

Abstract
Sleep-disordered breathing is common in heart failure patients and is associated with increased morbidity and mortality. Central sleep apnea occurs more commonly in heart failure-reduced ejection fraction, and obstructive sleep apnea occurs more frequently in heart failure with preserved ejection fraction. Although the two types of sleep-disordered breathing have distinct pathophysiologic mechanisms, both contribute to abnormal cardiovascular consequences. Treatment with continuous positive airway pressure for obstructive sleep apnea in heart failure has been well defined, whereas treatment strategies for central sleep apnea in heart failure continue to evolve. Unilateral transvenous neurostimulation has shown promise for the treatment of central sleep apnea. In this paper, we examine the current state of knowledge of treatment options for sleep-disordered breathing in heart failure.
Keywords: Sleep-disordered breathing, sleep apnea, central sleep apnea, obstructive sleep apnea, heart failure
Sleep-disordered breathing (SDB) is common in heart failure (HF) patients and is associated with increased morbidity and mortality. Abnormal sleep patterns are often characterised by cycles of significant pauses in breathing and partial neurological arousals that lead to maladaptive neurohormonal activation. SDB is broadly classified into two types: obstructive sleep apnea (OSA) and central sleep apnea (CSA). The former occurs in both the general and HF populations, whereas the latter is more often associated with HF.[1,2] Whereas the benefits of treatment for OSA have been repeatedly confirmed in the literature, the effectiveness and safety of treatments for CSA in HF remains controversial.[3,4] This brief review will explore the pathophysiology of SDB in HF, and discuss the effects of various treatment options specifically in this patient population.
Definition and Epidemiology
An apnea is defined as the absence of inspiratory airflow for at least 10 seconds. A hypopnea is a lesser decrease in airflow, associated with a drop in arterial oxygen saturation and/or an arousal. Apneas and hypopneas are classified according to type of SDB in which they occur: OSA occurs when upper airway occlusion occurs with continued activity of inspiratory thoracic pump muscles; CSA occurs when there is a reduction in neural stimulus to thoracic respiratory muscles (diaphragm and intercostal muscles), leading to a reduction/or absence of the breathing rhythm, without upper airway obstruction.[5] The apnea-hypopnea index (AHI), defined as the mean number of apnea and/or hypopnea episodes that occur during sleep divided by the number of hours of sleep, expressed as events/h, defines severity of SDB. Mild severity is defined as an AHI between 5 and 15 events/h, moderate severity as an AHI ≥15 events/h but <30 events/h, and severe sleep apnea as ≥30 events/h. Numerous studies have shown that mortality rises as the AHI increases.[6,7]
HF is one of the most common underlying conditions for SDB in adults, and more than 50 % of HF patients have SDB.[8,9] This disorder occurs in both HF with reduced ejection fraction (HFrEF), as well as HF with preserved ejection fraction (HFpEF). In a meta-analysis of several real-world sleep studies, the combined incidence of sleep apnea in HF is estimated at 53 % in HFrEF and 48 % in HFpEF patients. CSA occurs more frequently in HFrEF (34 % of patients), and OSA occurs more frequently in HFpEF (25 % of patients).[10] The OSA phenotype occurs more commonly in the general population as well, with 34 % men and 17 % women being affected.[11] Obesity and advancing age are the major risk factor for OSA.[12] Fluid overload states have also been identified as a risk factor because nocturnal fluid shifts to the neck and chest can cause collapse of the pharynx.[13] The CSA phenotype predominates in HF patients, with estimated rates of 30–50 %. This prevalence is likely to be underestimated because symptoms of CSA may be indistinguishable from those of underlying HF.[2] A number of risk factors have been identified for the development of CSA in HF, including male sex, higher New York Heart Association (NYHA) functional class, lower ejection fraction, higher B-type natriuretic peptide levels, waking hypocapnia (arterial partial pressure of carbon dioxide [PaCO[2] ] <38 mmHg), higher prevalence of atrial fibrillation and frequent nocturnal ventricular arrhythmias.[9,12,14] Patients with CSA are often distinct from those with OSA, in that they are often not obese, often have no history of snoring, and yet have more daytime fatigue.[9,10] Although no screening tool has been validated to identify CSA in HF, this SDB should be suspected when one or more of the above abnormalities are present.[15]
Pathophysiology
Obstructive Sleep Apnea
The pathogenesis of OSA stems from a complex interaction between unfavourable anatomic upper airway susceptibility and sleep-related changes in upper airway function. Sleep is associated with a decreased metabolic rate, loss of the wakefulness drive to breathe, and a subsequent decrease in ventilatory neural output to respiratory muscles, including upper airway muscles.[16] In patients with unfavourable anatomy, such as alterations in craniofacial structures, enlarged tonsils, upper airway oedema, decreased lung volume due to pulmonary oedema and obesity, vulnerability to upper airway obstruction is more common.[5] With reduction in the activity of the genioglossus muscle at the onset of sleep, the tongue falls backward, and individuals with altered mechanical properties of the upper airway are prone to upper airway obstruction.[5,17] Non-anatomic factors, such as upper airway dilator muscle dysfunction, heightened chemosensitivity to CO[2] and low arousal threshold, have also been implicated.[5]
Central Sleep Apnea
Commonly seen in HF patients, CSA is distinguished by the temporary withdrawal of central (brainstem-mediated) respiratory drive that results in the cessation of respiratory muscle activity and airflow. The SDB pattern that subsequently results in CSA commonly manifests in the form of Cheyne–Stokes respiration, a form of periodic breathing with recurring cycles of crescendo decrescendo ventilation that culminates in a prolonged apnea or hypopnea episode.[1] The pathogenesis of CSA in HF is complex and remains incompletely understood. However, a substantial body of research suggests that an increased respiratory control response to changes in PaCO[2] above and below the apneic threshold is central to the pathogenesis of CSA in HF.[18,19] The respiratory control system maintains tight regulation of levels of O[2] and CO[2] , and during sleep PaCO[2] becomes the primary stimulus for ventilation. Therefore, any increase in PaCO[2] will stimulate ventilation, whereas any decrease in PaCO[2] will suppress it. Respiration can cease altogether if PaCO[2] falls below the tightly regulated level called the apneic threshold. Normally, at the onset of sleep, ventilation decreases and PaCO[2] increases. This keeps the prevailing level of PaCO[2] well above the apneic threshold, allowing normal, rhythmic breathing to continue throughout the night. However, it is important to consider that it may not be the absolute value of steady state PaCO[2] that increases the likelihood of developing central apnea, but rather the absolute difference between the prevailing PaCO[2] and apneic threshold PaCO[2] that is more important.[20] Furthermore, not only may there be static hyperventilation in HF, alterations in various components of the negative feedback system that control breathing also increase the likelihood of developing periodic breathing, during both sleep and wakefulness. Factors such as prolonged circulatory time, increased chemoreceptor gain, and exaggerated responses to ventilation provoke instability in the negative feedback loop and consequent abnormal periodic breathing and CSA.[21]
The neurohormonal and haemodynamic alterations occurring in HF also contribute to the development and progression of CSA. The three key factors leading to CSA in HF include hyperventilation, circulatory delay, and brain responses to altered concentrations of O[2] and CO[2] .[1] Factors leading to chronic hyperventilation typical of HF patients include pulmonary interstitial congestion due to rostral fluid displacement occurring in the supine position, activation of pulmonary stretch receptors stimulating increase in ventilation, and activation of peripheral chemoreceptors triggering an exaggerated response to lowered CO[2] levels, a mechanism which contributes to the cyclical pattern of hyperventilation – hypoventilation and apnea.[1] Reduced cardiac output in HF patients delays detection of changes in blood gases between the peripheral and the central chemoreceptors, further exacerbating the cyclical pattern of periodic breathing and increasing the duration of apneic events seen with CSA.[22] Cerebrovascular reactivity is directly influenced by changes in PaCO[2] , and blunted responses are noted in HF and CSA patients, leading to an ineffective ability to dampen ventilatory hypoventilation or hyperventilation overshoot, perpetuating episodes of CSA.[23]
Pathological Consequences
The repeated episodes of apnea, hypoxia, re-oxygenation, and arousal throughout the night have serious pathophysiological consequences, including further sympathetic nervous system (SNS) activation, oxidative stress, systemic inflammation and endothelial dysfunction. Repeated bursts of sympathetic activity are noted in patients with SDB, manifesting with increased urinary nocturnal norepinephrine secretion as well as increased daytime muscle sympathetic nerve activity.[24,25] The link between increased SNS activity and higher mortality in HF is well known.[26–29] Arterial blood gas abnormalities, excessive arousals, and large intrathoracic pressure swings are known to occur during SDB.[5] These pressure changes may increase left ventricular afterload, increase myocardial oxygen demand and impede stroke volume. The exaggerated intrathoracic pressure changes during SDB can lead to increased transmural pressure exposure to the thin walled atria, leading to atrial stretch and susceptibility of atrial fibrillation.[30] Increased oxidative stress and development of reactive oxygen species in the setting of repeated hypoxia-reoxygenation episodes have been postulated to occur with SDB.[31] Several studies have demonstrated that patients with sleep apnea have increased levels of pro-inflammatory cytokines, cellular adhesion molecules, and activated circulating neutrophils.[31–33] These mechanisms can lead to chronic inflammation in SDB, which has been postulated to contribute to pulmonary oedema as well as to the anorexia and cachexia that frequently occurs in patients with advanced HF.[34,35]
OSA is a known risk factor for the development of arterial hypertension, and is associated with an increased incidence of stroke, metabolic syndrome, and coronary heart disease.[36–38] The occurrence of SDB complicated by recurrent episodes of oxygen desaturation has also been associated with an almost twofold increase in the risk of sudden death, independent of known risk factors.[39] Both OSA and CSA have now been shown in prospective longitudinal studies to be independent predictors of incident HF.[40,41] In fact, in patients with HFpEF, obstructive apnea events have been shown to cause increases in pulmonary capillary pressure, and OSA has been associated with an increase in LV mass and the development of diastolic dysfunction.[10] These findings suggest that SDB is not simply a marker of HF, but may be a mediating factor contributing to the onset and progression of clinically overt HF. These negative cardiovascular consequences highlight the critical need for safe and effective treatment of SDB in HF patients.
Treatment
Obstructive Sleep Apnea
Continuous positive airway pressure (CPAP) ventilation is the most widely used treatment option for OSA. In multiple studies, this therapy has been shown to produce several cardiovascular benefits, including reduction in blood pressure, risk of stroke/transient ischaemic attack and arrhythmias.[5,42,43] Several studies have been performed in patients with HF and OSA. Among 24 patients with left ventricular dysfunction and OSA, compared to controls, CPAP therapy markedly reduced AHI, systolic blood pressure, and average heart rate. Furthermore, compared to no treatment, the use of CPAP reduced left ventricular end systolic dimension (54.5 ± 1.8 to 51.7 ± 1.2 mm [p=0.009]), and improved the left ventricular ejection fraction (25.0 ± 2.8 % to 33.8 ± 2.4 % [p<0.001])[44] (see Figure 1). Reductions in overnight urinary norepinephrine excretion and improvements in quality of life also occurred with the treatment of OSA with CPAP in HF patients.[45] In the largest retrospective cohort study of a US Medicare database of 30,719 patients with newly diagnosed HF between 2003 and 2005, treatment of SDB was associated with decreased readmission, healthcare cost and mortality among subjects who were diagnosed and treated, with an improved 2-year survival rate in those who were treated compared to those who were not (hazard ratio: 0.49, 95 % CI: 0.29–0.84, p=0.009).[46] Hypoglossal nerve stimulation for the treatment of OSA has also been shown to reduce AHI and oxygen desaturation events, and now has reported long-term sustained benefits in patient reported outcomes.[47,48] This technology that consists of an implantable pulse generator with sensing and stimulation leads to prevent airway collapse during sleep has been approved by the FDA for commercial use in the USA for patients with moderate to severe OSA who have failed or are unable to tolerate CPAP therapies.
Figure 1: Effect of Continuous Positive Airway Pressure Versus Control on Left Ventricular Ejection Fraction in Twenty Four Patients with Obstructive Sleep Apnea.
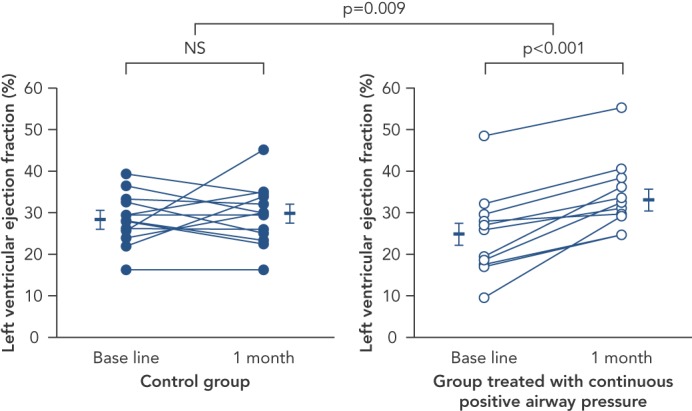
A statistically-significant increase in ejection fraction is noted with CPAP treatment as compared to control (p=0.009) (absolute increase in EF of 8.8 ± 1.6 %, relative increase in EF of 35 % [p<0.001]). CPAP = continuous positive airway pressure; EF = ejection fraction; NS = not significant. Source: Kaneko, et al., 2003, published with permission.44.
Central Sleep Apnea
In HF patients with CSA, optimisation of medical therapy and effective decongestion are the first and foremost steps in the treatment of this sleep disorder. Treatment with cardiac resynchronisation therapy has been shown to effectively reduce AHI events in patients with CSA.[49] Despite strict adherence to guideline-directed medical therapy, CSA remains a significant comorbidity in patients with HF.
In contrast to OSA, where the safety and effectiveness of CPAP are no longer questioned, the role of this therapy in patients with CSA remains the subject of controversy. Early trials of CPAP for CSA and HF patients showed some positive effects, including a reduction in central apnea/hypopnea events, ventricular ectopic beats, and nocturnal urinary and daytime plasma norepinephrine levels, and a trend towards a reduction in mortality and need for cardiac transplantation.[50–52] In the Canadian Positive Airway Pressure Trial for Patients with Congestive HF and Central Sleep Apnea (CANPAP) trial in 258 optimally treated HF patients with an LVEF <40 % and AHI >15 events/h, compared to control, CPAP did not prolong transplant-free survival, despite reduction of AHI from 40 to 19 events/h, improvement of nocturnal oxygenation, exercise tolerance and decrease in plasma norepinephrine levels after 3 months of CPAP therapy.[53] In fact, the trial was stopped early for futility given a diverging trend towards increased mortality early on in the CPAP group, and yet the overall event rates of death or transplant did not differ after 18 months. Notably, in CANPAP the average duration of CPAP was 3.6 h/night and CSA was not adequately suppressed in 43 % of the study subjects.[1] However, a post-hoc analysis showed that, compared to inadequately treated patients, those in whom CPAP decreased AHI below 15 events/h had significant prolongation of transplant-free survival.[54] These findings suggest that mask-based therapeutic strategies may be limited by poor patient compliance.
Adaptive pressure support servo-ventilation (ASV), an alternative noninvasive ventilatory support modality, was developed to make positive airway pressure more tolerable to patients with SDB. ASV delivers a baseline continuous positive airway pressure similar to CPAP, and yet can also detect episodes of central apneas and deliver several breaths at the tidal volume and respiratory rate previously determined to match the patient’s minute ventilation during stable breathing. The goal of ASV therapy is to prevent the increase in PaCO[2] during apnea and the hyperventilation that follows, thereby breaking the abnormal periodic breathing cycle. Several small preliminary studies have suggested that ASV is better tolerated than CPAP, and may be more effective than CPAP in the treatment of CSA in HF.[55] However, in a large randomised trial, the Treatment of Predominant Central Sleep Apnea by Adaptive Servo Ventilation in Patients With HF (SERVE-HF), ASV did not reduce the primary combined endpoint of all cause death, cardiac transplantation or ventricular assist device implantation, sudden cardiac arrest or HF hospitalisation. In fact, as compared to the controls, ASV was associated with an increase in cardiovascular mortality (hazard ratio for cardiovascular death: 1.34; 95 % CI: 1.09–1.65; p=0.006)[56] (see Figure 2).
Figure 2: Cumulative Incidence Curves for the Primary End Point, Death from Any Cause, and Cardiovascular Death in the SERVE-HF Trial.
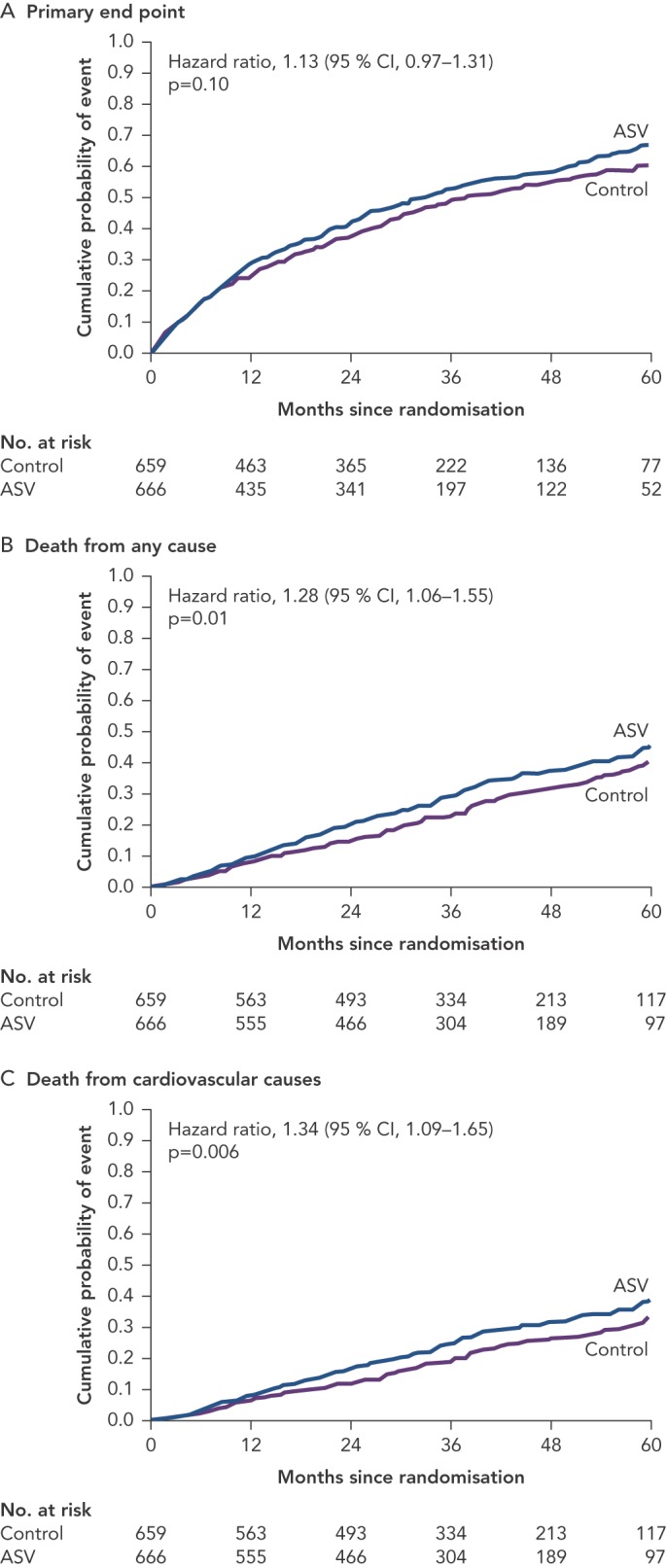
ASV treated patients had higher incidence of death from any cause and cardiovascular death. ASV = Adaptive servo ventilation. Source: Cowie, et al., 2015, published with permission.56.
Several postulates of the disconcerting results of SERVE-HF have been presented, including the possibility that positive airway pressure may further reduce cardiac output in a population that may be vulnerable with limited cardiac reserve, or that central sleep apnea could be a beneficial compensatory mechanism in patients with advanced HF. This latter theory appears inherently flawed, as the intermittent hypoxia and norepinephrine release associated with central sleep apnea events make it unlikely that this sleep disorder confers any long-term benefits to patients with HF. The first-generation ASV device used in SERVE-HF, no longer manufactured by the sponsor, had limited technology with fixed settings that may have applied pressures that were too low for some patients and excessive for others, leading to adverse cardiovascular consequences. Newer generation devices that incorporate novel algorithms with dynamic settings may allow for the prevention of excessive positive airway pressure and its potential detrimental cardiac effects.[57] Ultimately, further trials will be required to mediate these discrepant results, and studies with newer generation ASV devices in HFrEF and SDB are ongoing.[58]
Phrenic Nerve Stimulation
Transvenous unilateral neurostimulation is a unique physiological approach to the treatment of central sleep apnea. The remedē® System (Respicardia Inc) aims to stimulate a nerve to cause diaphragmatic movement producing changes in carbon dioxide concentrations and tidal volumes similar to normal breathing. Unilateral transvenous neurostimulation does not produce a ‘hiccup-type’ diaphragmatic response, such as that noted occasionally with direct stimulation of the diaphragm with cardiac resynchronisation therapy. Instead, the device provides neurostimulation pulses configured to smoothly engage the diaphragm, like normal breathing. The neurostimulator is placed in either the left or right pectoral region; the stimulation lead is placed in either the left pericardiophrenic or right brachiocephalic vein to stimulate the phrenic nerve. The sensing lead is placed in a thoracic vein, such as the azygos vein, to sense respiration by thoracic impedance. The system aims to automatically stimulate the phrenic nerve during the scheduled time at night when the patient is asleep and in a reclining position, which is detected by a position and motion sensor present in the device. Transvenous neurostimulation improved apnea indices, quality of life, and had an acceptable safety profile in pilot studies. In the remedē System Pivotal Trial, 151 eligible patients underwent device implantation and were then randomly assigned to initiate neurostimulation either 1 month later (treatment, n=73), or after the 6-month primary efficacy endpoint evaluation (control, n=68).
Significantly more patients in the treatment group (51 %) had an AHI reduction from baseline of 50 % or greater at 6 months than had those in the control group (11 %). The difference between groups was 41 % (95 % CI 25–54, p<0.0001) (see Figure 3). One hundred and thirty-eight (91 %) of 151 patients had no serious-related adverse events at 12 months. Seven (9 %) cases of related-serious adverse events occurred in the control group and six (8 %) cases were reported in the treatment group. Seven patients died (unrelated to implant, system, or therapy): four deaths (two in the treatment group and two in the control group) during the 6-month randomisation period and three deaths between 6 months and 12 months. Thirty-seven percent of the treatment group patients reported non-serious therapy-related discomfort that was resolved with simple system reprogramming in all but one patient. All prespecified hierarchically tested secondary sleep and quality of life measure endpoints were improved in the treatment group compared to control.59 Exploratory evaluation of the 64 % of the study subjects who had underlying HF revealed that, compared to HF controls, the HF treatment group included a greater percentage of patients who had a reduction in AHI>50 % at 6 months (63 % versus 4 %, p<0.0001). In the HF group, tolerability and safety were similar to those of the overall population.[59]
Figure 3: Percentage Change in Apnea-hypopnea Index at 6 Months’ Follow up Compared with Baseline.
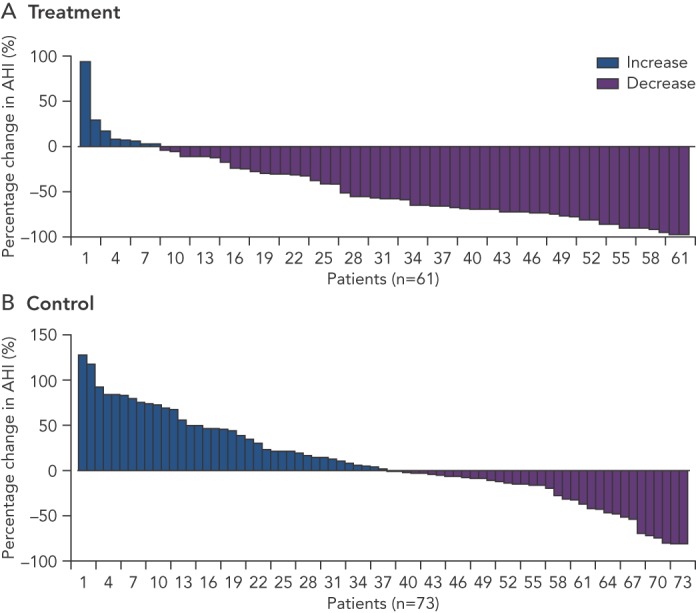
More patients in the treatment group had a decrease in AHI as opposed to the control group. AHI = apnea-hypopnea index. Source: Costanzo, et al., 2016, published with permission.59.
The results of this trial indicate that transvenous neurostimulation produces significant improvements in reducing the severity of central sleep apnea, as measured by several pre-specified sleep indices obtained during polysomnography and scored by masked investigators in a core laboratory. Improvements were observed with use of the device in the arousal index, REM sleep, PGA scores, and ESS quality of life measures at 6 months of follow up. The therapy was well tolerated, with only two patients who were unable to adjust to therapy, and first implant success was high. Procedural complications, including lead dislodgements, were comparable with other implantable devices using transvenous lead technology.
Results from the SERVE-HF trial showed an unexpected increase in the risk of cardiovascular mortality (p=0.006), despite a significant reduction in AHI from baseline to 12 months of follow up. However, it might not be appropriate or valid to assume that the effects of ASV on the outcomes of patients with advanced HF also apply to the effects of neurostimulation in a different population. Although 96 (64 %) patients in this study had previous HF, only 59 (39 %) had HF severity and left ventricular dysfunction similar >to the population in the SERVE-HF trial. Exploratory post-hoc analyses of the pivotal trial suggested that indeed the effects of neurostimulation in the subgroup of patients with HF were consistent with the findings in the overall trial population.[59] Importantly though, the mechanism of action of neurostimulation remains distinctly different from that of ASV. Specifically, while ASV delivers positive airway pressure, the diaphragmatic contraction triggered by neurostimulation generates negative intrathoracic pressure. In fact, unilateral transvenous stimulation has been the only therapy to show a reduction in sleep-related arousals, which are a manifestation of acute neurohormonal activation. Neurostimulation was associated with an improvement in quality of life measures, whereas ASV was not, suggesting clinical benefits beyond just that of improved sleep variables. Additional investigation will be warranted to study the hemodynamic effects of negative intrathoracic pressure in patients with HF, as well as trials focusing on cardiovascular outcomes to provide further supportive data.
Supplemental Oxygen
Observational studies in patients with HFrEF have shown that nocturnal nasal oxygen improves CSA, with data suggesting improvement in exercise capacity; decreases in nocturnal urinary norepinephrine excretion; improvement in ventricular arrhythmias, and quality of life.[60] Nocturnal hypoxemia is known to be an independent predictor of all-cause mortality in HFrEF patients, and an O[2] sat <78 % during SDB is a strong predictor of sudden cardiac death.[61,62] Supplemental oxygen is indicated for patients with CSA who have confirmed hypoxemia during sleep. It can be used along with positive airway pressure therapy, or may also be considered for patients who do not tolerate or fail positive airway pressure therapy. However, oxygen therapy remains contraindicated in patients without hypoxemia, as there can be theoretical detrimental effects to this treatment strategy, such as lengthening apnea duration and accelerating CO[2] retention.[63] Overall studies have been small with short-term duration of follow up. Further randomised controlled trials are needed to determine the role of oxygen in treating sleep apnea in HFrEF.
Pharmacologic Therapies
Patients who do not tolerate positive airway pressure therapy during sleep may consider treatment with a respiratory stimulant, such as acetazolamide or theophylline. Acetazolamide is a carbonic anhydrase inhibitor and a weak diuretic. It causes mild metabolic acidosis, which stimulates respiration and is shown to decrease the frequency of central apnea episodes.[20] Theophylline, a methylxanthine drug that acts as a nonselective adenosine receptor antagonist, at therapeutic plasma concentration levels (11 μg/mL, range 7–15 μg/mL), has shown to reduce AHI events in patients with HF and CSA.[64] Ultimately, there are no long-term data available on either of these medications, as well as narrow therapeutic ranges, and these therapies can have harmful side-effects that need to be monitored closely.
Knowledge Gaps
Randomised control trials are needed to provide further assessment of the role of any sleep apnea intervention on cardiovascular associated morbidity and mortality. Low adherence to CPAP therapy has been a major limitation in clinical trials, and continues to be a dilemma with clinical application of mask-based therapies. Treatment of CSA with ASV could be harmful, and further trials are needed to determine whether newer generation devices may be of benefit. Unilateral transvenous neurostimulation shows promise for the treatment of CSA, but will need further validation for long-term cardiovascular outcome benefit.
Conclusion
Our understanding of the causes and subsequent pathological consequences of SDB in HF has been greatly expanded over the past few decades. SDB is now recognised as an important, independent risk factor for the development of incident HF, worsening HF status, and reduced survival in patients with HF. Unfortunately, SDB is often under-recognised, and not tested for routinely; and yet, we know that treatment can improve outcomes in these patients. CPAP therapy for OSA in HF patients can improve AHI, improve blood pressure, and even improve left ventricular ejection fraction. Survival is also improved in observational studies of CPAP treatment in HF patients. Vigilance in diagnosis, testing, and treatment is paramount in this population.
Therapies for CSA remain more complex. Because the pathological consequences of CSA are known to worsen HF, treatment strategies remain vital to management. Optimising medical therapy as first approach remains of utmost importance, as research has shown that often when HF is clinically improved, CSA often improves as well.[65,66] In cases where CSA persists despite aggressive treatment of HF, further therapeutic interventions should be considered. The CANPAP trial did not reveal benefits of treatment with CPAP therapy for CSA. Results of the SERVE-HF trial suggest that treatment of CSA with ASV could be harmful. However, limitations of this study with utilisation of older generation devices and limited treatment algorithms still pose several questions.[57] Transvenous unilateral neurostimulation is now recognised as another treatment option of CSA known to reduce AHI, but does not carry the negative consequence of increasing intrathoracic pressure. Future randomised control trials with unilateral transvenous neurostimulation for HF should be powered to determine cardiovascular outcomes.
References
- 1.Costanzo MR, Khayat R, Ponikowski P et al. Mechanisms and clinical consequences of untreated central sleep apnea in heart failure. J Am Coll Cardiol. 2015;65:72–84. doi: 10.1016/j.jacc.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oldenburg O, Lamp B, Faber L et al. Sleep-disordered breathing in patients with symptomatic heart failure: a contemporary study of prevalence in and characteristics of 700 patients. Eur J Heart Fail. 2007;9:251–7. doi: 10.1016/j.ejheart.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Garcia MA, Campos-Rodriguez F, Catalan-Serra P et al. Cardiovascular mortality in obstructive sleep apnea in the elderly: role of long-term continuous positive airway pressure treatment: a prospective observational study. Am J Respir Crit Care Med. 2012;186:909–16. doi: 10.1164/rccm.201203-0448OC. [DOI] [PubMed] [Google Scholar]
- 4.Montserrat JM, Ferrer M, Hernandez L et al. Effectiveness of CPAP treatment in daytime function in sleep apnea syndrome: a randomized controlled study with an optimized placebo. Am J Respir Crit Care Med. 2001;164:608–13. doi: 10.1164/ajrccm.164.4.2006034. [DOI] [PubMed] [Google Scholar]
- 5.Javaheri S, Barbe F, Campos-Rodriguez F et al. Sleep apnea: types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69:841–58. doi: 10.1016/j.jacc.2016.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jilek C, Krenn M, Sebah D et al. Prognostic impact of sleep disordered breathing and its treatment in heart failure: an observational study. Eur J Heart Fail. 2011;13:68–75. doi: 10.1093/eurjhf/hfq183. [DOI] [PubMed] [Google Scholar]
- 7.Javaheri S, Shukla R, Zeigler H, Wexler L. Central sleep apnea, right ventricular dysfunction, and low diastolic blood pressure are predictors of mortality in systolic heart failure. J Am Coll Cardiol. 2007;49:2028–34. doi: 10.1016/j.jacc.2007.01.084. [DOI] [PubMed] [Google Scholar]
- 8.Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3:141–63. doi: 10.1002/cphy.c110057. [DOI] [PubMed] [Google Scholar]
- 9.Javaheri S, Parker TJ, Liming JD et al. Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations. Circulation. 1998;97:2154–9. doi: 10.1161/01.cir.97.21.2154. [DOI] [PubMed] [Google Scholar]
- 10.Kryger MH, Roth T, Dement WC. Principles and Practice of Sleep Medicine E-Book: Elsevier Health Sciences. 2015.
- 11.Peppard PE, Young T, Barnet JH et al. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol. 2013;177:1006–14. doi: 10.1093/aje/kws342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sin DD, Fitzgerald F, Parker JD et al. Risk factors for central and obstructive sleep apnea in 450 men and women with congestive heart failure. Am J Respir Crit Care Med. 1999;160:1101–6. doi: 10.1164/ajrccm.160.4.9903020. [DOI] [PubMed] [Google Scholar]
- 13.Lyons OD, Bradley TD. Heart failure and sleep apnea. Can J Cardiol. 2015;31:898–908. doi: 10.1016/j.cjca.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 14.Calvin AD, Somers VK, van der Walt C et al. Relation of natriuretic peptide concentrations to central sleep apnea in patients with heart failure. Chest. 2011;140:1517–23. doi: 10.1378/chest.10-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khayat R, Small R, Rathman L et al. Sleep-disordered breathing in heart failure: identifying and treating an important but often unrecognized comorbidity in heart failure patients. J Card Fail. 2013;19:431–44. doi: 10.1016/j.cardfail.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010 Jan;90((1)):47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dempsey JA, Veasey SC, Morgan BJ, Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90:47–47. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanly P, Zuberi N, Gray R. Pathogenesis of Cheyne-Stokes respiration in patients with congestive heart failure. Relationship to arterial PCO2. Chest. 1993;104:1079–84. doi: 10.1378/chest.104.4.1079. [DOI] [PubMed] [Google Scholar]
- 19.Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2005;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- 20.Javaheri S. Acetazolamide improves central sleep apnea in heart failure: a double-blind, prospective study. Am J Respir Crit Care Med. 2006;173:234–7. doi: 10.1164/rccm.200507-1035OC. [DOI] [PubMed] [Google Scholar]
- 21.Javaheri S. A mechanism of central sleep apnea in patients with heart failure. N Engl J Med. 1999;341:949–54. doi: 10.1056/NEJM199909233411304. [DOI] [PubMed] [Google Scholar]
- 22.Hall MJ, Xie A, Rutherford R et al. Cycle length of periodic breathing in patients with and without heart failure. Am J Respir Crit Care Med. 1996;154:376–81. doi: 10.1164/ajrccm.154.2.8756809. [DOI] [PubMed] [Google Scholar]
- 23.Xie A, Skatrud JB, Khayat R et al. Cerebrovascular response to carbon dioxide in patients with congestive heart failure. Am J Respir Crit Care Med. 2005;172:371–8. doi: 10.1164/rccm.200406-807OC. [DOI] [PubMed] [Google Scholar]
- 24.Spaak J, Egri ZJ, Kubo T et al. Muscle sympathetic nerve activity during wakefulness in heart failure patients with and without sleep apnea. Hypertension. 2005;46:1327–32. doi: 10.1161/01.HYP.0000193497.45200.66. [DOI] [PubMed] [Google Scholar]
- 25.Naughton MT, Benard DC, Liu PP et al. Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1995;152:473–9. doi: 10.1164/ajrccm.152.2.7633695. [DOI] [PubMed] [Google Scholar]
- 26.Brunner-La Rocca HP, Esler MD, Jennings GL, Kaye DM. Effect of cardiac sympathetic nervous activity on mode of death in congestive heart failure. Eur Heart J. 2001;22:1136–43. doi: 10.1053/euhj.2000.2407. [DOI] [PubMed] [Google Scholar]
- 27.Kaye DM, Lefkovits J, Jennings GL et al. Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol. 1995;26:1257–63. doi: 10.1016/0735-1097(95)00332-0. [DOI] [PubMed] [Google Scholar]
- 28.Cohn JN, Levine TB, Olivari MT et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–23. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 29.Triposkiadis F, Karayannis G, Giamouzis G et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. 2009;54:1747–62. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 30.Raman D, Kaffashi F, Lui LY et al. Polysomnographic heart rate variability indices and atrial ectopy associated with incident atrial fibrillation risk in older community-dwelling men. JACC Clin Electrophysiol. 2017;3:451–60. doi: 10.1016/j.jacep.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am J Respir Crit Care Med. 2002;165:934–9. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- 32.Ciftci TU, Kokturk O, Bukan N, Bilgihan A. The relationship between serum cytokine levels with obesity and obstructive sleep apnea syndrome. Cytokine. 2004;28:87–91. doi: 10.1016/j.cyto.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Schulz R, Mahmoudi S, Hattar K et al. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am J Respir Crit Care Med. 2000;162:566–70. doi: 10.1164/ajrccm.162.2.9908091. [DOI] [PubMed] [Google Scholar]
- 34.Levine B, Kalman J, Mayer L et al. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 35.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–98. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 36.Drager LF, Bortolotto LA, Figueiredo AC et al. Obstructive sleep apnea, hypertension, and their interaction on arterial stiffness and heart remodeling. Chest. 2007;131:1379–86. doi: 10.1378/chest.06-2703. [DOI] [PubMed] [Google Scholar]
- 37.Drager LF, Togeiro SM, Polotsky VY, Lorenzi-Filho G. Obstructive sleep apnea: a cardiometabolic risk in obesity and the metabolic syndrome. J Am Coll Cardiol. 2013;62:569–76. doi: 10.1016/j.jacc.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyons OD, Ryan CM. Sleep apnea and stroke. Can J Cardiol. 2015;31:918–27. doi: 10.1016/j.cjca.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 39.Gami AS, Olson EJ, Shen WK et al. Obstructive sleep apnea and the risk of sudden cardiac death. J Am Coll of Cardiol. 2013;62:610–6. doi: 10.1016/j.jacc.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Javaheri S, Blackwell T, Ancoli-Israel S et al. Sleep-disordered breathing and incident heart failure in older men. Am J Respir Crit Care Med. 2016;193:561–8. doi: 10.1164/rccm.201503-0536OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gottlieb DJ, Yenokyan G, Newman AB et al. Prospective study of obstructive sleep apnea and incident coronary heart disease and heart failure: the sleep heart health study. Circulation. 2010;122:352–60. doi: 10.1161/CIRCULATIONAHA.109.901801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ryan CM, Usui K, Floras JS, Bradley TD. Effect of continuous positive airway pressure on ventricular ectopy in heart failure patients with obstructive sleep apnoea. Thorax. 2005;60:781–5. doi: 10.1136/thx.2005.040972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McEvoy RD, Antic NA, Heeley E et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375:919–31. doi: 10.1056/NEJMoa1606599. [DOI] [PubMed] [Google Scholar]
- 44.Kaneko Y, Floras JS, Usui K et al. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med. 2003;348:1233–41. doi: 10.1056/NEJMoa022479. [DOI] [PubMed] [Google Scholar]
- 45.Mansfield DR, Gollogly NC, Kaye DM et al. Controlled trial of continuous positive airway pressure in obstructive sleep apnea and heart failure. Am J Respir Crit Care Med. 2004;169:361–6. doi: 10.1164/rccm.200306-752OC. [DOI] [PubMed] [Google Scholar]
- 46.Javaheri S, Caref EB, Chen E et al. Sleep apnea testing and outcomes in a large cohort of Medicare beneficiaries with newly diagnosed heart failure. Am J Respir Crit Care Med. 2011;183:539–46. doi: 10.1164/rccm.201003-0406OC. [DOI] [PubMed] [Google Scholar]
- 47.Strollo PJ, Soose RJ, Maurer JT et al. Upper-airway stimulation for obstructive sleep apnea. N Engl J Med. 2014;370:139–49. doi: 10.1056/NEJMoa1308659. [DOI] [PubMed] [Google Scholar]
- 48.Gillespie MB, Soose RJ, Woodson BT et al. Upper airway stimulation for obstructive sleep apnea: patient-reported outcomes after 48 months of follow-up. Otolaryngol Head Neck Surg. 2017;156:765–71. doi: 10.1177/0194599817691491. [DOI] [PubMed] [Google Scholar]
- 49.Lamba J, Simpson CS, Redfearn DP et al. Cardiac resynchronization therapy for the treatment of sleep apnoea: a meta-analysis. Europace. 2011;13:1174–9. doi: 10.1093/europace/eur128. [DOI] [PubMed] [Google Scholar]
- 50.Naughton MT, Liu PP, Bernard DC, Goldstein RS, Bradley TD. Treatment of congestive heart failure and Cheyne-Stokes respiration during sleep by continuous positive airway pressure. Am J Respir Crit Care Med. 1995;151:92–7. doi: 10.1164/ajrccm.151.1.7812579. [DOI] [PubMed] [Google Scholar]
- 51.Javaheri S. Effects of continuous positive airway pressure on sleep apnea and ventricular irritability in patients with heart failure. Circulation. 2000;101:392–7. doi: 10.1161/01.cir.101.4.392. [DOI] [PubMed] [Google Scholar]
- 52.Sin DD, Logan AG, Fitzgerald FS et al. Effects of continuous positive airway pressure on cardiovascular outcomes in heart failure patients with and without Cheyne-Stokes respiration. Circulation. 2000;102:61–6. doi: 10.1161/01.cir.102.1.61. [DOI] [PubMed] [Google Scholar]
- 53.Bradley TD, Logan AG, Kimoff RJ et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2005;353:2025–33. doi: 10.1056/NEJMoa051001. [DOI] [PubMed] [Google Scholar]
- 54.Arzt M, Floras JS, Logan AG et al. Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure. Circulation. 2007;115:3173. doi: 10.1161/CIRCULATIONAHA.106.683482. [DOI] [PubMed] [Google Scholar]
- 55.Philippe C, Stoica-Herman M, Drouot X et al. Compliance with and effectiveness of adaptive servoventilation versus continuous positive airway pressure in the treatment of Cheyne-Stokes respiration in heart failure over a six month period. Heart. 2006;92:337–42. doi: 10.1136/hrt.2005.060038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cowie MR, Woehrle H, Wegscheider K et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. N Engl J Med. 2015;373:1095–105. doi: 10.1056/NEJMoa1506459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Javaheri S, Brown LK, Randerath W, Khayat R. SERVE-HF: More questions than answers. Chest. 2016;149:900–4. doi: 10.1016/j.chest.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 58.Lyons OD, Floras JS, Logan AG et al. Design of the effect of adaptive servo-ventilation on survival and cardiovascular hospital admissions in patients with heart failure and sleep apnoea: the ADVENT-HF trial. Eur J Heart Fail. 2017;19:579–87. doi: 10.1002/ejhf.790. [DOI] [PubMed] [Google Scholar]
- 59.Costanzo MR, Ponikowski P, Javaheri S et al. Transvenous neurostimulation for central sleep apnoea: a randomised controlled trial. Lancet. 2016;388:974–82. doi: 10.1016/S0140-6736(16)30961-8. [DOI] [PubMed] [Google Scholar]
- 60.Javaheri S. Pembrey’s dream: the time has come for a long-term trial of nocturnal supplemental nasal oxygen to treat central sleep apnea in congestive heart failure. Chest. 2003;123:322–5. doi: 10.1378/chest.123.2.322. [DOI] [PubMed] [Google Scholar]
- 61.Oldenburg O, Wellmann B, Buchholz A et al. Nocturnal hypoxaemia is associated with increased mortality in stable heart failure patients. Eur Heart J. 2016;37:1695–703. doi: 10.1093/eurheartj/ehv624. [DOI] [PubMed] [Google Scholar]
- 62.Gami AS, Olson EJ, Shen WK et al. Obstructive sleep apnea and the risk of sudden cardiac death: a longitudinal study of 10,701 adults. J Am Coll Cardiol. 2013;62:610–6. doi: 10.1016/j.jacc.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehta V, Vasu TS, Phillips B, Chung F. Obstructive sleep apnea and oxygen therapy: a systematic review of the literature and meta-analysis. J Clin Sleep Med. 2013;9:271–9. doi: 10.5664/jcsm.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu K, Li Q, Yang J et al. The effect of theophylline on sleep-disordered breathing in patients with stable chronic congestive heart failure. Chin Med J. 2003;116:1711–6. [PubMed] [Google Scholar]
- 65.Walsh JT, Andrews R, Starling R et al. Effects of captopril and oxygen on sleep apnoea in patients with mild to moderate congestive cardiac failure. Br Heart J. 1995;73:237–41. doi: 10.1136/hrt.73.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dark DS, Pingleton SK, Kerby GR et al. Breathing pattern abnormalities and arterial oxygen desaturation during sleep in the congestive heart failure syndrome. Improvement following medical therapy. Chest. 1987;91:833–6. doi: 10.1378/chest.91.6.833. [DOI] [PubMed] [Google Scholar]