

Abstract
Objective
The first aim was to demonstrate a previously hypothesized increased sensitivity of corticostriatal glutamatergic terminals in the rodent with brain iron deficiency (BID), a pathogenetic model of Restless Legs Syndrome (RLS). The second aim was to determine if these putative hypersensitive terminals could constitute a significant target for drugs effective in RLS, including dopamine agonists (pramipexole and ropinirole) and α2δ ligands (gabapentin).
Methods
A recently introduced in vivo optogenetic-microdialysis approach was used, which allows the measurement of the extracellular concentration of glutamate upon local light-induced stimulation of corticostriatal glutamatergic terminals. The method also allows to analyze the effect of local perfusion of compounds within the same area being sampled for glutamate.
Results
BID rats showed hypersensitivity of corticostriatal glutamatergic terminals (lower frequency of optogenetic stimulation to induce glutamate release). Both hypersensitive and control glutamatergic terminals were significant targets for locally perfused pramipexole, ropinirole and gabapentin, which significantly counteracted optogenetically-induced glutamate release. The use of selective antagonists demonstrated the involvement of dopamine D4 and D2 receptor subtypes on the effects of pramipexole.
Interpretation
Hypersensitivity of corticostriatal glutamatergic terminals can constitute a main pathogenetic mechanism of RLS symptoms. Selective D4 receptor agonists, by specifically targeting these terminals, should provide a new efficient treatment with less secondary effects.
INTRODUCTION
Restless Legs Syndrome (RLS) is a common neurologic disorder1 characterized by a rest-induced, movement-responsive, mostly nocturnal, urge to move the legs commonly associated with periodic leg movements during sleep (PLMS) and hyperarousal.2–4 Altered dopamine function plays an important role in PLMS symptomatology, which is supported by the remarkable therapeutic response to L-dopa and dopamine receptor agonists (such as pramipexole and ropinirole), and the repeated demonstration of biochemical changes related to the dopamine system.5 On the other hand, glutamate mechanisms seem to be involved in both PLMS and in the hyperarousal component of RLS. This is supported by the efficacy of ligands of the α2δ subunits of the calcium channels (such as gabapentin) for both types of disturbances.6–8 Thus, α2δ-containing calcium channels are preferentially localized in neuronal glutamate terminals.9
The clinical success of dopamine agonists and α2δ ligands is utterly empirical and there is no consensus about the identity (receptor subtypes) and localization (neuronal elements, circuits) of their main therapeutic targets. Both supra-spinal and spinal mechanisms have been invoked to be involved in the pathophysiology of RLS.10 Supra-spinal mechanisms favor a predominant subcortical, striatal, impairment of sensorimotor integration.11,12 The striatum is the brain area with the highest dopamine innervation and highest density of dopamine receptors and the main point of interaction of dopamine within the cortical-striatal-thalamic-cortical circuits. Furthermore, the main two extrinsic striatal inputs are dopaminergic mesencephalic inputs and glutamatergic cortical, limbic and thalamic inputs.13
Brain iron deficiency (BID) is now well recognized as a main initial pathogenetic mechanism in the development of RLS. This is based on extensive research studies using cerebrospinal fluid, autopsy material, and brain imaging indicating reduced regional brain iron content,5,14 and is further supported by the efficacy of iron therapy for RLS6,8,15,16, including RLS refractory to other treatments.17 Animal models have allowed establishing a causal relation between BID and altered dopamine function in RLS. BID during the postweaning period in rats or mice produces changes in the dopamine system that parallel those found in RLS, therefore representing a valuable pathophysiological model of RLS (see Discussion).18
We have recently shown that the brain iron-deficient rodent is associated with specific alterations in adenosine neurotransmission that can provide a pathogenetic link between BID and the glutamate mechanisms involved in the PLMS and hyperarousal of RLS.19,20 Those include changes in the density of striatal adenosine receptors which modulate corticostriatal glutamate release, which would be expected to increase the sensitivity of corticostriatal glutamatergic neurotransmission. We therefore postulated that an increased sensitivity of corticostriatal glutamatergic neurotransmission can be a pathogenetic factor in the development of PLMS in RLS.8,19
Using a recently introduced in vivo optogenetic-microdialysis approach,21,22 we can now demonstrate the existence of hypersensitivity of corticostriatal glutamatergic terminals in rodents with BID. Significantly, hypersensitive and control glutamatergic terminals were targeted by pramipexole, ropinirole and gabapentin. Using specific dopamine receptor antagonists, we demonstrate the involvement of D2 and D4 receptors (D2R and D4R) on the effect of pramipexole, specially indicating that D4R-selective agonists may provide a better treatment for RLS.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley albino rats (Charles River Laboratories, Wilmington, MA), weighing 80–100 grams and 250–350 g at the time of the first and second surgeries, respectively, were used in the experiments. Animals were housed 2 per cage and kept on a 12/12-h dark/light cycle with food and water available ad libitum. All animals used in the study were maintained in accordance with the guidelines of the National Institutes of Health Animal Care and the animal research conducted to perform this study was approved by the NIDA IRP Animal Care and Use Committee (protocol #: 15-BNRB-73). The rats were divided into two groups: the control group was fed with a diet containing an essential amount of iron (48 ppm iron, Catalog TD.80394, Harlan-Teklad, Madison, WI) and the BID group was fed with a diet containing a low iron concentration (4 ppm iron, Catalog TD.80396, Harlan-Teklad). The other contents of the diet were the same. Diets were started immediately after weaning and continued for 21 days, when microdialysis experiments were performed.
Assessment of systemic and brain iron deficiency
Systemic iron deficiency was assessed by measuring the hematocrit content from blood collected before perfusion at the end of the microdialysis experiments. Blood was collected in heparin-coated capillary glass tubes and hematocrit content was measured after centrifugation. As in previous studies, BID was assessed by analyzing the density of transferrin receptor (TfR) in the brain (lateral striatum) by Western blot, as described in detail elsewhere.19 It is well established that an increase in TfR density in the brain constitutes a reliable indicator of chronic exposure to low levels of iron in the brain.23,24 Since tissue processing used for viral vector expression assessment prevents the use of the Western blot technique, the efficacy of the low-iron diet to produce TfR upregulation in animals included in the optogenetic-microdialysis experiments was confirmed in parallel control groups of animals fed with the two different diets. Demonstration of the same decrease in hematocrit content was then used to demonstrate the same efficacy of the low-iron diet in this experimental group.
Surgical procedures
Four weeks before the microdialysis experiments, the animals, weighing between 80 to 100 grams, were anesthetized with 3 ml/kg of Equithesin (4.44 g of chloral hydrate, 0.972 g of Na pentobarbital, 2.124 g of MgSO4, 44.4 ml of propylene glycol, 12 ml of ethanol and distilled H2O up to 100 ml of final solution; NIDA Pharmacy, Baltimore, MD). A unilateral injection of an adeno-associated virus (AAV) encoding Channelrhodopsin 2 (ChR2) fused to enhanced yellow fluorescence protein (EYFP) under control of the CaMKII neuronal promoter (AAV-CaMKIIa-hChR2(H134R)-EYFP; University of North Carolina core vector facility) was delivered in the agranular motor cortex; coordinates of injection were 2.5 mm anterior, 3 mm lateral and -5 mm ventral with respect to bregma). Virus (0.5 μl of purified and concentrated AAV; 1 × 1012 infectious units/ml) was injected using a 105 μm-thick silica tubing injector coupled directly to a 1 μl-syringe driven by an infusion pump. Virus suspension was injected over a 10-min period at a rate of 50 nl/min and the injector was left in place for an additional ten minutes to allow diffusion of the suspension. Four weeks after virus injection, with the rats weighing 250 to 325 grams, a modified microdialysis probe (optogenetic-microdialysis probe) with an embedded light-guiding optic fiber (see below and ref. 21) was implanted into the lateral striatum under Equithesin anesthesia (3 ml/kg); coordinates were 0 mm anterior, 4.5 lateral and −7 mm ventral with respect to bregma. The probes were fixed to the skull with stainless steel screws and dental glassionomer cement.
Intracranial optogenetic stimulation
An optogenetic-microdialysis probe with a 125 μm-diameter optic fiber (0.22 numerical aperture) embedded in a microdialysis probe was used for optogenetic stimulation of glutamate release by cortico-striatal terminals21. The tip of the optic fiber was given a conical shape to allow a local light dispersion through and around the working portion of the dialyzable membrane. The conical sculpted tip with the cladding fused to the core was obtained by pulling the fiber with a Flamming-Brown pipette puller (Sutter Instruments, Novato, CA), fitted with a custom platinum heating filament of circular cross-section (1 mm in diameter) and a holder designed for the small diameter of the optic fiber. Optical stimulation was delivered coupling the light guiding port of the implanted optogenetic-microdialysis probe to a 473-nm solid-state laser module driven by the electrical stimulator (Grass S88 stimulator). Light was applied over a 20-min period in 160-ms trains of 1-ms pulses at 100 Hz at 5–8 mW at the probe tip (one train/s). Light intensity at the probe tip was measured before implantation using an integrating sphere silicon photodiode power sensor designed for optical power measurements independent of beam shape and divergence (model S144C, Thor Labs, Newton, NJ).
In vivo microdialysis
Microdialysis sampling was performed during optogenetic stimulation to analyze the extracellular concentrations of striatal glutamate of freely moving rats 24 h after probe implantation. An artificial cerebrospinal solution (aCSF; 144 NaCl, 4.8 KCl, 1.7 CaCl2, and 1.2 MgCl2, in mM) with or without the dopamine receptor agonists pramipexole or ropinirole (1 μM), the α2δ ligand gabapentin (1 μM) and the selective D4R antagonist L745–870 (10 μM), the non-selective D2R-D3R antagonist raclopride (10 μM) or the selective D3R antagonist VK4–116 (10 μM), was pumped through the optogenetic-micordialysis probe at a constant rate of 1 μl/min. After a washout period of 90 min, dialysate samples were collected for 60 min of baseline sampling at 20-min intervals. After baseline sampling, optogenetic stimulation was applied for 20 min and 20-min samples were taken for 80 additional min after the beginning of the stimulation. Glutamate content was measured by HPLC coupled to a glutamate oxidase enzyme reactor and electrochemical detector (Eicom, San Diego, CA). Estimation of diffusion of the compounds through the optogenetic-microdialysis probe was assessed by in vitro recovery. Probes were placed in an Eppendorf tube containing 1 ml of a 10-μM concentration of either L745–870, raclopride or N-(4-(4-(3-Chloro-5-ethyl-2-methoxyphenyl)piperazin-1-yl)-3-hydroxybutyl)-1H-indole-2-carboxamide (VK4–116; compound 19 in ref. 32) dissolved in aCSF. A syringe drive set at 1 μl/min was used to pump aCSF through the probes and three different probes were used for each different compound (with three replicate samples). Samples were analyzed by HPLC reverse phase separation and UV detection on a Thermo Ultimate 3000 HPLC equipped with a diode array detector (Bio-Techne, Minneapolis, MI). At the end of the microdialysis experiment, animals were deeply anesthetized with Equithesin and perfused transcardially with 0.1 M phosphate buffered saline (PBS), followed by 4% formaldehyde in 0.1 M PBS, pH 7.4. Brains were kept in the same fixative for 2 h and then stored in 20% sucrose/0.1 M sodium phosphate, pH 7.4, solution for 48 h at 4°C. Forty-μm coronal sections were cut in a Leica (Nussloch, Germany) CM3050S cryostat at −20°C, collected in PBS, and stored in antifreeze-buffered solution (20% ethylene glycol, 10% glycerol, and 10% sucrose in PBS) at −80°C until processing. Sections were then evaluated for probe localization and ChR2-EYFP expression using a Typhoon multimode laser scanner (GE life Sciences, Piscataway, NJ). Confocal fluorescence microscopy images were acquired with a Zeiss microscope (Examiner Z1, Zeiss, Gottingen, Germany) fitted with a confocal laser module (LSM-710, Zeiss).
RESULTS
Effect of BID on optogenetically-induced corticostriatal glutamate release
In the parallel groups for control of low-iron diet efficiency, the hematocrit content (expressed as percentage of total volume) from animals with and without low-iron diet was, in mean ± S.E.M, 18.1 ± 1.0% and 47.7 ± 0.5% (n = 20/group), respectively. In the low-iron diet group, striatal TfR density showed a significant increase of 23.7 ± 2.9% (p < 0.001; unpaired t test). In the two groups included in the optogenetic-microdialysis experiments, the hematocrit content from animals with and without low-iron diet was, in mean ± S.E.M, 17.8 ± 0.8% (n = 30), 47.8 ± 0.3% (n = 22), respectively
In our previous studies with the in vivo optogenetic-microdialysis approach, we studied the ability of corticostriatal terminals in the ventromedial striatum to release glutamate upon optogenetic stimulation.21,22 In the present study, we aimed at a more motor-involved striatal area, the dorsal striatal area that receives innervation from the agranular motor cortex (Fig 1A). This corticostriatal projection has been anatomically well defined from different studies analyzing striatal neuronal activation upon cortical-electrical stimulation.20,25,26 A significant glutamate release could be obtained in both iron-deprived animals and controls when using a frequency of stimulation of 100 Hz (Fig 1D), found to be optimal in previous studies of cortical-electrical and striatal optogenetic stimulation.20,21,25. Importantly, when decreasing the frequency of stimulation to 60 Hz, a significant glutamate release could only be observed in the rats with iron deficiency (Fig 1E). These results therefore confirmed our hypothesis of a higher sensitivity of corticostriatal terminals to depolarization-induced glutamate release in the rodent brain with iron deficiency.
Figure 1. Effect of BID on optogenetically-induced corticostriatal glutamate release.
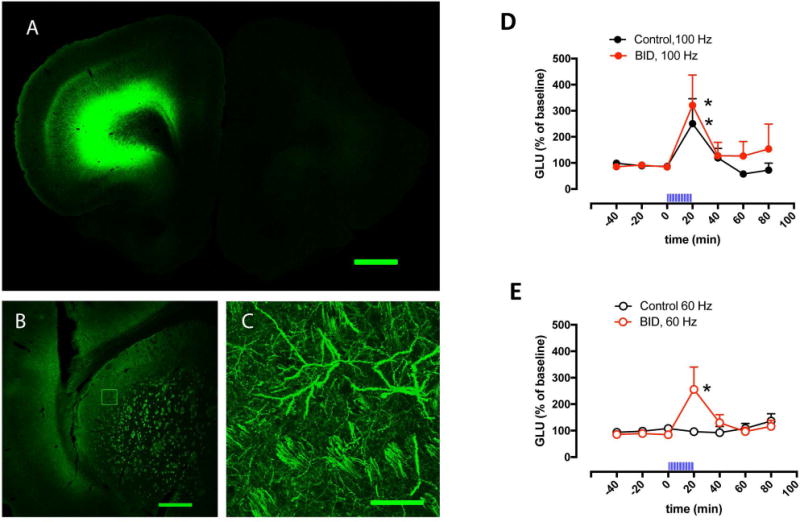
(A–C) Confocal laser microscopy of coronal brain sections showing the localization of ChR2-EYFP after unilateral AA microinjection in the agranular motor cortex. (A) Unilateral expression of ChR2-EYFP in the agranular motor cortex; coronal section at 3.0 mm anterior from bregma; scale bar, 1 mm. (B) Expression of ChR2-EYFP in the ipsilateral lateral striatum; coronal section at 0 mm anterior from bregma; scale bar, 0.5 mm. (C) Superposition of 5 adjacent confocal planes (5 μm-think planes of a 25 μm-think section) from the framed field in (B), showing corticostriatal terminals; scale bar, 0.05 mm. (D,E) Effect of local optogenetic stimulation at high-frequency (100 Hz, D) and low-frequency (60 Hz, E) on the extracellular levels of glutamate in the lateral striatum of BID rats (red plot) and controls (black plot); time ‘0’ represents the values of samples prior to stimulation; the period of stimulation (20 min) is represented as a train of vertical lines; results are expressed as means + S.E.M. of percentage of the average of three values before stimulation (n = 9–12 per group). *: p<0.05, as compared to value of the last sample before the stimulation, respectively (paired t test).
Inhibition of optogenetically-induced corticostriatal glutamate release by the adenosine A2A receptor antagonist MSX-3, the dopamine receptor agonists pramipexole and ropinirole and the α2δ ligand gabapentin
We have recently shown that the optogenetic-microdialysis method allows the study of the functional role of a GPCR localized in striatal glutamatergic terminals in the modulation of glutamate release.21,22 It could then be demonstrated that adenosine A2A receptors (A2AR) play a strong facilitatory role, since perfusion through the microdialysis probe of the selective A2AR antagonist MSX-3, completely counteracted optogenetically-induced corticostriatal glutamate release.21 In agreement with a functional upregulation of A2AR being involved with the increased sensitivity of corticostriatal terminals induced by BID (see Discussion and refs. 8 and 19), perfusion of MSX-3 (1 μM) counteracted glutamate release induced by optogenetic stimulation both in controls (at 100 Hz) and in iron-deprived animals (at 60 Hz) (Fig 2A). We could then confirm that perfusion of clinically efficient RLS drugs, either the dopamine agonists pramipexole or ropinirole (1 μM in both cases) or the α2δ ligand gabapentin (1 μM), blocked glutamate release induced by optogenetic stimulation, both in controls (at 100 Hz) and in animals with BID (at 60 Hz) (Figs 2B–2D).
Figure 2. Inhibition of optogenetically-induced corticostriatal glutamate release by MSX-3, pramipexole, ropinirole and gabapentin.
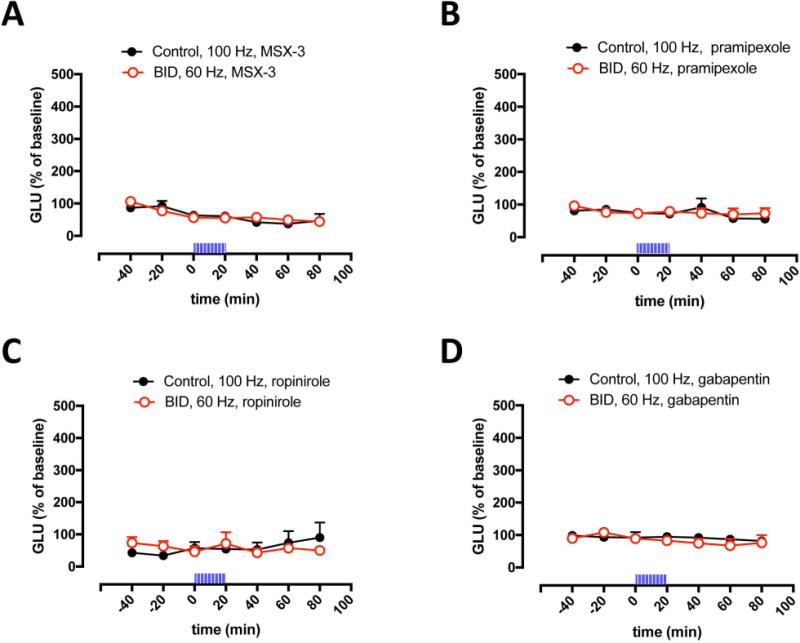
(A–D) Effect of perfusion of the adenosine A2AR antagonist MSX-3, the dopamine receptor agonists pramipexole and ropinirole and the α2δ ligand gabapentin (1 μM in all cases) on optogenetically-induced glutamate release in the lateral striatum of BID rats (red plot) and controls (black plot); time ‘0’ represents the values of samples prior to stimulation; the period of stimulation (20 min) is represented as a train of vertical lines; results are expressed as means + S.E.M. of percentage of the average of three values before stimulation (n = 6–9 per group). *: p<0.05, as compared to value of the last sample before the stimulation, respectively (paired t test).
Effect of D2-like receptor antagonists on pramipexole-mediated inhibition of optogenetically-induced corticostriatal glutamate release
We then questioned the identity of the dopamine receptor subtypes involved in the pharmacological effect of the dopamine receptor agonists. We have recently reported results obtained with the optogenetic-microdialysis technique in knock-in mice expressing the long intracellular domain of D4.7, the product of a polymorphic variant of the D4R gene (DRD4) associated with attention deficit hyperactivity disorder (ADHD).22 When compared with the wild-type mouse D4R, the expanded intracellular domain of the humanized D4R conferred a gain of function, blunting optogenetically-induced corticostriatal glutamate release.22 These results confirmed a key role of striatal D4R localized in glutamatergic terminals in the control of corticostriatal transmission. In addition, previous studies have also shown evidence for the presence of D2R, particularly its short isoform (D2SR), in striatal glutamatergic terminals and their involvement in the control of glutamate release,27 possibly by establishing molecular and functional interactions with D4R.28 Three D2-like receptor antagonists were then chosen to disclose the possible contribution of D4R and D2S in the effect of pramipexole: L745–870, with selective high affinity for D4R; 29–31 raclopride, with high affinity for D2R and D3R but low affinity for D4R; 30–31 and VK4–116, a recently introduced compound with selective high affinity for D3R.32 The in vitro recovery of these compounds through the optogenetic-microdialysis probe at the same flow rate used in the in vivo experiments (1 μl/min), as analyzed by HPLC (see Materials and Methods), was 12.0 ± 0.7%, 8.2 ± 0.2% and 20.6 ± 0.6% for L745–870, raclopride and VK4–116, respectively. These figures provide an approximation to the effective extracellular concentration of drugs administered through the microdialysis probe (reverse dialysis). Thus, in vivo recovery should be lower than the in vitro recovery because of reduced fluid volume, increased tortuosity and several other factors.33 The concentration of the D2-like receptor antagonists in the perfusion media (10 μM) should therefore yield extracellular concentrations around the probe of around 1 μM or lower, which would be insufficient for L745–870 to bind significantly to D2R or D3R, for raclopride to bind to D4R and for VK4–116 to bind to D2R or D4R.29–32 Co-perfusion with 10 μM of L745–870 or raclopride, but not VK4–116, significantly counteracted the effect of pramipexole (1 μM) and the optogenetic stimulation could still increase glutamate release both in controls (at 100 Hz) and in animals with BID (at 60 Hz) (Fig 3A and 3B). The results therefore imply a participation of both D4R and D2SR, but not D3R, in the effect of pramipexole.
Figure 3. Effect D2-like receptor antagonists on pramipexole-mediated inhibition of optogenetically-induced corticostriatal glutamate release.
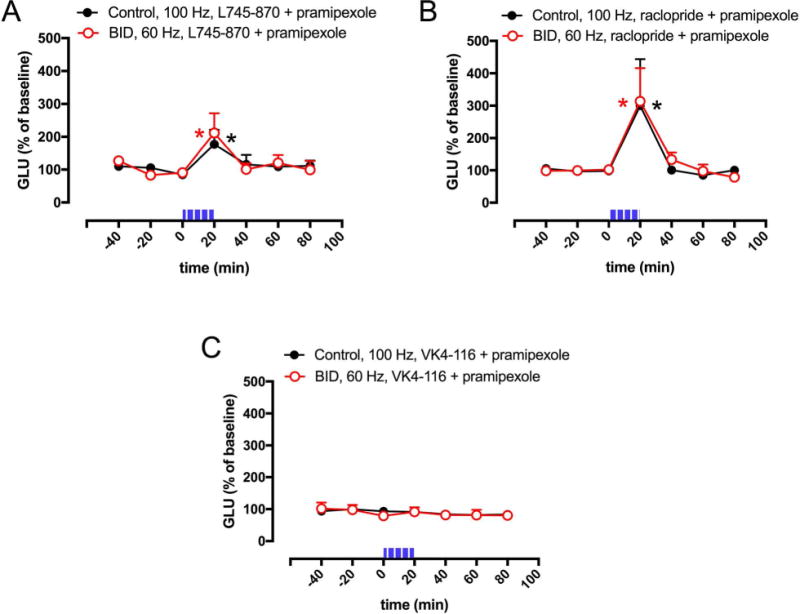
(A–C) Effect of co-perfusion of pramipexole (1 μM) with the selective D4R antagonist L745–870, the non-selective D2R-D3R antagonist raclopride or the D3R antagonist VK4–116 (10 μM in all cases) on optogenetically-induced glutamate release in the lateral striatum of BID rats (red plot) and controls (black plot); time ‘0’ represents the values of samples prior to stimulation; the period of stimulation (20 min) is represented as a train of vertical lines; results are expressed as means + S.E.M. of percentage of the average of three values before stimulation (n = 7–9 per group). *: p<0.05, as compared to value of the last sample before the stimulation, respectively (paired t test).
DISCUSSION
The present study first demonstrates that BID in rodents is associated with hypersensitive corticostriatal terminals, which show an increased sensitivity to depolarization-induced glutamate release. An association between iron deficiency and RLS was originally identified by Nordlander in the 1950s.34 Further studies showed a higher prevalence of RLS symptoms in conditions that compromise iron availability.35 A recent study in a population of patients with iron-deficiency anemia reported finding a 31.5% prevalence of RLS,36 which is six times higher than the general USA population prevalence for RLS.1 Most patients with RLS, however, do not have systemic iron deficiency. Nevertheless, as already proposed by Nordlander,34 an iron insufficient state exists in the brains of RLS patients and all studies of CNS iron have consistently shown BID in RLS (see Introduction). This brain-specific deficit in iron may be a consequence of the tight regulation of iron transportation by the blood–brain barrier.5 In rodents, BID can be consistently induced by providing a severe iron-deficient diet during the post-weaning period. Even though it does not show motor alterations that would imitate PLMS, the post-weaning, diet-induced BID rodent represents a well-accepted pathophysiological model of RLS.5,12,37 In fact, it provides a biological model for the understanding of the iron-dopamine connection in RLS, since it reproduces the main alterations in dopaminergic transmission observed in RLS patients. Those include an increase in striatal extracellular concentrations of dopamine, a reduction in the density of striatal D2R and an increased tyrosine hydroxylase activity in the ventral midbrain.12,37
In view of the validity of the rodent with BID as a pathogenetic model of RLS, the present results imply that hypersensitive corticostriatal terminals might represent a key mechanism responsible for the deficit of sensorimotor integration responsible for PLMS in RLS. That being the case, it would be expected that drugs with clinical efficacy for PLMS in RLS target corticostriatal terminals, counteracting depolarization-induced glutamate release. In fact, the two classes of drugs used as a first alternative in RLS, dopamine agonists and α2δ ligands,6 were very effective at counteracting optogenetically-induced glutamate release by both normal and hypersensitive corticostriatal terminals.
In previous studies, we provided evidence for alterations in adenosine neurotransmission in rats with BID, which could represent a main factor responsible for the increase in the sensitivity of corticostriatal terminals. In fact, those changes led us to hypothesize, first, that rodents with BID should show hypersensitive corticostriatal terminals and, second, that this could represent a main pathogenetic factor in the development of PLMS in RLS.8,19 Specifically, we found functional upregulation of A2AR and downregulation of adenosine A1 receptors (A1R).19,20 In the striatum, both receptors are co-localized presynaptically in glutamatergic terminals, where they form A1R-A2AR heteromers that play a fine-tune modulation of glutamate release.38,39 Since activation of A1R and A2AR lead to the opposite effect, inhibition and facilitation of striatal glutamate release,38,39 upregulation of A2AR and downregulation of A1R would be expected to increase sensitivity of corticostriatal terminals to release glutamate, decreasing the filtering of multiple cortical signals that converge in the striatum and therefore impairing striatal sensory-motor integration (Fig 4). In fact, in the present study, blockade of A2AR with the perfusion of a selective antagonist also counteracted optogenetically-induced glutamate release by both normal and hypersensitive corticostriatal terminals. These results would predict that A2AR antagonists could be useful in RLS. However, the highest density of striatal A2AR is found postsynaptically, in the GABAergic striatopallidal neurons, and postsynaptic A2AR were also found to be up-regulated in rats with BID.19 Unfortunately, blockade of postsynaptic A2AR could be expected to increase RLS symptomatology by potentiating postsynaptic D2LR signaling (see below). Nevertheless, striatal postsynaptic A2AR form heteromers with D2LR and it has already been established that heteromerization can determine changes in the affinity of ligands offering a strategy for ligand selectivity.40 In fact, by screening several A2AR antagonist, we found that the A2AR antagonist SCH-442416 shows lower affinity for postsynaptic A2AR (A2AR-D2LR heteromers) than for presynaptic A2AR (A1R-A2AR heteromers).39 Presynaptic A2AR antagonists are not yet introduced into the clinic, but they could provide a new treatment for RLS.
Figure 4. Schematic representation of a corticostriatal glutamatergic terminal and their modulatory dopamine and adenosine receptors.
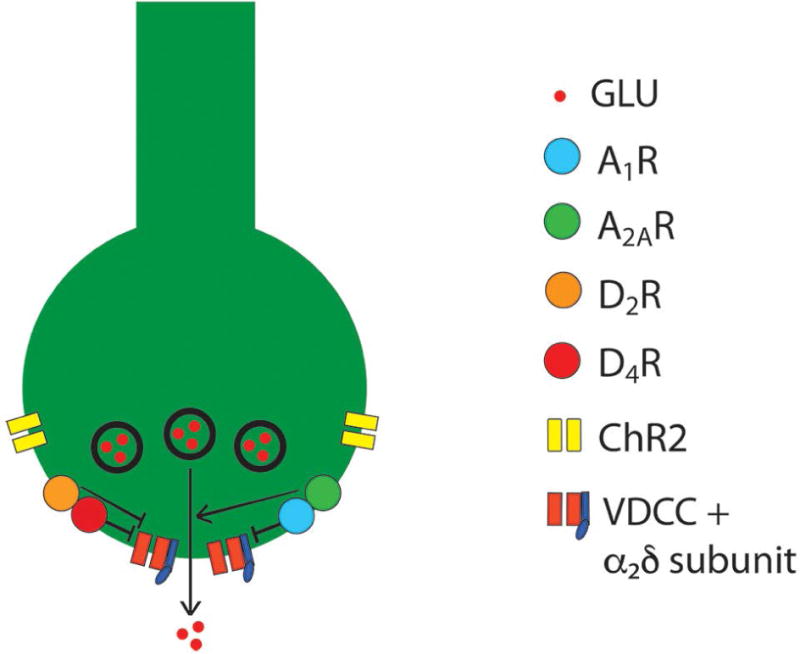
Dopamine and adenosine modulate corticostriatal glutamate release by acting on A1R-A2AR and D2R-D4R heteromers. The A1R-A2AR heteromer act as an adenosine concentration-dependent switch, by which a low adenosine concentration activates preferentially A1R, which produces inhibition of glutamate release, and a high adenosine concentration also activates A2AR, which shuts down A1R signaling and promotes and A2AR-mediated stimulation of glutamate release. The D2R-D4R heteromer provides a dopamine concentration-dependent stepwise inhibitory mechanism of glutamate release, that depends on the higher affinity of dopamine for the D4R and on a D4R-mediated-increase of D2R signaling. The BID-dependent increased in the excitability of the glutamatergic terminal to release glutamate seems to depend on functional downregulation of A1R and upregulation of A2AR, which can be counteracted by A2AR antagonists D2R or D4R agonists and α2δ ligands (see text). The function of voltage-dependent calcium channels (VDCC), which activation promotes vesicular fusion and neurotransmitter release, is regulated by Gi-coupled receptors (ßγ-mediated inhibition), including A1R, D2R and D4R, as well as by accessory α2δ subunits, the targets of gabapentin-like compounds. ChR2: channelrhodopsin 2 (see text).
Different to α2δ ligands, like gabapentin, which are already assumed to act by decreasing glutamatergic neurotransmission, the ability of the dopamine agonists pramipexole and ropinirole to strongly modulate the function of corticostriatal glutamatergic terminals imply a conceptual change in their generally assumed therapeutic mechanism. Pramipexole and ropinirole are ligands with preferential affinity for D2-like versus D1-like receptors and, among the different D2-like receptor subtypes, they both have preferential affinity for D3R, as compared to D2SR, D2LR and D4R.41–43 This has led to the assumption that D3R is the main target involved in the therapeutic effect of dopamine receptor agonists in RLS.44,45 However, as mentioned by Varga et al.,44 it remained to be determined if the relative D3R selectivity of pramipexole and ropinirole had pharmacological relevance at the doses used clinically. The present study does not support the D3R hypothesis. First, striatal D3R are only localized postsynaptically,46 indicating that they cannot target corticostriatal glutamate terminals. Instead, dopamine D2SR and D4R are the D2-like receptor subtypes preferentially localized in corticostriatal glutamatergic terminals and involved in a direct modulation of striatal glutamate release (Fig 4).22,27,28 In fact, the ability of pramipexole to counteract optogenetically-induced glutamate release in rats with BID and in controls was counteracted by the selective D4R antagonist L745–870 and by raclopride, a D2-like receptor antagonist with very low affinity for D4R, while it was not modified by the selective D3R antagonist VK4–116.
The present results suggest that searching for more selective D2SR and D4R agonists could be a promising strategy for treating RLS. D4R can be particularly seen as the main therapeutic target, since it is more selectively expressed by corticostriatal neurons than D2SR and it has been recently shown to play a main role as a mediator of dopamine modulation of corticostriatal neurotransmission.22 On the other hand, D2SR has a more widespread expression, including the dopamine cells, where it acts as autoreceptor.47 In addition, a selective D2SR ligand devoid of activity for the striatal postsynaptic D2LR, might be difficult to obtain. Activation of D2LR might in fact contribute to the non-wanted side effects in RLS, such as the augmentation of the symptoms upon prolonged treatment with dopamine receptor agonists. The clinical study by Manconi et al.,45 which finds a much higher efficacy of pramipexole as compared to bromocriptine in RLS, can be quite demonstrative of the therapeutic value of D4R ligands. Thus, although the results were interpreted as related to the different affinities for D3R, the most dramatic pharmacological difference between both drugs is in fact the very low affinity of bromocriptine for D4R.43
In summary, the present study provides preclinical evidence for a main pathogenetic mechanism of PLMS in RLS, hypersensitive corticostriatal glutamatergic terminals. It also provides a plausible explanation for the previously unknown common mechanism responsible for the therapeutic effect of α2δ ligands and dopamine receptor agonists in RLS, the inhibition of corticostriatal glutamate release. Finally, these data support a switch in the dopamine receptor subtype as a therapeutic target for RLS, from the D3R to the D4R. This switch implies the need for further investigation of selective D4R agonists for the treatment of RLS.
Acknowledgments
Work supported by the intramural funds of the National Institute on Drug Abuse and RLS Foundation. We acknowledge Dr. Vivek Kumar, Medicinal Chemistry Section, NIDA-IRP, for synthesizing the compound VK4–116 used in these studies.
Footnotes
Author Contributions
XG, AHN, RPA, CJE, CQ and SF contributed to the conception and design of the study; GY, WR and CQ contributed to the performance of experiments, acquisition and analysis of data; GY, RPA, CJE, CQ and SF contributed to drafting the text and preparing the figures.
Potential Conflict of Interests
The authors declare no potential conflict of interests
References
- 1.Allen RP, Walters AS, Montplaisir J, et al. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med. 2005;165:1286–1292. doi: 10.1001/archinte.165.11.1286. [DOI] [PubMed] [Google Scholar]
- 2.Allen RP, Stillman P, Myers AJ. Physician-diagnosed restless legs syndrome in a large sample of primary medical care patients in western Europe: Prevalence and characteristics. Sleep Med. 2010;11:31–37. doi: 10.1016/j.sleep.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Ferri R, Cosentino FI, Manconi M, et al. Increased electroencephalographic high frequencies during the sleep onset period in patients with restless legs syndrome. Sleep. 2014;37:1375–1381. doi: 10.5665/sleep.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferri R, Rundo F, Zucconi M, et al. An evidence-based analysis of the association between periodic leg movements during sleep and arousals in restless legs syndrome. Sleep. 2015;38:919–924. doi: 10.5665/sleep.4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Earley CJ, Connor J, Garcia-Borreguero D, et al. Altered brain iron homeostasis and dopaminergic function in restless legs syndrome (Willis-Ekbom Disease) Sleep Med. 2014;15:1288–1301. doi: 10.1016/j.sleep.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 6.Silber MH, Becker PM, Earley C, et al. Willis-Ekbom Disease Foundation revised consensus statement on the management of restless legs syndrome. Mayo Clin Proc. 2013;88:977–986. doi: 10.1016/j.mayocp.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Borreguero D, Patrick J, DuBrava S, et al. Pregabalin versus pramipexole: effects on sleep disturbance in restless legs syndrome. Sleep. 2014;37(4):635–643. doi: 10.5665/sleep.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferré S, Earley C, Gulyani S, et al. In search of alternatives to dopaminergic ligands for the treatment of restless legs syndrome: iron, glutamate, and adenosine. Sleep Med. 2017;31:86–92. doi: 10.1016/j.sleep.2016.08.019. [DOI] [PubMed] [Google Scholar]
- 9.Dooley DJ, Taylor CP, Donevan S, Feltner D. Ca2+ channel alpha2delta ligands: novel modulators of neurotransmission. Trends Pharmacol Sci. 2007;28:75–82. doi: 10.1016/j.tips.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Trenkwalder C, Paulus W. Restless legs syndrome: pathophysiology, clinical presentation and management. Nat Rev Neurol. 2010;6:337–346. doi: 10.1038/nrneurol.2010.55. [DOI] [PubMed] [Google Scholar]
- 11.Tergau F, Wischer S, Paulus W. Motor system excitability in patients with restless legs syndrome. Neurology. 1999;52:1060–1063. doi: 10.1212/wnl.52.5.1060. [DOI] [PubMed] [Google Scholar]
- 12.Rizzo V, Aricò I, Liotta G, et al. Impairment of sensory-motor integration in patients affected by RLS. J Neurol. 2010;257:1979–1985. doi: 10.1007/s00415-010-5644-y. [DOI] [PubMed] [Google Scholar]
- 13.Gerfen CR. Basal Ganglia. In: Paxinos G, editor. The Rat Nervous System. Amsterdam: Elsevier Academic Press; 2004. pp. 445–508. [Google Scholar]
- 14.Rizzo G, Manners D, Testa C, et al. Low brain iron content in idiopathic restless legs syndrome patients detected by phase imaging. Mov Disord. 2013;28:1886–1890. doi: 10.1002/mds.25576. [DOI] [PubMed] [Google Scholar]
- 15.Cho YW, Allen RP, Earley CJ. Clinical efficacy of ferric carboxymaltose treatment in patients with restless legs syndrome. Sleep Med. 2016;25:16–23. doi: 10.1016/j.sleep.2016.06.021. [DOI] [PubMed] [Google Scholar]
- 16.Allen RP, Adler CH, Du W, et al. Clinical efficacy and safety of IV ferric carboxymaltose (FCM) treatment of RLS: A multi-centered, placebo-controlled preliminary clinical trial. Sleep Med. 2011;12:906–13. doi: 10.1016/j.sleep.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 17.Ondo WG. Intravenous iron dextran for severe refractory restless legs syndrome. Sleep Med. 2010;11:494–6. doi: 10.1016/j.sleep.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Connor JR, Wang XS, Allen RP, et al. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain. 2009;132:2403–2412. doi: 10.1093/brain/awp125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quiroz C, Pearson V, Gulyani S, et al. Up-regulation of striatal adenosine A(2A) receptors with iron deficiency in rats: effects on locomotion and cortico-striatal neurotransmission. Exp Neurol. 2010;224:292–298. doi: 10.1016/j.expneurol.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quiroz C, Gulyani S, Ruiqian W, et al. Adenosine receptors as markers of brain iron deficiency: Implications for Restless Legs Syndrome. Neuropharmacology. 2016;111:160–168. doi: 10.1016/j.neuropharm.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quiroz C, Orrú M, Rea W, et al. Local control of extracellular dopamine levels in the medial nucleus accumbens by a glutamatergic projection from the infralimbic cortex. J Neurosci. 2016;36:851–859. doi: 10.1523/JNEUROSCI.2850-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonaventura J, Quiroz C, Cai NS, et al. Key role of the dopamine D(4) receptor in the modulation of corticostriatal glutamatergic neurotransmission. Sci Adv. 2017;3:e1601631. doi: 10.1126/sciadv.1601631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lok CN, Loh TT. Regulation of transferrin function and expression: review and update. Biol Signals Recept. 1998;7:157–178. doi: 10.1159/000014542. [DOI] [PubMed] [Google Scholar]
- 24.Gulyani S, Earley CJ, Camandola S, et al. Diminished iron concentrations increase adenosine A(2A) receptor levels in mouse striatum and cultured human neuroblastoma cells. Exp Neurol. 2009;215:236–242. doi: 10.1016/j.expneurol.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sgambato V, Pagès C, Rogard M, et al. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerfen CR, Miyachi S, Paletzki R, et al. D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci. 2002;22:5042–5054. doi: 10.1523/JNEUROSCI.22-12-05042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Centonze D, Gubellini P, Usiello A, et al. Differential contribution of dopamine D2S and D2L receptors in the modulation of glutamate and GABA transmission in the striatum. Neuroscience. 2004;129:157–166. doi: 10.1016/j.neuroscience.2004.07.043. [DOI] [PubMed] [Google Scholar]
- 28.González S, Rangel-Barajas C, Peper M, et al. Dopamine D4 receptor, but not the ADHD-associated D4.7 variant, forms functional heteromers with the dopamine D2S receptor in the brain. Mol Psychiatry. 2012;17:650–662. doi: 10.1038/mp.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel S, Freedman S, Chapman KL, et al. Biological profile of L-745,870, a selective antagonist with high affinity for the dopamine D4 receptor. J Pharmacol Exp Ther. 1997;283:636–647. [PubMed] [Google Scholar]
- 30.Newman-Tancredi A, Audinot V, Chaput C, et al. [35S]Guanosine-5′-O-(3-thio)triphosphate binding as a measure of efficacy at human recombinant dopamine D4.4 receptors: actions of antiparkinsonian and antipsychotic agents. J Pharmacol Exp Ther. 1997;282:181–191. [PubMed] [Google Scholar]
- 31.Gazi L, Schoeffter P, Nunn C, et al. Cloning, expression, functional coupling and pharmacological characterization of the rat dopamine D4 receptor. Naunyn Schmiedebergs Arch Pharmacol. 2000;361:555–564. doi: 10.1007/s002100000236. [DOI] [PubMed] [Google Scholar]
- 32.Kumar V, Bonifazi A, Ellenberger MP, et al. Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: New leads for opioid dependence treatment. J Med Chem. 2016;59:7634–7650. doi: 10.1021/acs.jmedchem.6b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chefer VI, Thompson AC, Zapata A, et al. Overview of brain microdialysis. Curr Protoc Neurosci. 2009 doi: 10.1002/0471142301.ns0701s47. Chapter 7:Unit7.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nordlander NB Therapy in restless legs. Acta Med Scand. 1953;145:453–457. [PubMed] [Google Scholar]
- 35.Allen RP, Earley CJ. The role of iron in restless legs syndrome. Mov Disord. 2007;18:S440–S448. doi: 10.1002/mds.21607. [DOI] [PubMed] [Google Scholar]
- 36.Allen RP, Auerbach S, Bahrain H, et al. The prevalence and impact of restless legs syndrome on patients with iron deficiency anemia. Am J Hematol. 2013;88:261–264. doi: 10.1002/ajh.23397. [DOI] [PubMed] [Google Scholar]
- 37.Unger EL, Bianco LE, Jones BC, et al. Low brain iron effects and reversibility on striatal dopamine dynamics. Exp Neurol. 2014;261:462–468. doi: 10.1016/j.expneurol.2014.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciruela F, Casadó V, Rodrigues RJ, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orru M, Bakešová J, Brugarolas M, et al. Striatal pre- and postsynaptic profile of adenosine A(2A) receptor antagonists. PLoS One. 2011;6:e16088. doi: 10.1371/journal.pone.0016088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferré S, Casadó V, Devi LA, et al. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66:413–434. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wood M, Dubois V, Scheller D, Gillard M. Rotigotine is a potent agonist at dopamine D1 receptors as well as at dopamine D2 and D3 receptors. Br J Pharmacol. 2015;172:1124–1135. doi: 10.1111/bph.12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coldwell MC, Boyfield I, Brown T, et al. Comparison of the functional potencies of ropinirole and other dopamine receptor agonists at human D2(long), D3 and D4.4 receptors expressed in Chinese hamster ovary cells. Br J Pharmacol. 1999;127:1696–1702. doi: 10.1038/sj.bjp.0702673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newman-Tancredi A, Cussac D, Audinot V, et al. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor. J Pharmacol Exp Ther. 2002;303:805–814. doi: 10.1124/jpet.102.039875. [DOI] [PubMed] [Google Scholar]
- 44.Varga LI, Ako-Agugua N, Colasante J, et al. Critical review of ropinirole and pramipexole – putative dopamine D(3)-receptor selective agonists - for the treatment of RLS. J Clin Pharm Ther. 2009;34:493–505. doi: 10.1111/j.1365-2710.2009.01025.x. [DOI] [PubMed] [Google Scholar]
- 45.Manconi M, Ferri R, Zucconi M, et al. Preferential D2 or preferential D3 dopamine agonists in restless legs syndrome. Neurology. 2011;77:110–117. doi: 10.1212/WNL.0b013e3182242d91. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz JC, Diaz J, Bordet R, et al. Functional implications of multiple dopamine receptor subtypes: the D1/D3 receptor coexistence. Brain Res Brain Res Rev. 1998;26:236–242. doi: 10.1016/s0165-0173(97)00046-5. [DOI] [PubMed] [Google Scholar]
- 47.De Mei C, Ramos M, Iitaka C, Borrelli E. Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol. 2009;9:53–58. doi: 10.1016/j.coph.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]