in infants with maternal antibodies, and vaccine enhancement of disease7. We also lack a complete view of host-viral interactions that occur in response to hRSV infection8. As a consequence, limited effective prophylactic or therapeutic approaches are currently available to attenuate hRSV infections9.
hRSV is a member of the Pneumoviridae family and its genome encodes for 11 proteins (Supplementary Fig. 1a), including the non-structural proteins 1 (NS1) and 2 (NS2). hRSV NS1 and NS2 are multifunctional proteins that are important for host immune evasion. Sequence analysis of NS1 and NS2 shows that there are no counterparts for NS1 and NS2 in other non-segmented negative strand RNA viruses except for other members of the Orthopneumovirus genus (bovine RSV, ovine RSV, and pneumonia virus of mice) (Fig. 2). This observation highlights a unique evolutionary path taken by hRSV to achieve effective immune suppression. Functionally, NS1 has been implicated as an antagonist of type I IFN responses3–6. NS1 has been shown to inhibit IRF3/7 dependent gene expression6,10,11 and is involved in STAT2 degradation through a proteosome mediated process12. NS1 may serve as a specificity factor for the cullin-ring family of E3 ubiquitin ligases13,14. NS1 may also function as an inhibitor of dendritic cell (DC) maturation15,16 as an inhibitor of CD103+ CD8+ T-cells and Th17 cell proliferation, and as a stimulator of Th2 cells, which can all enhance disease severity15–17. Furthermore, NS1, in concert with NS2, is thought to inhibit DC maturation and T-cell responses through defects in the immunological synapse formation in infected cells18–20. However, it is not clear if these effects are solely due to suppression of type I IFNs or additional direct targeting of the host signaling pathways by NS1, including the disruption of multiple host gene expression programs as previously implicated21,22. Thus, the basis for these host-viral interactions mediated by hRSV NS1 that contributes dysregulation of host functions during infections remains incompletely defined.
Figure 2. hRSV NS1 is a structural paralog of hRSV matrix (M) protein.

a, The crystallographic asymmetric unit contains two molecules of hRSV NS1 (molecule A, magenta; molecule B, light blue). b, Structural alignment of hRSV NS1 (magenta) with hRSV M NTD (gray, PDB 2VQP). RMSD is 3.78 Å over 96 residues. Cartoon representation of hRSV c, NS1 and d, M N-terminal domain (NTD) highlighting similar secondary structural elements using color. Secondary structural elements not common between NS1 and M are colored in white. Electrostatic surface representation and topology diagrams of hRSV e–f, NS1 and g–h, M NTD, respectively. d, f top, hRSV NS1 and e, f bottom, hRSV M NTD. For e and g red, white, and blue represent negative, neutral, and positive electrostatic potential, respectively (−5 to +5 kBTe-1). For f and h, coloring scheme is the same in c–d.
To better understand the molecular basis for NS1-associated function, we solved the structure of hRSV NS1 to 2.22 Å resolution by X-ray crystallography using selenomethionine single wavelength anomalous dispersion (Se-SAD) (Supplementary Table 1). In the crystal, there are two molecules in the asymmetric unit (termed molecule A and molecule B) (Fig. 2a). Analysis of the crystal packing revealed that the C-terminal helix of one molecule is positioned against the beta strands of the second molecule. However, there is no significant surface area buried between the two molecules to support a true dimer interface. Consistent with this structural observation, hydrodynamic studies by size exclusion chromatography-multiangle light scattering (SEC-MALS) analysis reveals that NS1 is 17.5 ± 2.1 kDa in solution under physiological salt conditions (Supplementary Fig. 2a), suggesting that the predominant form of NS1 may be the monomer. The NS1 monomer is comprised of a β-sandwich flanked by three α-helices. Three β-strands are arranged in two orthogonal sheets to form the beta-sandwich, with a seventh beta strand β6 that runs antiparallel to β3 before twisting to run antiparallel to β1 (Fig. 2b). All the strands in the β-sandwich are antiparallel except β7, which forms a strand parallel to β4 (Fig. 2c). The two β-sheets are connected by a flexible linker (residues 99-108) for which little density is observed, suggesting that there is increased flexibility in this region. Helices α1 and α2 are arranged to one side of the β-sandwich whereas alpha helix α3 is extended from the core of the protein (Fig. 2b–c).
Comparisons of the hRSV NS1 structure to available structures in the Protein Data Bank (PDB) using the DALI server yielded a list of several viral matrix proteins, including hRSV matrix (M) protein (Z score 6.5; Supplementary Fig. 2b)23, the Ebola virus matrix protein VP40 (Z score 6.8)24, and Newcastle Disease virus matrix protein (Z score 5)25 (Table S2). Structural alignment of hRSV NS1 with the N-terminal domains of hRSV M protein (PDB: 2VQP; Fig. 2d–f) and Ebola virus VP40 (PDB: 1ES6) displayed remarkable alignment of the structures (backbone RMSD 3.78 Å over 96 residues and 4.15 Å over 88 residues, respectively). However, hRSV NS1 displays less resemblance to the matrix proteins of other non-segmented negative strand RNA viruses, such as Borna Disease virus and vesicular stomatitis virus (PDB 3F1J and 1LG7, respectively)26,27. Despite the structural similarities, the lack of sequence homology between hRSV NS1 and different Mononegavirales matrix (M) proteins suggest that the functional significance correlates with the structural fold and not the primary sequence. Based on our finding, it is tempting to speculate that a duplication event of the N-terminal domain of M followed by divergent evolution resulted in the current NS1 proteins (Fig. 2b). In contrast to the sequence similarity between hRSV NS1 and M, Mononegavirales M proteins share significant sequence and structural similarities and are thus considered homologs (Supplementary Fig. 1c)23. Collectively, these observations suggest that hRSV NS1 may be a paralog of hRSV M protein although convergent evolution cannot be ruled out.
While the structural similarities are striking, structural differences between hRSV NS1 and M can potentially reveal unique features of hRSV NS1 that contribute to NS1-specific functions, including immune suppressive functions. First, hRSV NS1 is comprised of a single domain whereas there are two domains with similar folds in the hRSV M protein (Supplementary Fig. 1). Second, a change in β-strand direction of NS1 β4 and β7 to parallel, which is similar to hRSV M β1 and β8, is absent in other M proteins (Fig. 2c–d & Fig. 2f–h). Third, there is an insertion of β5 strand and α3 helix in the hRSV NS1 structure, the functional significance of which are unknown. High sequence conservation among RSV sequences suggest that the helix α3 itself may be important for protein stability or a potential hotspot for intermolecular interactions that are required for hRSV NS1 function. Furthermore, comparison of the electrostatic surface potential reveals that hRSV NS1 lacks the extensive positively charged surface (Fig. 2e–g) that is present in full length hRSV M proteins23,28, which correspond to membrane associated functions23.
In order to test the importance of specific residues and helix α3 of NS1, which is a secondary structural element absent in M, and its role in hRSV mediated host immune evasion, we calculated the solvent accessible surface area (ASA) of NS1 with and without the terminal α3 helix and of the α3 helix alone (Fig. 3a). Results from the ASA calculations show that a large number of hydrophobic residues on the α3 helix are solvent accessible in the absence of the rest of the NS1 structure, suggesting that these residues may be involved in intra-protein interfacial interactions. Atomistic computer simulations indicate that the α3 helix is stable and likely displays cooperative folding, with higher stability of α3 helix in the context of the NS1 structure rather than the isolated helix (Fig. 3b–c). A subset of the interface residues between α3 helix and the rest of the NS1 structure and a α3 helix truncation mutants were generated and characterized by biochemical and functional assays in order to assess a functional role for α3 helix in NS1-mediated functions (Fig. 3d). Using circular dichroism (CD), we confirmed that the mutations Y125A, L132A/L133A, and 1-118 did not compromise the overall stability of the proteins (Fig. 3e–f). Deletion of the α3 helix in the NS1 1-118 construct reduced helical content, as expected, and but did not affect the overall stability of the protein in thermal measurements (Supplementary Fig. 2b–c). Overall, our analyses suggest that the α3 helix of NS1 is an important structural element that may also directly facilitate function.
Figure 3. hRSV NS1 mutations impact NS1 function.

a, Accessible surface area (ASA) analysis of NS1 residues 1-118 and 119-139 using Areaimol60 with a search probe radius used of 1.4Å. Increase in ASA of NS1 residues 1-118 in the absence of the C-terminal α3 helix is plotted (left). ASA for NS1 residues 1-118 within chain A and B of the crystal structure was calculated in the presence and absence of the helix α3 (119-139). The average of the ASA values of chains A and B were considered. The increase in ASA was calculated by subtracting the average ASA in the absence of helix α3 from that in the presence of α3 helix. For residues 119 to 139 within helix α3 (right), the increase in ASA was calculated in the presence and absence of the rest of the polypeptide (1-118). The dotted line represents average plus one standard deviation of ΔASA. b, Results based on atomistic simulations using the ABSINTH implicit solvation model and forcefield paradigm61 show overall helical propensity calculated as a probability of finding helical stretches within the α3 sequence with at least L consecutive residues in such a stretch. The light grey colors refer to the peptide in isolation and the dark gray colors are for α3 in the context of the remainder of NS1. c, Simulation results for the per-residue helical propensity in the isolated α3 peptide calculated as a probability of finding helical stretches within the sequence with at least L consecutive residues (see methods) in a stretch. d, Cartoon representation of hRSV NS1 structure highlighting the residues mutated in this study (yellow). e, CD wavelength scans and f, thermal denaturation of NS1 constructs. NS1 WT (black), NS1 F17A (red), NS1 F56A (green), and NS1 Y125A (blue) at 10 μM. g, IFN-b reporter activity upon Sendai virus (SeV) infection in 293T cells (bolded underline) for vector only (E), wildtype and mutants NS1 proteins. Ebola VP35 is used as the positive control. The firefly values are normalized with renilla reporter values. Fold induction is determined by setting mock treated vector values as 1. The experiment is representative of three independent experiments. Each bar represents mean of three replicates with error bars indicating standard deviation. The p-values are determined by one way Anova followed by tukey’s test, **** p<0.0001. The expression of each construct used in the luciferase assay is shown by the western blot using anti-flag antibody.
Previous studies have shown that the non-structural proteins NS1 and NS2 play unique roles in hRSV mediated host immune evasion as inhibitors of IFN responses. To test inhibition of IFN responses, we measured activation of the IFN-β promoter during infection with non-NS protein containing Sendai virus (SeV). Transfection of WT hRSV NS1 significantly inhibits the IFN-β response to levels similar to those observed for Ebola virus VP35 (eVP35), a well-characterized IFN antagonist (Fig. 3g). Furthermore, hRSV NS1 partially suppresses activation of IFN-β mediated by IRF3-5D, a constitutively activated phosphomimic of the transcription factor IRF3, suggesting that hRSV NS1 may function at multiple points of the IFN-β signaling pathway, including a nuclear function (Supplementary Fig. 2d). Mutation of residues Y125 and L132/L133 in the α3 helix or truncation of the α3 helix (1-118 construct) impacted the NS1 function. In contrast, mutation of residues F17 and F56 display near-wildtype inhibition of IFN-β promoter activity. Altogether, these results suggesting that Y125 and L132/L133 residues as well as the α3 helix may play a critical role in NS1-mediated IFN antagonism, whereas F15 and G56 residues outside of α3 helix do not.
Studies of mutant RSVs implicate NS1 as a suppressor of DC maturation. We sought to determine whether NS1 expression is sufficient to suppress DC maturation in response to RNA virus infection and to assess the functional relevance of the α3 helix and α3 helix proximal residues in the NS1 structure in DC suppression. Lentiviruses encoding hRSV NS1 mutations were generated and human monocyte-derived DCs (MDDCs) were transduced with control (empty and eVP35), NS1 WT, or NS1 L132A/L133A mutant. Transduced DCs were then mock-infected or infected with SeV in order to stimulate maturation. Relative to SeV-infected empty vector controls, WT NS1 suppressed IFN-β, cytokine (TNF-α) and representative IFN stimulated gene upregulation, whereas the L132A/L133A mutant was impaired for this suppression (Fig. 4a–d). Further, WT NS1 impaired the upregulation of cell surface markers of DC maturation, as assessed by flow cytometry (Fig. 4e–h). These observations further support a role for NS1 α3 helix as an important determinant in innate immune modulation.
Figure 4. NS1 unique regions are important for modulating host responses.

Human monocyte-derived dendritic cells (MDDCs) were transduced with either only empty lentiviral vectors (empty) or lentivirus vectors that express either EBOV-VP35, hRSV-NS1, hRSV-NS1-(1-118), hRSV-NS1-L132A/L133A. RNA was isolated from transduced MDDCs that were either mock-infected or infected with SeV. RNA was extracted at the indicated hours post-infection; and a, IFN-β, b, TNF-α, c, ISG54, and d, ISG56 mRNA levels were quantified RT-qPCR, normalizing their levels to that of β-actin mRNA. The graph indicates the fold-change relative to the mock-infected samples. For a–d, symbols are: ○, empty vector; ▲, hRSV NS1 1-118; ▼, hRSV NS1 L132A/L133A; ■, hRSV NS1 WT; and ●, EBOV VP35. hRSV NS1 inhibits upregulation of DC maturation markers. Transduced MDDCs were infected with SeV for 20h, harvested and stained for expression of CD40, CD80, CD83 and CD86. Fold change in mean fluorescence intensity (MFI) for the indicated proteins in transduced MDDCs where fold increase indicates comparisons of SeV-infected to uninfected MDDCs. Error bars indicate standard deviations from three independent experiments (*p< 0.05 relative to empty vector-transduced MDDCs at the same time point). e, CD40, f, CD80, g, CD83, and h, CD86 upon SeV infection. i, Growth curves of Vero cells (left) and A549 cells (right) infected with hRSVs encoding wildtype or mutant NS1 proteins. Virus RNA was isolated from medium at indicated time points and quantified by qPCR. Values presented as fold-change relative to 2h time point. Results are expressed as mean ± s.e.m. from three biological replicates.
 , NS1 wildtype;
, NS1 wildtype;
 , NS1 1-118;
, NS1 1-118;
 , NS1 Y125A; and
, NS1 Y125A; and
 , NS1 L132A/L133A. j, Heatmap representing genes with number of matching reads greater than five in A549 cells 96 hpi post infection with hRSV NS1 wildtype or mutant proteins. Colors represent expression values; red indicates higher expression and blue indicates lower expression within a gene expression profile. k, Heatmaps representing the members of the top set of genes in IFN induction (left panel), IFN response (middle panel), and antioxidative response (right panel) signaling pathways in A549 cells 96 hpi with hRSV NS1 wildtype or mutant proteins.
, NS1 L132A/L133A. j, Heatmap representing genes with number of matching reads greater than five in A549 cells 96 hpi post infection with hRSV NS1 wildtype or mutant proteins. Colors represent expression values; red indicates higher expression and blue indicates lower expression within a gene expression profile. k, Heatmaps representing the members of the top set of genes in IFN induction (left panel), IFN response (middle panel), and antioxidative response (right panel) signaling pathways in A549 cells 96 hpi with hRSV NS1 wildtype or mutant proteins.
RSV infections are characterized by airway inflammation and excess mucus production, and excessive cytokine production likely driven by Th2 cytokines29. To establish the structural basis for NS1-unique regions in promoting virus replication and inducing responses characteristic of hRSV disease, we generated recombinant RSV viruses (rRSVs) encoding NS1 WT or 1-118, L132A/L133A, or Y125A mutations. Each of these rRSVs mutants displayed similar syncytia formation in type I IFN-deficient Vero cells (Supplementary Fig. 3), indicating that the mutations had no effect on the capacity for viral replication in the absence of innate antiviral responses. In contrast, we observed an approximately two order of magnitude decrease in replication rates of rRSV mutants in IFN competent A549 cells compared to Vero cells (Fig. 4i). Corresponding RNA-seq studies using cells infected with mock, rRSV WT, or rRSV containing NS1 mutants (1-118, L132A/L133A, or Y125A) at low multiplicity of infection (MOI=0.01), revealed that WT rRSV infections result in higher levels of viral RNA relative to mock or rRSV containing NS1 mutant infected cells, whereas accumulation of mutant viral RNAs were significantly reduced even at 12 and 24 hpi (Supplementary Fig. 4a–b). This result supports other data that NS1 mutant viruses exhibit substantially attenuated replication in IFN competent cells and that the observed attenuation is linked to the function of NS1-unique regions of hRSV5,30. Next, analysis of differential gene expression (DGEs) between mock and WT revealed a significant upregulation of inflammatory and IFN pathways in cells infected with WT rRSV (Fig. 4j). Importantly, data at 96 hpi suggest that overall DGEs between mock and mutant rRSVs (118, L132A/L133A, or Y125A) appear significantly different from WT rRSV infected samples (Fig. 4j). In addition to direct comparison of DGEs, we identified the top 25 up- and down-regulated genes for WT and mutant rRSVs relative to mock infections and the resulting data also highlight significant differences between gene expression in cells infected with WT rRSV compared to cells infected with mutant rRSVs (Supplementary Fig. 5a–d). Further examination of the RNA-seq results suggests that key signaling pathways such as IFN induction (Fig 4k left panel & Supplementary Fig. 6), IFN response (Fig. 4k middle panel & Supplementary Fig. 7), and oxidative stress signaling pathways (Fig. 4k right panel & Supplementary Fig. 8) are all differentially expressed with significantly higher DGEs reported for WT rRSV infection. These observations regarding gene expression differences are consistent with previous studies of hRSV NS131,32 and our results suggest that these differences are likely due to a direct role for NS1-mediated global regulation of host gene expression. Because the viral loads at 96 hpi were higher for WT rRSV than compared to mutant rRSVs, it is possible that the differences in DGEs are due to differences in viral replication. Therefore, we also infected A549 cells with WT and mutant rRSVs and examined RNA-seq results for 12 and 24 hpi, which support viral infection for early time points as comparable viral transcripts are present for WT and mutant rRSVs (see Supplementary Fig. 4b). Furthermore, examination of the transcriptomic data for earlier timepoints (12, 24, and 48 hpi) reveal similar overall expression patterns in the IFN induction (Supplementary Fig. 9), IFN response (Supplementary Fig. 10), and oxidative stress signaling pathways (Supplementary Fig. 11) among the WT and mutant rRSV infected cells at 12 and 24 hpi, but higher DGEs in these signaling pathways for WT rRSV infection starting at 48 hpi.
Together, our data support a model wherein hRSV NS1-specific regions of (i.e. those areas that do not share structural features) that are distinct from those in RSV M protein, directly target critical components in order to modulating immune signaling pathways, leading to productive viral infection. Owing to the limited encoding capacity and cytosolic viral replication, many negative sense RNA viruses have potent counter mechanisms to host immune responses. Our studies of hRSV NS1 structure reveal that hRSV NS1 uses similar secondary and tertiary structural elements as the N-terminal domain of the hRSV M protein and is suggestive that this protein likely evolved as a structural paralog. Given the significant involvement of the hRSV NS1-unique regions as an important regulator of host gene expression, these NS1-unique regions also present previously unrecognized opportunities for development of hRSV therapies, including the potential to use helix α3 mutants as a means to attenuate hRSV. Such mutants may provide a mechanism of attenuation that are different from currently available attenuated viruses, including those with gene deletions of M2-2 or NS2 in clinical trials33. Moreover, such viruses may be used in parallel or in conjunction to elicit non-redundant host responses that may be beneficial for understanding confounding factors during RSV infections. Importantly, our studies point to a significant divergence of hRSV from other Pneumoviridae family members as well as from the larger non-segmented negative strand RNA viruses that resulted in the acquisition of NS1 and potentially NS2 open reading frames. Our findings also raise the possibility that the M protein in related family members may compensate for the lack of NS1 by utilizing their M protein to modulate host gene expression during viral infection. Future studies that address these questions may provide insights into the specific molecular mechanisms that are at play during hRSV infections, which is human pathogen with significant impact on a subset of the human population.
Methods
Cloning, expression, and purification
Full-length and mutant human respiratory syncytial virus non-structural 1 (NS1) proteins were subcloned into a modified pET15b vector (Novagen). Mutations in the resulting vectors were verified by sequencing prior to use. NS1 proteins were expressed in BL21(DE3) E. coli cells, harvested by centrifugation at 30,000 × g, and lysed using an Emulsiflex-C5 homogenizer (Avestin). NS1 proteins were purified using a series of chromatographic columns followed by a final purification step using a Superdex 75 16/60 column (GE Healthcare) into buffer containing 20 mM Tris pH 7.5, 150 NaCl, and 2 mM TCEP. Selenomethionine-labeled NS1 was expressed and purified using protocols similar to those used for the native protein.
IFN-β promoter assay
IFN-β promoter reporter gene activity was measured in the presence of NS1 mutants following infection with Sendai virus (SeV), as indicated. IFN-β-firefly luciferase activities were normalized to Renilla luciferase activity expressed from a cotransfected constitutive expression plasmid. % Activity was determined by setting vector with SeV as 100%. The error bars indicate standard deviation of three independent replicates.
Culturing, infection, and flow cytometry analysis of MDDCs
Human MDDCs were purified from blood purchased from New York Blood Center that has been collected from healthy anonymous human blood donors. The human MDDCs were generated from CD14 cells purified from concentrated leukocytes of healthy human donors as described previously31. The MDDCs were incubated at 37°C for 5 days in DC medium (RPMI containing 4% human AB serum [Fisher Scientific], 2 mM L-glutamine 1 mM sodium pyruvate, 100 U/ml penicillin–100 g/ml streptomycin, and 55 uM b-mercaptoethanol) supplemented with 500 U/ml human granulocyte-macrophage colony-stimulating factor (hGM-CSF; PeproTech) and 500 U/ml human interleukin-4 (hIL-4; PeproTech). Typically, we obtained approximately 107 MDDCs per 20 ml of culture. MDDCs were transduced by spinoculation at 1,850 rpm with lentiviruses expressing respective proteins (wt-eVP35, NS1-wt, NS1 1-118, L132A/L133A) for 2.5 h and then cultured in fresh medium for 72 h. Transduced MDDCs were then treated with sendai virus (30 HA units/ml) for 20h and the RNA from the MDDCs were harvested to assess gene expression by qPCR at the indicated timepoints. Alternatively, MDDCs were harvested and stained for cell surface expression of multiple markers of DC maturation by flow cytometry at 20 h after SeV infection. DCs were pelleted and resuspended in fluorescence-activated cell sorting buffer (1× phosphate-buffered saline, 1% bovine serum albumin, and 0.1% sodium azide) and then incubated for 30 min on ice in the dark with the antibodies. Antibodies, including anti-CD40-PE, anti-CD80-PE, anti-CD83-PE, and anti-CD86-PE, were purchased from eBioscience. Flow cytometry data were collected using an LSR II flow cytometer (BD Bioscience) and analyzed using FlowJo software (Tree Star, Ashland, OR).
Crystallization and diffraction data collection
Initial conditions for crystallization were identified using commercially available screens (Hampton Research) and subsequently optimized using in-house generated reagents. Diffraction data was collected at the Advanced Light Source (Beamline 4.2.2; Berkeley, CA) and at the Argonne National Laboratory (sector 19; Argonne, IL) at 100 K.
Structure determination, refinement, and figure generation
X-ray data were collected at the Structural Biology Center 19ID at the Advanced Photon Source (Argonne, IL). Data from a crystal of SeMet derivatized NS1 protein was collected at low remote energy to minimize radiation damage with an oscillation angle of 1 and a crystal-to-detector distance of 280 mm. Data were processed using HKL300034,35. The data statistics are presented in Table S1. Heavy atom positions were identified by ShelxD36, which identified 14 Se positions. Phasing was carried out by MLPHARE37 as implemented in HKL3000, which was followed by phase refinement and by density modification (solvent flattening) using DM38. Model building was carried out with ARP/wARP39,40 and Buccaneer41 with visual inspection with Coot42. Final refinement was carried out with Refmac43,44. Local NCS restraints, jelly body refinement, and in the later cycles with TLS refinement45. Structure quality was assessed with MolProbity46. Surface area was calculated using AREAIMOL as implemented in CCP4 program suite47. Structural alignment and structure figures were prepared using PyMOL48. Topology diagrams were generated by PDBSum49,50 and TopDraw51. Sequence alignment was performed using ClustalW52 and prepared using ESPript 3.053.
CD experiments
Circular dichroism (CD) wavelength scans were performed using a Chirascan CD spectrometer (Applied Photophysics). The change in molar ellipticity of each protein was monitored from 10 to 90 °C on samples containing 5–10 μM of NS1 WT and mutant proteins.
SEC-MALS
SEC-MALS experiments were performed using a DAWN-HELEOS II detector (Wyatt Technologies) coupled to a Superdex SD200 column (GE Healthcare) in buffer containing 10 mM Hepes, 150 mM NaCl, and 2 mM TCEP. 2 mg/ml sample was injected and raw data were analyzed using ASTRA 6 software (Wyatt Technologies) to determine the weight averaged molecular mass (MW). Protein concentrations were determined using the refractive index measured by an Optilab T-rEX (Wyatt Technologies) and a dn/dc = 0.185 ml3 g−1.
Cells, viruses
hRSV strain A2, line 19F was recovered using a reverse genetic system provided54 through BEI Resources, National Institute of Allergy and Infectious Diseases, National Institute of Health. 293T (ATCC CRL 3216), Hep2 (ATCC CCL-23), A549 (ATCC CCL-185), and VeroE6 (ATCC CRL-1586) cell lines were obtained from ATCC and were not further aunthenticated. Cells were negative for mycoplasma contamination using LookOut Mycoplasma PCR Detection Kit (Sigma) or MycoAlert kit (Lonza). Cells were maintained in Dulbecco’s Modified Eagle Medium (Life Technologies) supplemented with 10% fetal bovine serum.
Generation hRSV A2, line19F and hRSV NS1 mutants
A plasmid containing the full length genome sequence of hRSV A2, line 19F was generated from plasmid A2-mKate2-line19F by elimination of the Kate2 gene by using the BlpI restriction site54. All other fragments were relegated and the integrity and size of the obtained full length genome plasmid was verified with BlpI and several other restriction enzymes. Mutations were introduced into the NS1 gene by PCR amplification of the C-terminal end using oligonucleotides that contained corresponding mutations (Y125A, L132A/L133A) and a deletion mutant (1-118). PCR fragments with corresponding mutations were cloned in a shuttle plasmid. A plasmid clone with the correct sequence was identified by nucleotide sequencing and the fragment containing the part of NS1 with the corresponding mutation was returned into the full length genome plasmid. The NS1 gene in the final plasmid was sequenced again to verify the sequence of introduced mutations.
Virus recovery
Transfections were conducted as described previously4. Briefly, sub-confluent BHK-21/SinRep19-T7 cells constitutively expressing T7 polymerase from a Sindbis virus replicon were transfected with 5μg of antigenomic F-L plasmids (19F, 125, 132/133, Δ118) and four support plasmids (2 μg pN, 2 μg pP, 1 μg pM2-1, and 1 μg pL) that express hRSV N, P, M2 and L proteins under the control of the T7 promoter. Transfection was conducted using FuGENE 6 (Roche) in DMEM with 2% FBS in accordance with manufacturer’s instructions. After overnight incubation with transfection mixture, the medium was replaced with fresh DMEM 2%FBS. Transfected cells were split every 4–5 days until syncytia became visible, then the cells and medium were harvested and homogenized, and clarified supernatant was used for infection of Hep2 (wildtype virus) or VeroE6 cells (mutant viruses).
Virus titers were determined by plaque assay for 6 days in VeroE6 cells under 0.8% agarose followed by fixation with 10% paraformaldehyde. Plaques were developed by incubation of cells with biotinylated goat anti-RSV antibodies (Fitzerald) followed by VectastainABC-AK 5000 kit and alkaline phosphatase substrate kit VectorRed-SK-5100 (Vector Laboratories).
Growth curve
A subconfluent monolayer of A549 and VeroE6 cells was infected in triplicate with hRSV A2 line 19F (wildtype) and three mutants with multiplicity of infection (MOI) 0.1 in a 12 well plate. After 1h of adsorption, infected cells were washed three times with DPBS and DMEM with 2% FBS. Medium was harvested at 2, 48, 72, and 96 hours post infection and clarified by centrifugation. RNA was isolated from medium using Qiagen Viral RNA Mini Kit and hRSV titer was determined by qPCR and expressed as fold change compared to 2 hour time point.
Virus-specific real-time qPCR (RT-qPCR)
Quantitative qPCR was conducted by using TaqMan Fast Virus 1-Step Master Mixture (Applied Biosystem) according to the manufacturer’s instructions. The primers and probes for the hRSV large (L) polymerase gene (forward primer, 5′-TCCCTACGGTTGTGATCGATAGA -3′; reverse primer, 5′-TGATGGGAAGTAGTAGTGTAAAGTTGGT -3′; MGB-probe, 5′-(FAM)- AGGTAATACAGCCAAATC - nonfluorescent quencher (NFQ) on the 3′ end) were obtained from Applied Biosystem. Standard to quantitate virus specific RNA was plasmid DNA with cloned portion of L gene.
RNA sequencing
mRNA was prepared using Dynabeads mRNA kit (Invitrogen) and fragmented by heat. For cDNA synthesis, custom oligo-dT primers were used with a barcode and an adapter-linker sequence (CAG ACG TGT GCT CTT CCG ATC T—XXXXXXXX-T15) and with the Affinity Script enzyme (Agilent). After first strand synthesis, samples were pooled together based on ACTB qPCR values. RNA-DNA hybrids was degraded using consecutive acid-alkali treatment. A second sequencing linker (AGA TCG GAA GAG CGT CGT GTAG) was then ligated using T4 ligase (NEB). The mixture was then PCR enriched for 12 cycles and purified to yield final strand specific RNA-seq libraries as previously described55. Libraries were sequenced using a HiSeq 2500 (Illumina) using 50bp × 25bp pair-end sequencing.
RNA-seq analysis
Fastq files for each sample were aligned to hg38 genome (Release 23 Gencode, GRCh38.p3) using STAR56. Each sample was evaluated according to a variety of both pre- and post-alignment QC measures with Picard tools. To assess batch effects in data set we used hierarchical clustering (average linkage algorithm with 1 minus the Pearson correlation coefficient as the dissimilarity measure) and principal component analysis. Aligned reads were quantified using quant3p script (github.com/ctlab/quant3p) to account for specifics of 3′ sequencing: higher dependency on good 3′ annotation and lower level of sequence specificity close to 3′ end. Actual read contings were carried out by HTSeq57 with enriched genome annotation and alignment with fixed multimapper flags.
For analysis, two or three replicates from each condition were used, due to presence of outliers for some conditions. Performed PCA (Fig. S12) supports the ability to use either two or three replicates. Undetected genes (number of matching reads < 5 across all samples) were excluded. DESeq258 was used for analysis of differential gene expression. Pre-ranked GSEA59 was used to identify pathway enrichments.
Data availability
The hRSV NS1 crystal structure and reflection data have been deposited in the Protein Data Bank under accession code 5VJ2. Other data supporting the findings of this study are available upon request.
Supplementary Material
Figure 1. Sequence alignment of NS1 proteins from the genus Orthopneumovirinae.
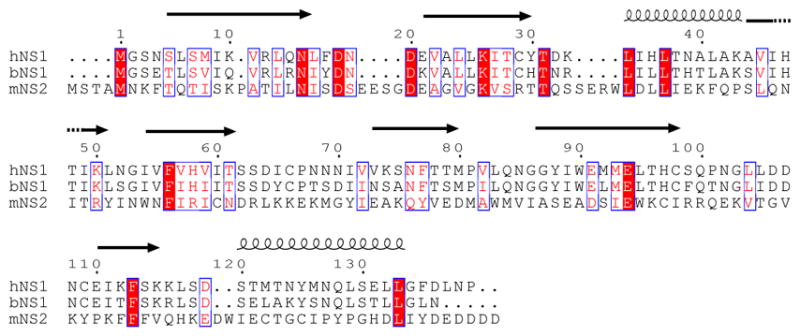
Sequences NS1 from human (hNS1; P04544), bovine (bNS1; Q65694), and NS2 from pneumonia virus of mice (mNS2; Q6PWL3) were aligned by ClustalW. Secondary structural elements for hRSV NS1 are shown above. Residues conserved across all family members are highlighted red, partially conserved are red with a blue box, and not conserved are not highlighted.
Acknowledgments
We would like to thank Drs. H. Virgin, M. Diamond, T. Ellenberger, and J. Payton and Evan E.L. Amarasinghe for discussion and J. Huh for technical support. Work in our laboratories are supported by in part by NIH grants (R01AI107056 (Leung), R01AI123926 (Amarasinghe), R01AI114654 (Basler), U191099565 (Ting, PI) to G.K.A., U19AI109945 (Basler), U19AI109664 (Basler), U19AI070489 (Holtzman), R01AI111605 (Holtzman), R01 AI130591 (Holtzman), R01AI087798 (Moore), U19AI095227 (Moore), and T32-CA09547-37 (Allen, PI) to D.S.J; Department of the Defense, Defense Threat Reduction Agency grants HDTRA1-16-0033 (Basler) and HDTRA1-16-0033 (Basler); the National Science Foundation MCB-1121867 (Pappu); Children’s Discovery Institute PD-II-2013-272 (Amarasinghe). S.C. is funded in part by American Heart Association Postdoctoral Fellowship (15POST25140009). The content of the information does not necessarily reflect the position or the policy of the federal government and no official endorsement should be inferred.
Footnotes
Author contributions: GKA and DWL conceived and designed the overall study with input from co-authors. SC, PL, EE, EA, BCY, DMB, MRE, AM, JH, PR, SJ, GKA, and DWL performed research. MLM provided the wildtype virus. All co-authors analyzed results. RP, MJH, MLM, MA, CFB, GKA, and DWL designed and coordinated studies within each group. CFB, GKA and DWL wrote manuscript with input from all co-authors. All authors analyzed results, read and approved manuscript for submission.
References
- 1.Hall CB. The burgeoning burden of respiratory syncytial virus among children. Infectious disorders drug targets. 2012;12:92–97. doi: 10.2174/187152612800100099. [DOI] [PubMed] [Google Scholar]
- 2.Stockman LJ, Curns AT, Anderson LJ, Fischer-Langley G. Respiratory syncytial virus-associated hospitalizations among infants and young children in the United States, 1997–2006. The Pediatric infectious disease journal. 2012;31:5–9. doi: 10.1097/INF.0b013e31822e68e6. [DOI] [PubMed] [Google Scholar]
- 3.Bitko V, et al. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J Virol. 2007;81:1786–1795. doi: 10.1128/JVI.01420-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol. 2005;79:9315–9319. doi: 10.1128/JVI.79.14.9315-9319.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spann KM, Tran KC, Chi B, Rabin RL, Collins PL. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected] J Virol. 2004;78:4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spann KM, Tran KC, Collins PL. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol. 2005;79:5353–5362. doi: 10.1128/JVI.79.9.5353-5362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munoz FM. Respiratory syncytial virus in infants: is maternal vaccination a realistic strategy? Current opinion in infectious diseases. 2015;28:221–224. doi: 10.1097/QCO.0000000000000161. [DOI] [PubMed] [Google Scholar]
- 8.Collins PL, Melero JA. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. 2011;162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simoes EA, et al. Challenges and opportunities in developing respiratory syncytial virus therapeutics. The Journal of infectious diseases. 2015;211(Suppl 1):S1–S20. doi: 10.1093/infdis/jiu828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bossert B, Marozin S, Conzelmann KK. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol. 2003;77:8661–8668. doi: 10.1128/JVI.77.16.8661-8668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren J, et al. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J Gen Virol. 2011;92:2153–2159. doi: 10.1099/vir.0.032987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goswami R, et al. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013;23:1025–1042. doi: 10.1038/cr.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliott J, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol. 2007;81:3428–3436. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Straub CP, et al. Mutation of the elongin C binding domain of human respiratory syncytial virus non-structural protein 1 (NS1) results in degradation of NS1 and attenuation of the virus. Virol J. 2011;8:252. doi: 10.1186/1743-422X-8-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munir S, et al. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. 2011;7:e1001336. doi: 10.1371/journal.ppat.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munir S, et al. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J Virol. 2008;82:8780–8796. doi: 10.1128/JVI.00630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Nouen C, et al. Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology. 2009;385:169–182. doi: 10.1016/j.virol.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker Y. Respiratory syncytial virus(RSV)-induced allergy may be controlled by IL-4 and CX3C fractalkine antagonists and CpG ODN as adjuvant: hypothesis and implications for treatment. Virus Genes. 2006;33:253–264. doi: 10.1007/s11262-006-0063-y. [DOI] [PubMed] [Google Scholar]
- 19.Becker Y. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy--a review. Virus Genes. 2006;33:235–252. doi: 10.1007/s11262-006-0064-x. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez PA, et al. Respiratory syncytial virus impairs T cell activation by preventing synapse assembly with dendritic cells. Proc Natl Acad Sci U S A. 2008;105:14999–15004. doi: 10.1073/pnas.0802555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hastie ML, et al. The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol Cell Proteomics. 2012;11:108–127. doi: 10.1074/mcp.M111.015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu W, et al. The interactome of the human respiratory syncytial virus NS1 protein highlights multiple effects on host cell biology. J Virol. 2012;86:7777–7789. doi: 10.1128/JVI.00460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Money VA, McPhee HK, Mosely JA, Sanderson JM, Yeo RP. Surface features of a Mononegavirales matrix protein indicate sites of membrane interaction. Proc Natl Acad Sci U S A. 2009;106:4441–4446. doi: 10.1073/pnas.0805740106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dessen A, Volchkov V, Dolnik O, Klenk HD, Weissenhorn W. Crystal structure of the matrix protein VP40 from Ebola virus. EMBO J. 2000;19:4228–4236. doi: 10.1093/emboj/19.16.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Battisti AJ, et al. Structure and assembly of a paramyxovirus matrix protein. Proc Natl Acad Sci U S A. 2012;109:13996–14000. doi: 10.1073/pnas.1210275109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gaudier M, Gaudin Y, Knossow M. Crystal structure of vesicular stomatitis virus matrix protein. EMBO J. 2002;21:2886–2892. doi: 10.1093/emboj/cdf284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann P, et al. Crystal structure of the Borna disease virus matrix protein (BDV-M) reveals ssRNA binding properties. Proc Natl Acad Sci U S A. 2009;106:3710–3715. doi: 10.1073/pnas.0808101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forster A, Maertens GN, Farrell PJ, Bajorek M. Dimerization of matrix protein is required for budding of respiratory syncytial virus. J Virol. 2015;89:4624–4635. doi: 10.1128/JVI.03500-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holtzman MJ, Byers DE, Alexander-Brett J, Wang X. The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nature reviews. Immunology. 2014;14:686–698. doi: 10.1038/nri3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valarcher JF, et al. Role of alpha/beta interferons in the attenuation and immunogenicity of recombinant bovine respiratory syncytial viruses lacking NS proteins. J Virol. 2003;77:8426–8439. doi: 10.1128/JVI.77.15.8426-8439.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dave KA, et al. A comprehensive proteomic view of responses of A549 type II alveolar epithelial cells to human respiratory syncytial virus infection. Mol Cell Proteomics. 2014;13:3250–3269. doi: 10.1074/mcp.M114.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez I, Lombardia L, Garcia-Barreno B, Dominguez O, Melero JA. Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J Gen Virol. 2007;88:570–581. doi: 10.1099/vir.0.82187-0. [DOI] [PubMed] [Google Scholar]
- 33.Karron RA, et al. A gene deletion that up-regulates viral gene expression yields an attenuated RSV vaccine with improved antibody responses in children. Sci Transl Med. 2015;7:312ra175. doi: 10.1126/scitranslmed.aac8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscilation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 35.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta crystallographica. Section D, Biological crystallography. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 36.Sheldrick GM. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta crystallographica. Section D, Biological crystallography. 2010;66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otwinowski Z. Maximum likelihood refinement of heavy atom parameters. In: Wolf W, EPR, Leslie AGW, editors. CCP4 Study Weekend. Science and Engineering Research Council; 1991. pp. 80–86. [Google Scholar]
- 38.Cowtan K, Main P. Miscellaneous algorithms for density modification. Acta crystallographica. Section D, Biological crystallography. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 39.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nature protocols. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. ARP/wARP and molecular replacement. Acta crystallographica. Section D, Biological crystallography. 2001;57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 41.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta crystallographica. Section D, Biological crystallography. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 42.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica. Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 43.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica. Section D, Biological crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ. Efficient anisotropic refinement of macromolecular structures using FFT. Acta crystallographica. Section D, Biological crystallography. 1999;55:247–255. doi: 10.1107/S090744499801405X. [DOI] [PubMed] [Google Scholar]
- 45.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta crystallographica. Section D, Biological crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis IW, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.COLLABORATIVE COMPUTATIONAL PROJECT, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 48.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 49.Laskowski RA. Enhancing the functional annotation of PDB structures in PDBsum using key figures extracted from the literature. Bioinformatics. 2007;23:1824–1827. doi: 10.1093/bioinformatics/btm085. [DOI] [PubMed] [Google Scholar]
- 50.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 51.Bond CS. TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
- 52.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Current protocols in bioinformatics/editoral board, Andreas D. Baxevanis … [et al.] 2002;Chapter 2(Unit 2):3. doi: 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- 53.Gouet P, Robert X, Courcelle E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hotard AL, et al. A stabilized respiratory syncytial virus reverse genetics system amenable to recombination-mediated mutagenesis. Virology. 2012;434:129–136. doi: 10.1016/j.virol.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jha AK, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 56.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. Journal of molecular biology. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 61.Vitalis A, Pappu RV. ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. Journal of computational chemistry. 2009;30:673–699. doi: 10.1002/jcc.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The hRSV NS1 crystal structure and reflection data have been deposited in the Protein Data Bank under accession code 5VJ2. Other data supporting the findings of this study are available upon request.