

Abstract
The traceless Petasis borono-Mannich reaction of enals, sulfonylhydrazines, and allylboronates catalyzed by chiral biphenols results in an asymmetric reductive transposition of the in situ generated allylic diazene. Acyclic 1,4-diene products bearing alkyl- or aryl- substituted benzylic stereocenters are afforded in excellent yields and enantiomeric ratios up to 99:1. The use of crotylboronates in the reaction results in concomitant formation of two stereocenters in a 1,4-syn or anti relationship from the corresponding E- or Z-crotylboronate used in the reaction. The use of beta mono-substituted enals in the asymmetric traceless Petasis borono-Mannich reaction of crotylboronates installs tertiary methyl-bearing stereocenters in good yields and high enantioselectivities.
Keywords: asymmetric synthesis, organocatalysis, diazene, allylation, hydrazones
Leave no trace
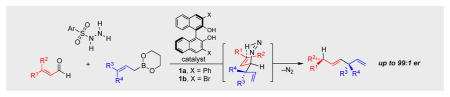
Tertiary stereogenic centers are installed by an asymmetric traceless reductive allylboration catalyzed by chiral biphenols. The reaction proceeds via transient enantioenriched allyic diazene species that break down through a sigmatropic rearrangement to generate 1,4-dienes bearing aryl- or alkyl-substituted tertiary stereocenters with high enantio- and diastereoselectivities.
The retroene[1] rearrangement of diazenes proceeds with the loss of dinitrogen via a concerted pericyclic reaction pathway.[2] The stereospecific nature of the transformation results in the efficient transfer of chirality in a 1,3-atom transfer process.[3] We reported the catalytic enantioselective traceless Petasis reaction, a reaction that uses 1,3-chirality transfer from an enantioenriched propargylic diazene to generate an axially chiral allene.[4] Herein we describe the reaction of allylic diazenes to give rise to products bearing tertiary alkyl stereocenters,[5] following the allylic diazene rearrangement. The intermediate diazene is formed in situ by fragmentation of the corresponding allylic sulfonylhydrazide, synthesized by a chiral biphenol-catalyzed Petasis borono-Mannich reaction of sulfonylhydrazides, enals, and allylboronates (Scheme 1).[6][7] Subsequent rearrangement of the enantioenriched diazene, with concomitant extrusion of molecular nitrogen, results in the allylic transposition of chirality from the diazene stereocenter to the allylic, sp2 hybridized carbon center. When a γ-substituted allylboronate (R3 or R4 = methyl) is employed, two tertiary stereocenters in a 1,4-relationship are generated in a stereoselective manner.
Scheme 1.
Asymmetric traceless Petasis borono-Mannich reactions of enals resulting in the reductive transpositions of allylic diazenes. The reaction generates 1,4-dienes bearing tertiary carbon stereocenters with high levels of enantioselectivity.
Strategies employing 1,3-chirality transfer by the allylic diazene rearrangement have been elegantly utilized in asymmetric synthesis, notably for the installation of stereocenters within cyclic systems.[8] For this approach, methods to access the requisite sulfonyl hydrazine precursor include trapping of carbonium ions[8a], displacement of terminal alcohols[8b, 8c] by sulfonylhydrazides, asymmetric hydride reduction controlled by cyclic substrates,[8d–j] and Diels–Alder cycloadditions of 1-hydrazino dienes.[8k, 8l] While these advances are significant, an enantioselective catalytic reaction that provides access to acyclic allylic diazenes has been elusive. Only two methods describing the asymmetric synthesis of tertiary stereocenters via acyclic allylic diazenes exist. First, McIntosh and co-workers developed a diastereoselective reduction of an acyclic tosylhydrazone facilitated by a proximal α-alkoxy stereocenter, resulting in a methyl-substituted tertiary stereocenter.[9] Second, Movassaghi and co-workers reported a palladium-catalyzed conversion of enantioenriched allylic epoxides to diazenes, which afforded methyl-substituted tertiary stereocenters adjacent to secondary alcohols.[10] Despite the efficient acyclic stereocontrol achieved by these methods, the formation of the enantioenriched allylic diazenes prior to the chirality transfer relied on the use of optically active starting materials containing oxygenated stereocenters. In order to access the desired chiral secondary hydrazine, we designed an approach that would directly generate acyclic allylic diazenes from achiral substrates using a BINOL-catalyzed traceless Petasis reaction.
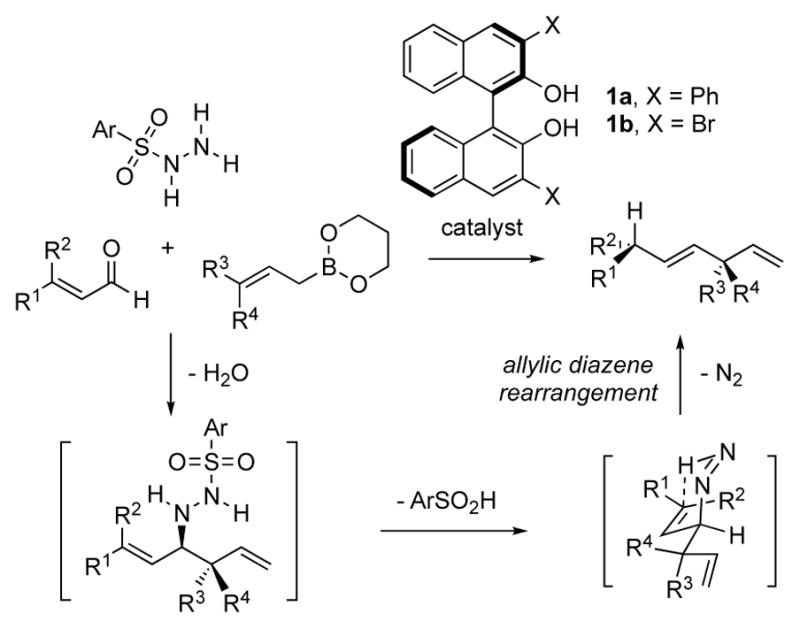
We began by investigating the asymmetric Petasis allylation with β-methyl enals using chiral biphenols catalysts. The 3,3′-Ph2-BINOL catalyst 1a proved ideal for enantiomeric control, consistent with our previous observations in the asymmetric allylation of imines (Scheme 2).[6a, 6e] In addition, the electron-deficient hydrazide 2a was identified as optimal for producing the highest yields of the desired rearranged products 5. The multicomponent reaction proceeded with initial generation of the hydrazone by condensation of enals 3 with hydrazide 2a in the presence of 3 Å molecular sieves. After removal of the solvent in vacuo, the unpurified hydrazone was dissolved in toluene and to this mixture was added t-BuOH (3 equivalents), catalyst (R)-1a (7 mol%) and allyl boronate 4 at room temperature. The reaction generated the corresponding enantioenriched 1,4-dienes 5 after stirring at room temperature for 48 hours. The reaction tolerated β-aryl-β-methyl enal substrates, furnishing allylated products bearing benzylic stereocenters in good yields and with excellent enantioselectivities (Scheme 2, 5a–5e). The observed absolute stereochemistry was consistent with the previously reported biphenol-catalyzed borono-Mannich allylation reaction (see Supporting Information for absolute stereochemical determination). [6e] 3-(Thiophen-2-yl)but-2-enal (3f) was also a suitable substrate, affording the product 5f in 73% isolated yield and an enantiomeric ratio (e.r.) of 99:1. (Z)-β-Silyl-β-methyl enal 3g yielded the corresponding chiral silane 5g in high enantiomeric purity with the use of 5 equivalents of t-BuOH in the absence of toluene. The addition of t-BuOH as an additive was found to accelerate the catalytic pathway while impeding the uncatalyzed reaction, consistent with previous observations.[11] Geranial (3h), a β-alkyl-β-methyl enal, proved to be a challenging substrate, affording product 5h with modest enantioselectivity (85:15 e.r.). The lack of enantioselectivity observed was due to facile E/Z isomerization of the corresponding hydrazone intermediate, as either hydrazone isomer would give rise to opposite enantiomers.[12]
Scheme 2.
Enantioselective reductive allylation of β-methyl enals.[a] [a] All reactions were run on 0.4 mmol scale. Yields of isolated products are given. The e.r. values were determined by HPLC analysis using chiral stationary phases. [b] Reaction was run at 1 M concentration. [c] 5 equivalents of t-BuOH were used in the absence of toluene. The (Z)-silyl enal 3g was used in the reaction. See Supplementary Information for experimental details and stereochemical proof.
We sought to expand the scope of the reaction by placing other functional groups at the β-position of the enal (Scheme 3). Under the same conditions used for β-methyl enals, however, β-ethyl enal 6a afforded the corresponding product in lower yield and with reduced enantioselectivity (41% yield, 82:18 e.r.). The nature of the sulfonylhydrazide and the biphenol catalyst proved crucial to the reactivity and selectivity of the traceless Petasis allylation. In a two-dimensional evaluation of arylsulfonyl hydrazides and catalysts (see Supplementary Information for details), 2-NO2-benzenesulfonyl hydrazide 2b and 3,3′-Br2-BINOL catalyst 1b proved to be optimal for β,β-disubstituted enals with substituents larger than a methyl group (Scheme 3). Under these conditions, the corresponding products were thereby accessed with excellent enantioselectivity (Scheme 3, 7a-7c). The allylation product 7d containing a stereogenic benzylic CF3 group was formed in good yield and with high selectivity (85% yield, 97:3 e.r.), an approach complementary to existing strategies.[13] The monofluoromethyl-substituted product 7e was also produced in good yield and with high enantiopurity. Under these reaction conditions, the olefin geometry of the enals determined the stereochemical outcome. Thus, the reaction of the Z-enal catalyzed by (R)-Br2-BINOL (1b) afforded the (S)-product 7c in 82% yield and with a 98:2 enantiomeric ratio, while the same product [i.e., (S)-7c] was obtained by using (E)-enal 8 with the enantiomeric catalyst (S)-1b, albeit in lower yield and with slightly diminished enantiomeric purity (60% yield, 94:6 e.r.).
Scheme 3.

Enantioselective reductive allylation of β,β-disubstituted enals.[a] [a] All reactions were run at 0.4 mmol scale. Yields of isolated products are given. The e.r. values were determined by HPLC analysis using chiral stationary phases. [b] (S)-Br2-BINOL (ent-1b) and enal (E)-8 were used. [c] 5 equivalents of t-BuOH was employed in the absence of toluene. See Supplementary Information for experimental details.
Tertiary diarylmethane stereocenters are a moiety that appears in natural products and pharmaceuticals.[14] While there have been recent notable advances in accessing these structures enantioselectively,[15] we envisaged that use of the traceless Petasis reaction would provide access to diarylmethane tertiary stereocenters through a complementary strategy. β,β-Diaryl enals proved to be good substrates for the optimized reductive allylation conditions. The corresponding 1,4-dienes bearing diaryl-substituted tertiary stereocenters were obtained with excellent yields and enantioselectivities, irrespective of the steric or electronic properties of the aryl substituents (Scheme 3, 7f–7k).
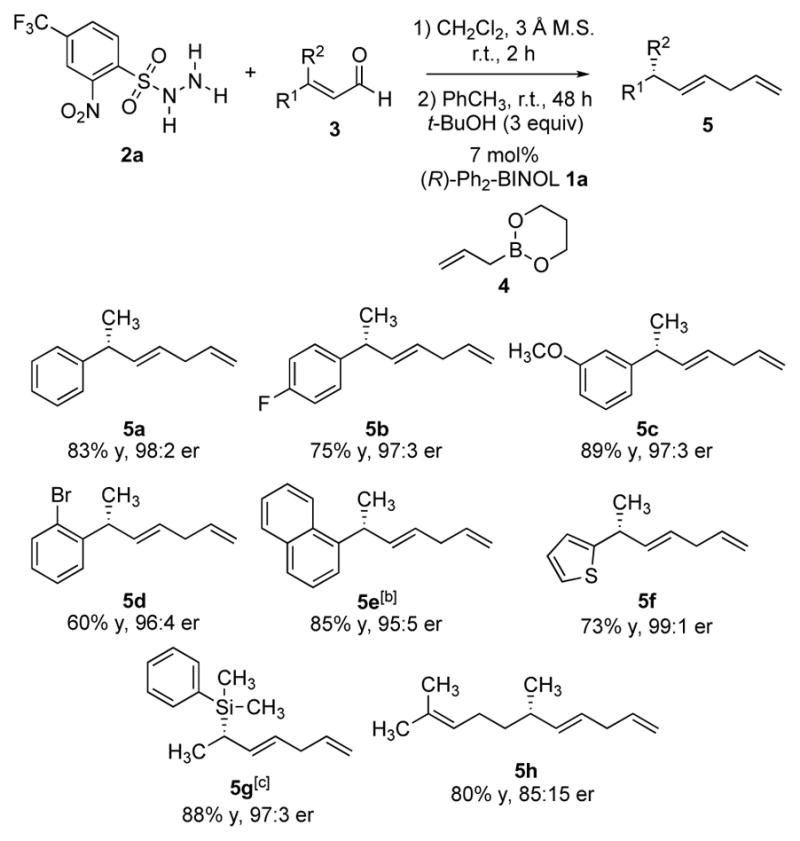
We have previously disclosed that both syn and anti α-methyl amines were accessible by an asymmetric Petasis borono-Mannich crotylation of aldehydes and amines.[6e] To investigate related reactivity in the reductive allylation, β-methyl cinnamaldehyde 3a was subjected to the crotylation conditions using either (E)-crotylboronate 9 or (Z)-crotylboronate 10 (Scheme 4). The Petasis products were postulated to provide access to both syn and anti α-methyl diazene intermediates after the elimination of sulfinic acid. As anticipated, the reaction afforded diene products bearing two methyl-substituted tertiary stereogenic centers with a 1,4-syn or anti stereochemistry (Scheme 4, 11a and 12a). β,β-Diaryl enal 6k also proved to be a viable substrate, providing access to both 1,4-disubstituted acyclic products 11b and 12b with good selectivity. The observed sense of enantio- and diastereoselectivity was consistent with the stereochemical outcome from the previously reported asymmetric Petasis crotylations.[6e] The lower diastereoselectivity for 12a and 12b resulting from (Z)-crotylboronate was presumably due to an unfavorable 1,3-diaxial interaction between the terminal methyl group from the boronate and the arylsulfonyl group on the nitrogen arising from a cyclic transition state.[16] Notably, this method achieved high levels of stereocontrol in acyclic hydrocarbons bearing 1,4-stereocenters without using any directing functional groups. The results compare favorably with existing methods to directly access tertiary alkyl stereocenters in a 1,4-relationship using asymmetric cycloadditions.[8l, 17] The observed diastereoselectivity was consistent with the previously reported biphenol-catalyzed borono-Mannich allylation reaction.[6e]
Scheme 4.
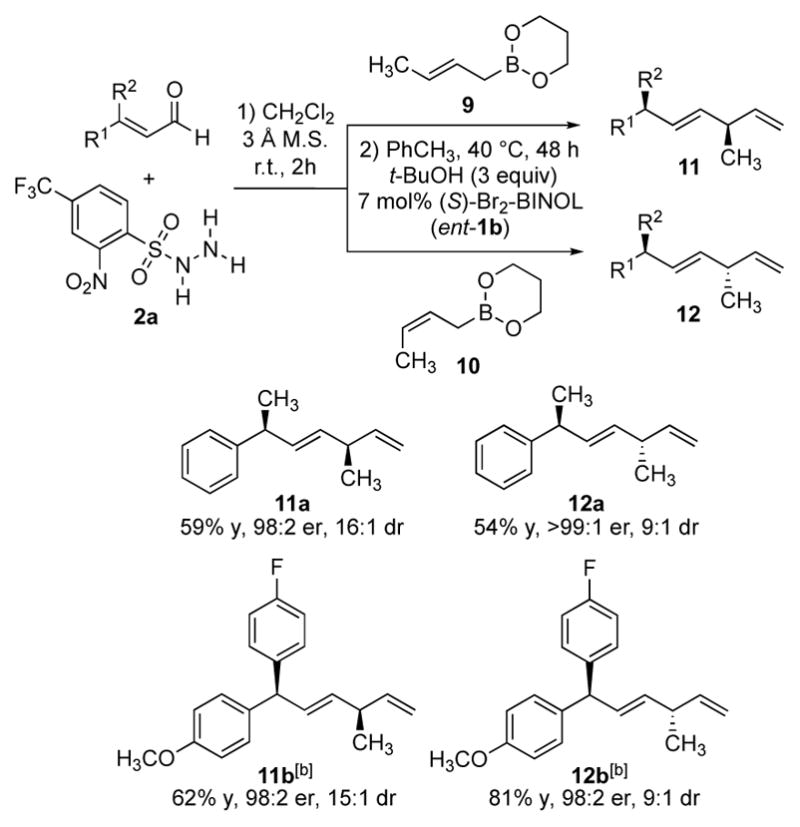
Diastereoselective reductive crotylations of β,β-disubstituted enals.[a] [a] All reactions were run on 0.4 mmol scale. Yields of isolated products are given. The values of e.r. and diastereomeric ratio (d.r.) were determined by HPLC analysis using chiral stationary phases. [b] 10 mol% catalyst was employed. See Supplementary Information for experimental details.
The methyl-substituted stereocenters generated in the crotylation reaction (Scheme 4) encouraged us to further explore the possibility of introducing branched methyl groups in 1,4-dienes. We investigated this strategy by subjecting cinnamaldehyde 13a to the crotylboration conditions using hydrazide 2a, 3,3′-Ph2-BINOL catalyst 1a, and (E)-crotylboronate 9 (Scheme 5). The desired methyl-branched hydrocarbon product 14a was isolated in 85% yield with a 97:3 enantiomeric ratio. Other cinnamaldehyde derivatives were successfully converted to the corresponding enantioenriched 1,4-dienes with excellent selectivities (Scheme 5, 14b–14e). Enantioenriched 1,4,7-triene (14g) was also obtained under similar conditions albeit in moderate yield (54% yield, 98:2 e.r.). Similarly, β-alkyl enal 13h displayed attenuated reactivity, affording the diene product 14h in modest yield but with excellent enantioselectivity when using 1b as the catalyst (54% yield, 99:1 e.r.). Given the operational ease of the reaction and the generally high level of selectivities, we anticipate that this methodology will find useful applications in the total synthesis of the natural products bearing the methyl-substituted stereocenters.[18]
Scheme 5.
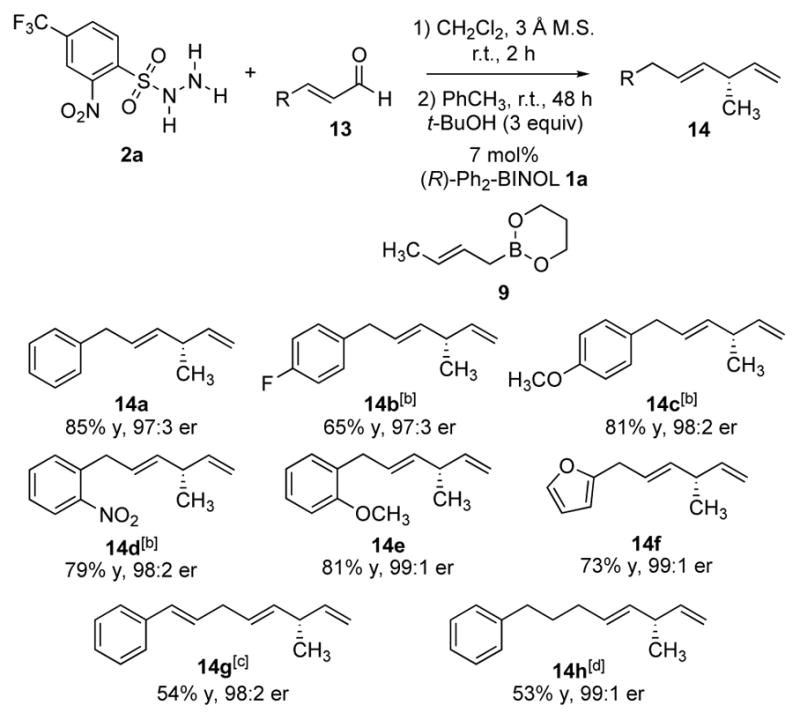
Enantioselective reductive crotylation of non-branched enals.[a] [a] All reactions were run on 0.4 mmol scale. Yields of isolated products are given. The e.r. values were determined by HPLC analysis using chiral stationary phases. [b] Reactions were run at 50 °C for 24 h. [c] 14 mol% of catalyst was employed. [d] 14 mol % of (R)-1b was employed. See Supplementary Information for experimental details and stereochemical proofs.
In summary, we have developed an asymmetric reductive allylation of aldehydes that affords tertiary alkyl stereocenters using chiral biphenol catalysts. The reaction proceeds via the sigmatropic rearrangement of a transient enantioenriched allylic diazene intermediate; a process mechanistically distinct from the reductive coupling of boronic acids with tosylhydrazones via diazo intermediates.[19] Both allylation and crotylation reactions provide access to 1,4-dienes bearing tertiary alkyl-substituted stereocenters in excellent yields and high enantioselectivities. Key aspects of the methodology include the exclusive generation of (E)-alkenes by allylic transposition, and the traceless installation of two stereocenters within acyclic systems. Further exploration of the observed reactivity within the context of asymmetric catalysis is underway and will be reported in due course.
Supplementary Material
Acknowledgments
The research was supported by the National Institutes of Health (R01 GM078240 to SES) and the National Science Foundation (CHE1361173 to RJT).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Hoffmann HMR. Angew Chem Int Ed. 1969;8:556–577. [Google Scholar]; Hoffmann HMR. Angew Chem. 1969;81:597–618. [Google Scholar]; b) Oppolzer W, Snieckus V. Angew Chem Int Ed. 1978;17:476–486. [Google Scholar]; Oppolzer W, Snieckus V. Angew Chem. 1978;90:506–516. [Google Scholar]; c) Boyd GV. In: Double-Bonded Functional Groups (1989) Patai S, editor. John Wiley & Sons, Inc; Chichester, UK: 2010. pp. 477–525. [Google Scholar]
- 2.Ripoll J-L, Vallée Y. Synthesis. 1993;1993:659–677. [Google Scholar]
- 3.Jabbari A, Sorensen EJ, Houk KN. Org Lett. 2006;8:3105–3107. doi: 10.1021/ol0612049. [DOI] [PubMed] [Google Scholar]
- 4.a) Jiang Y, Diagne AB, Thomson RJ, Schaus SE. J Am Chem Soc. 2017;139:1998–2005. doi: 10.1021/jacs.6b11937. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mundal DA, Lutz KE, Thomson RJ. J Am Chem Soc. 2012;134:5782–5785. doi: 10.1021/ja301489n. [DOI] [PubMed] [Google Scholar]; c) Diagne AB, Li S, Perkowski GA, Mrksich M, Thomson RJ. ACS Comb Sci. 2015;17:658–662. doi: 10.1021/acscombsci.5b00131. [DOI] [PubMed] [Google Scholar]
- 5.Kleemann A, Engel J. Pharmaceutical Substances: Syntheses, Patents, Applications. 4. Thieme; Stuttgart: 2001. [Google Scholar]
- 6.a) Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398–15404. doi: 10.1021/ja075204v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lou S, Schaus SE. J Am Chem Soc. 2008;130:6922–6923. doi: 10.1021/ja8018934. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bishop JA, Lou S, Schaus SE. Angew Chem Int Ed. 2009;48:4337–4340. doi: 10.1002/anie.200901023. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bishop JA, Lou S, Schaus SE. Angew Chem. 2009;121:4401–4404. doi: 10.1002/anie.200901023. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Muncipinto G, Moquist PN, Schreiber SL, Schaus SE. Angew Chem Int Ed. 2011;50:8172–8175. doi: 10.1002/anie.201103271. [DOI] [PMC free article] [PubMed] [Google Scholar]; Muncipinto G, Moquist PN, Schreiber SL, Schaus SE. Angew Chem. 2011;123:8322–8325. doi: 10.1002/anie.201103271. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Jiang Y, Schaus SE. Angew Chem Int Ed. 2017;56:1544–1548. doi: 10.1002/anie.201611332. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang Y, Schaus SE. Angew Chem. 2017;129:1566–1570. [Google Scholar]
- 7.a) Han WY, Wu ZJ, Zhang XM, Yuan WC. Org Lett. 2012;14:976–979. doi: 10.1021/ol203109a. [DOI] [PubMed] [Google Scholar]; b) Shi X, Kiesman WF, Levina A, Xin Z. J Org Chem. 2013;78:9415–9423. doi: 10.1021/jo4016425. [DOI] [PubMed] [Google Scholar]; c) Alam R, Diner C, Jonker S, Eriksson L, Szabó KJ. Angew Chem Int Ed. 2016;55:14417–14421. doi: 10.1002/anie.201608605. [DOI] [PMC free article] [PubMed] [Google Scholar]; Alam R, Diner C, Jonker S, Eriksson L, Szabó KJ. Angew Chem. 2016;128:14629–14633. doi: 10.1002/anie.201608605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Wood JL, Porco JA, Taunton J, Lee AY, Clardy J, Schreiber SL. J Am Chem Soc. 1992;114:5898–5900. [Google Scholar]; b) Myers AG, Zheng B. Tetrahedron Lett. 1996;37:4841–4844. [Google Scholar]; c) Siegel DS, Piizzi G, Piersanti G, Movassaghi M. J Org Chem. 2009;74:9292–9304. doi: 10.1021/jo901926z. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Girotra NN, Wendler NL. Tetrahedron Lett. 1982;23:5501–5504. [Google Scholar]; e) Corey EJ, Virgil SC. J Am Chem Soc. 1990;112:6429–6431. [Google Scholar]; f) Chu M, Coates RM. J Org Chem. 1992;57:4590–4597. [Google Scholar]; g) Tanaka T, Maeda K, Mikamiyama H, Funakoshi Y, Uenaka K, Iwata C. Tetrahedron. 1996;52:4257–4268. [Google Scholar]; h) Chai Y, Vicic DA, McIntosh MC. Org Lett. 2003;5:1039–1042. doi: 10.1021/ol034052f. [DOI] [PubMed] [Google Scholar]; i) Hutchison JM, Lindsay HA, Dormi SS, Jones GD, Vicic DA, McIntosh MC. Org Lett. 2006;8:3663–3665. doi: 10.1021/ol061072j. [DOI] [PubMed] [Google Scholar]; j) Anada M, Tanaka M, Shimada N, Nambu H, Yamawaki M, Hashimoto S. Tetrahedron. 2009;65:3069–3077. [Google Scholar]; k) Sammis GM, Flamme EM, Xie H, Ho DM, Sorensen EJ. J Am Chem Soc. 2005;127:8612–8613. doi: 10.1021/ja052383c. [DOI] [PubMed] [Google Scholar]; l) Xie H, Sammis GM, Flamme EM, Kraml CM, Sorensen EJ. Chem Eur J. 2011;17:11131–11134. doi: 10.1002/chem.201102394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Qi W, McIntosh MC. Org Lett. 2008;10:357–359. doi: 10.1021/ol702921x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shrestha ML, Qi W, McIntosh MC. J Org Chem. 2017;82:8359–8370. doi: 10.1021/acs.joc.7b00428. [DOI] [PubMed] [Google Scholar]
- 10.Movassaghi M, Ahmad OK. Angew Chem Int Ed. 2008;47:8909–8912. doi: 10.1002/anie.200802921. [DOI] [PubMed] [Google Scholar]; Movassaghi M, Ahmad OK. Angew Chem. 2008;120:9041–9044. [Google Scholar]
- 11.Barnett DS, Moquist PN, Schaus SE. Angew Chem Int Ed. 2009;48:8679–8682. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]; Barnett DS, Moquist PN, Schaus SE. Angew Chem. 2009;121:8835–8838. doi: 10.1002/anie.200904715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Appella DH, Moritani Y, Shintani R, Ferreira EM, Buchwald SL. J Am Chem Soc. 1999;121:9473–9474. [Google Scholar]; b) Tolstoy P, Engman M, Paptchikhine A, Bergquist J, Church TL, Leung AWM, Andersson PG. J Am Chem Soc. 2009;131:8855–8860. doi: 10.1021/ja9013375. [DOI] [PubMed] [Google Scholar]; c) Ouellet SG, Tuttle JB, MacMillan DWC. J Am Chem Soc. 2005;127:32–33. doi: 10.1021/ja043834g. [DOI] [PubMed] [Google Scholar]; d) Yang JW, Hechavarria Fonseca MT, Vignola N, List B. Angew Chem Int Ed. 2005;44:108–110. doi: 10.1002/anie.200462432. [DOI] [PubMed] [Google Scholar]; Yang JW, Hechavarria Fonseca MT, Vignola N, List B. Angew Chem. 2005;117:110–112. doi: 10.1002/anie.200462432. [DOI] [PubMed] [Google Scholar]
- 13.a) Dong K, Li Y, Wang Z, Ding K. Angew Chem Int Ed. 2013;52:14191–14195. doi: 10.1002/anie.201307903. [DOI] [PubMed] [Google Scholar]; Dong K, Li Y, Wang Z, Ding K. Angew Chem. 2013;125:14441–14445. [Google Scholar]; b) Nishimine T, Fukushi K, Shibata N, Taira H, Tokunaga E, Yamano A, Shiro M, Shibata N. Angew Chem Int Ed. 2014;53:517–520. doi: 10.1002/anie.201308071. [DOI] [PubMed] [Google Scholar]; Nishimine T, Fukushi K, Shibata N, Taira H, Tokunaga E, Yamano A, Shiro M, Shibata N. Angew Chem. 2014;126:527–530. doi: 10.1002/anie.201308071. [DOI] [PubMed] [Google Scholar]; c) Liang Y, Fu GC. J Am Chem Soc. 2015;137:9523–9526. doi: 10.1021/jacs.5b04725. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Massolo E, Benaglia M, Orlandi M, Rossi S, Celentano G. Chem Eur J. 2015;21:3589–3595. doi: 10.1002/chem.201405730. [DOI] [PubMed] [Google Scholar]
- 14.Ameen D, Snape TJ. Med Chem Commun. 2013;4:893–907. [Google Scholar]
- 15.a) Cherney AH, Kadunce NT, Reisman SE. Chem Rev. 2015;115:9587–9652. doi: 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tollefson EJ, Hanna LE, Jarvo ER. Acc Chem Res. 2015;48:2344–2353. doi: 10.1021/acs.accounts.5b00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774–7854. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]; b) Diner C, Szabó KJ. J Am Chem Soc. 2017;139:2–14. doi: 10.1021/jacs.6b10017. [DOI] [PubMed] [Google Scholar]
- 17.a) Wender PA, Dyckman AJ. Org Lett. 1999;1:2089–2092. [Google Scholar]; b) Peng F, Dai M, Angeles AR, Danishefsky SJ. Chem Sci. 2012;3:3076–3080. doi: 10.1039/C2SC20868G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Goren MB, Brokl O, Das BC, Lederer E. Biochemistry. 1971;10:72–81. doi: 10.1021/bi00777a012. [DOI] [PubMed] [Google Scholar]; b) Balieu S, Hallett GE, Burns M, Bootwicha T, Studley J, Aggarwal VK. J Am Chem Soc. 2015;137:4398–4403. doi: 10.1021/ja512875g. [DOI] [PubMed] [Google Scholar]; c) Amat M, Llor N, Guignard G, Bosch J. Synthesis. 2016;48:2705–2720. [Google Scholar]; d) Son S, Fu GC. J Am Chem Soc. 2008;130:2756–2757. doi: 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]; e) Davidson BS. J Org Chem. 1991;56:6722–6724. [Google Scholar]; f) Shi Y, Jung B, Torker S, Hoveyda AH. J Am Chem Soc. 2015;137:8948–8964. doi: 10.1021/jacs.5b05805. [DOI] [PubMed] [Google Scholar]; g) Gouiffes D, Juge M, Grimaud N, Welin L, Sauviat MP, Barbin Y, Laurent D, Roussakis C, Henichart JP, Verbist JF. Toxicon. 1988;26:1129–1136. doi: 10.1016/0041-0101(88)90297-8. [DOI] [PubMed] [Google Scholar]; h) Claraz A, Sahoo G, Berta D, Madarász Á, Pápai I, Pihko PM. Angew Chem Int Ed. 2016;55:669–673. doi: 10.1002/anie.201509302. [DOI] [PubMed] [Google Scholar]; i) Mori K. Bioorg Med Chem. 2007;15:7505–7523. doi: 10.1016/j.bmc.2007.08.040. [DOI] [PubMed] [Google Scholar]; j) Ando T, Yamakawa R. Nat Prod Rep. 2015;32:1007–1041. doi: 10.1039/c4np00138a. [DOI] [PubMed] [Google Scholar]; k) Bello JE, McElfresh JS, Millar JG. Proc Natl Acad Sci U S A. 2015;112:1077–1082. doi: 10.1073/pnas.1417605112. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Motais dNM, d’Ettorre P, van ZJS, Wenseleers T, Bello JE, Millar JG. J Exp Biol. 2016;219:1632–1638. doi: 10.1242/jeb.136069. [DOI] [PubMed] [Google Scholar]
- 19.a) Barluenga J, Tomás-Gamasa M, Aznar F, Valdés C. Nat Chem. 2009;1:494–499. doi: 10.1038/nchem.328. [DOI] [PubMed] [Google Scholar]; b) Perez-Aguilar MC, Valdes C. Angew Chem Int Ed. 2012;51:5953–5957. doi: 10.1002/anie.201200683. [DOI] [PubMed] [Google Scholar]; Pérez-Aguilar MC, Valdés C. Angew Chem. 2012;124:6055–6059. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.