

Summary
During assembly of the bacterial flagellum, protein subunits that form the exterior structures are exported through a specialized secretion apparatus energized by the proton gradient. This category of protein transport, together with the similar process that occurs in the injectisomes of gram-negative pathogens, is termed type-III secretion. The membrane-embedded part of the flagellar export apparatus contains five essential proteins: FlhA, FlhB, FliP, FliQ, and FliR. Here, we have undertaken a variety of experiments that together support the proposal that the protein-conducting conduit is formed primarily, and possibly entirely, by FliP. Chemical modification experiments demonstrate that positions near the center of certain FliP trans-membrane (TM) segments are accessible to polar reagents. FliP expression sensitizes cells to a number of chemical agents, and mutations at predicted channel-facing positions modulate this effect. Multiple assays are used to show that FliP suffices to form a channel that can conduct a variety of medium-sized, polar molecules. Conductance properties are strongly modulated by mutations in a methionine-rich loop that is predicted to lie at the inner mouth of the channel, which might form a gasket around cargo molecules undergoing export. The results are discussed in the framework of an hypothesis for the architecture and action of the cargo-conducting part of the type-III secretion apparatus.
Keywords: Membrane transport, energy transduction, injectisome, pathogenesis
Graphical abstract
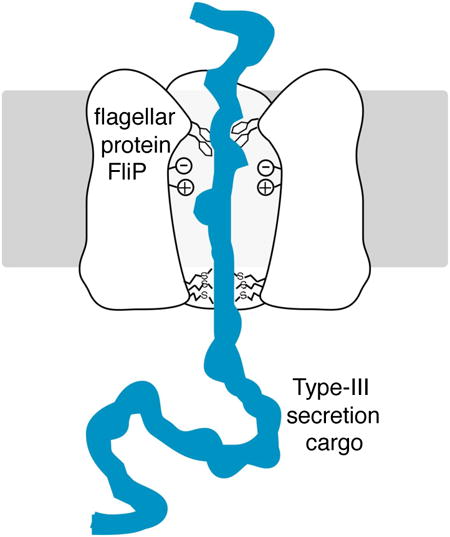
Introduction
Many species of bacteria swim by means of rotating flagella that obtain energy from the membrane ion gradient [1-3]. A flagellum comprises a thin, fairly rigid helical filament that functions as propeller, a flexible hook serving as universal joint, and a membrane-embedded structure termed the basal body consisting of several rings mounted on a central rod [4, 5]. The flagellum is formed from about 20 different proteins, in copy numbers ranging from many thousands (the filament protein FliC) to just a handful (e.g., the filament-capping protein FliD). To reach their sites of installation in the structure, protein subunits forming the rod, hook, and filament must be actively transported through a specialized secretion apparatus at the bottom of the basal body [6, 7]. In addition to the protein subunits that compose the structure, assembly and operation of the flagellum requires several proteins that serve export or regulatory roles, as well as the proteins of the chemotaxis pathway that regulate its direction of rotation. The flagellar regulon comprises more than 60 genes, hierarchically regulated in a scheme that is tied to events in flagellar assembly as well as being responsive to environmental factors such as temperature and nutritional status [6, 8-12].
The bacterial flagellum is structurally and functionally related to the ‘injectisome’ apparatus used by many pathogens to inject virulence factors into host cells [13-15]. Like the flagellum, the injectisome contains a specialized secretion apparatus, which in this case functions to export virulence effectors into host cells. The protein-exporting systems of the flagellum and injectisome are homologous, as evidenced by strong sequence conservation in several membrane-associated components. Transport is thus expected to occur by similar mechanisms in the two systems, and is collectively termed type III secretion, or T3S [7, 13, 16]. Transport through T3S systems is notably rapid; the flagellar apparatus can secrete several 55-kDa flagellin subunits per second in the early stages of filament assembly [17, 18], and injectisomes deliver most of their complement of virulence factors within minutes of contacting the host [19]. The systems must also be capable of recognizing the appropriate export substrates from among the many hundreds of proteins in the cell. Although the basis of this specificity has not been fully defined, export substrates appear to share amino-terminal regions that are structurally disordered prior to their assembly into the structure [20], and certain substrates (in the case of the flagellum, the late substrates) are targeted to the apparatus by specialized secretion chaperones [21, 22].
Essential membrane components of the flagellar secretion apparatus include FliP, FliQ, FliR, FlhA, and FlhB; FliO contributes to assembly of the apparatus [23] but is not essential for transport per se [24]. Efficient export also depends on the cytoplasmic components FliH, FliI, and FliJ, which form a complex that binds substrates and targets them to the inner mouth of the apparatus [25]. Cargo subunits are delivered to the mouth of the apparatus in a process involving ATP hydrolysis by FliI, and subsequent translocation across the membrane is energized by the membrane proton gradient [26-30]. The core of the secretion apparatus is thus a proton-energized protein pump. Recent studies gave evidence that FlhA is the protein most likely to couple to the proton gradient [31, 32]; a recent study of the FliP homolog SpaP provided structural and functional indications that it might form the cargo-conducting element [33]. The molecular mechanism by which cargo is transported across the membrane is not understood.
Here, we have undertaken several experiments to test that proposal that the trans-membrane channel for exported cargo subunits is formed from the protein FliP. Positions near the middle of some trans-membrane (TM) segments of FliP were found to be accessible to chemical modification by polar reagents. Expression of FliP sensitized cells to a variety of toxic agents, and this effect was modulated by mutations at predicted channel-facing positions in FliP membrane segments. Other assays of solute movement indicate that FliP suffices to conduct a variety of medium-sized, polar molecules. Conductance by FliP is strongly modulated by mutations in a methionine-rich loop that is predicted to lie at the inner mouth of the channel. The results are discussed in the framework of an hypothesis for the architecture and function of the cargo-conducting part of the T3S apparatus.
Results
Sequence alignments show strong conservation in the membrane segments of FliP, particularly in TM3 and TM4. Invariant residues include Asp197 near the middle of TM3, Lys222 in TM4, and a stretch of methionine residues in the loop connecting TM3 to TM4, which is predicted to lie on the cytoplasmic side of the membrane (Figure 1 and Figure S1). In a mutational study of conserved charged residues of the apparatus, replacements of Asp197 and Lys222 caused a substantial reduction in function as assayed on motility plates [31]. The strong sequence conservation in TM3 and TM4 and the significant motility defects of Asp197 and Lys222 mutants indicate that these parts of FliP contribute in an important way to the transport process. We hypothesized that TM3 and TM4 of FliP might be involved in forming the cargo-conducting pore, with the invariant Asp197 and Lys222 residues occurring at solvent-exposed positions in the interior of the pore.
Figure 1.
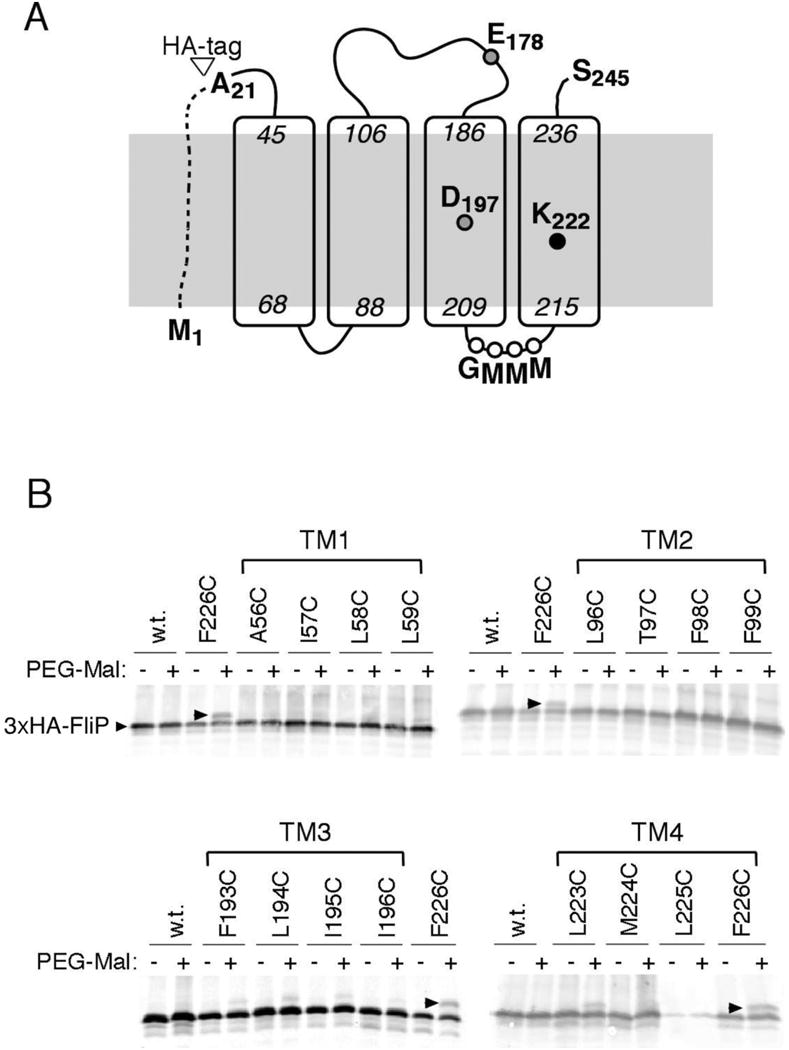
(A) Topology and key features of FliP. The site of attachment of the HA tag used in some experiments is indicated. (B) PEG-maleimide modification of FliP molecules with individual Cys residues at the indicated positions. Nominal molecular weight of the PEG reagent was 1 Kd. Cys-substituted, HA-tagged FliP proteins were expressed from a plasmid (variants of pMS89, each mutated to contain a single Cys codon) induced with 2.5 μM sodium salicylate. The four gels show results for each of the four TM segments of FliP. Position 226 in TM4 showed the highest yield of chemical modification and is included on each gel as a positive control.
To probe the solvent accessibility of positions in the membrane segments of FliP, single Cys replacements were made in four consecutive positions in each of the segments, and reactivity with a large sulfhydryl reagent (PEG-maleimide, MW ∼1 Kd) was examined. FliP was detected on immunoblots using a haemagglutinin (HA) tag at the amino terminus of the mature protein (i.e., lacking the signal sequence) (Figure 1). The HA-tagged FliP supported motility comparable to wild type in complementation experiments with a ΔfliP strain (Figure S2), and all of the Cys-replacement variants functioned as well as wild type with the exception of L225C, which showed poor motility in soft-agar assays (Figure S3) and low levels of the protein on immunoblots. Labeling experiments used a ΔflhDC strain that expresses no other flagellar proteins and were carried out with spheroplasts to maximize FliP access by the reagent. Products were visualized on anti-HA immunoblots; results are shown in Figure 1 (panel B). Certain of the Cys replacements in TM3 and TM4, but not in TM1 or TM2, showed significant labeling with PEG-maleimide. Labeling was greatest at position 194 in TM3 and position 226 in TM4, which would lie on roughly the same helix faces as the invariant Asp197 and Lys222 residues. These surfaces of TM3 and TM4 thus appear to be solvent-accessible to at least some extent.
Previous estimates of FliP stoichiometry suggested the presence of 4-5 copies per basal body [34], and structural studies of a sub-domain of FliP similarly indicate the likelihood of FliP multimers [35]. To further test the proposal that the apparatus contains multiple, jointly functioning copies of FliP, we examined inter-subunit complementation between FliP mutations that were previously found to cause fairly severe motility impairments individually [31]. Selected fliP alleles were transferred from the CmR plasmid onto a plasmid encoding KmR, then transformed into the ΔfliP strain in pairs and tested for function in motility plates. Motility of the E178A mutant was increased more than two-fold by co-expression of the D197N allele, even though the D197N mutation alone nearly abolishes motility (Figure 2). The occurrence of such inter-subunit complementation supports the view that multiple copies of FliP function together within the apparatus.
Figure 2.
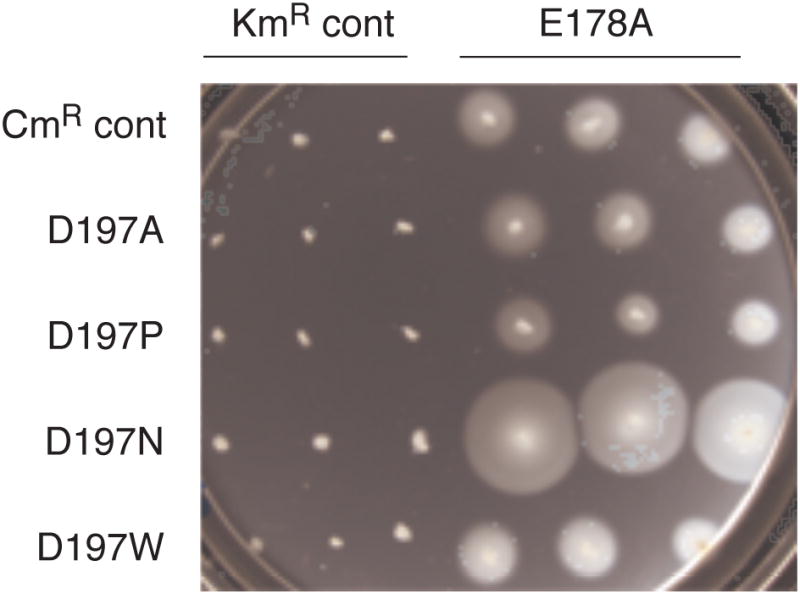
Improved motility of the E178A mutant when the D197N allele is co-expressed. Control experiments (indicated) used empty vectors encoding only the needed antibiotic resistance. The plate contained tryptone, 0.26% agar, antibiotics (25 μg/ml each Cm and Km), and 10 μM salicylate to induce expression of the FliP variants. The plate was spotted with 3 μl of overnight cultures and incubated for 7.5 h at 32° C.
In mutants with Cys at exposed positions in FliP, flagellar assembly might be affected by Cd2+, which has some affinity for the Cys side-chain. To test this, cells of the ΔfliP strain expressing either wild-type or Cys-mutant FliP (or no FliP as a negative control) were cultured in the presence of varying concentrations of CdCl2, and effects on motility and growth were examined. Motility was not strongly affected by Cd2+. Growth of cells, however, was more strongly inhibited by Cd 2+ in cells expressing FliP than in the vector-only control (Figure 3). Cd2+ sensitivity was measured in each of the 16 Cys-mutant strains and was found to be significantly increased in the F226C mutant (Figure 3, panel B). Residue Phe226 is fully conserved in T3S systems and would lie on roughly the same face of TM4 as the invariant Lys222 (assuming α-helical structure). Residue 193 in TM3 is also an invariant Phe and would lie on roughly the same helix face as Asp197. A F193C single-replacement mutant did not show increased sensitivity to Cd2+, but a F193C/F226C double replacement showed greater sensitivity than F226C or any of the other single-Cys mutants (Figure S4). To determine whether the sensitization to Cd2+ is due to FliP alone or requires the participation of other flagellar proteins, similar experiments were done in the ΔflhDC background. Sensitization to Cd2+ was comparable to that observed in the ΔfliP background (Figure 3, panels C and D). The sensitization effect is not related to the presence of the HA-tag on FliP, because it was also observed with the native (non-HA-tagged) protein (Figure S6, panel A). Another toxic divalent ion, Mn2+, showed similar effects; cells became sensitive to Mn2+ upon expression of FliP, and more so with the F226C mutant protein (Figure S5, panels A and B).
Figure 3.
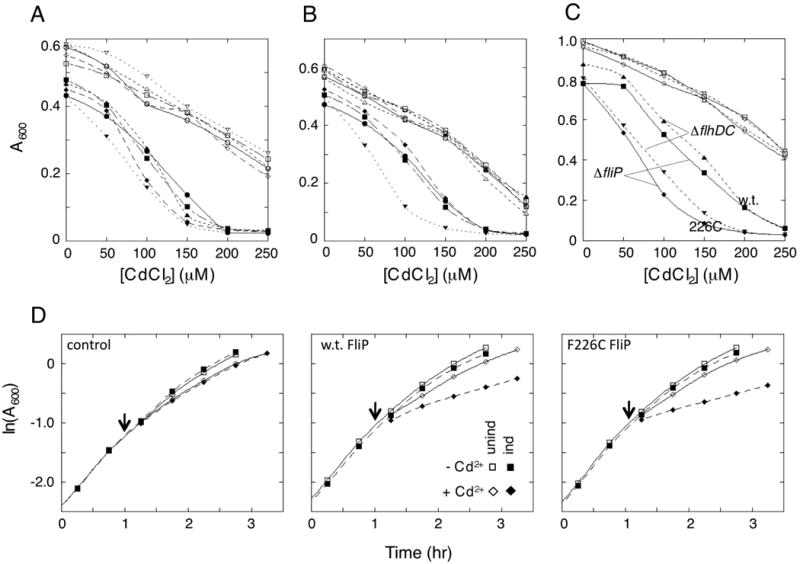
Sensitization of cells to cadmium ion (Cd2+) upon expression of FliP. In all panels, open symbols are for uninduced cultures and closed symbols for cultures induced with salicylate (2.5 μM). (A) Cd2+ sensitization in cells expressing wild-type FliP or FliP with Cys replacements near the middle of TM3. Filled symbols, induced; open symbols, uninduced. ●, wild type; ■, F193C; ◆, L194C; ▲, I195C; ▼, I196C. (B) Cd2+ sensitization in cells expressing FliP proteins with Cys replacements near the middle of TM4. ●, wild type; ■, L223C; ◆, M224C; ▲, L225C; ▼, F226C. Sensitivity to Cd2+ was increased by the Cys replacement at position 226. Sensitivity was decreased by the Cys replacement at position 225, which decreased the accumulation of the protein in cells (Figure 1). Cys replacements in TM1 and TM2 (not shown) did not affect the Cd2+ induced growth impairment. Results of representative individual experiments are shown; errors in absorbance measurements are comparable to symbol sizes. An additional example of the F226C result is shown in Figure S4. (C) Comparison of the Cd2+ sensitization in the ΔfliP strain where cells assemble flagella (with either wild-type or F226C FliP expressed from a plasmid) and the ΔflhDC strain that expresses no other flagellar proteins (and that does not assemble flagella). Filled symbols, induced; open symbols, uninduced. Sensitivity to Cd2+ is comparable in the two strains and is stronger with the F226C mutant than the wild type in both backgrounds. (D) Growth curves showing the rapid onset of the Cd2+ effect and the lack of substantial growth impairment prior to addition of the stressor. Experiments were carried out in the ΔflhDC background, with FliP (w.t. or F226C mutant) expressed from salicylate-regulated plasmids (pMS89 for w.t. and pMS301 for the F226C mutant). Induction was with 1.5 μM salicylate, similar to the level required for complementation of a ΔfliP mutant (Figure S6). CdCl2 addition (to final concentration 200 μM) is indicated by the arrows.
The inhibitory effects of Cd2+ and Mn2+ do not appear to be due to a FliP-induced decrease in general health of the cells such as might occur if, for example, FliP jammed the translocon: FliP expression did not significantly impact cell growth prior to addition of the stressing agent (Figure 3 and figure S5), and pronounced sensitization to Cd2+ and Mn2+ was observed at relatively low FliP expression levels (1-1.5 μM salicylate) comparable to that needed for complementation of a ΔfliP mutant (∼1 μM salicylate; Figure S6). In contrast to Cd2+ and Mn2+, the addition of ethanol (to 2.8%), which can readily cross membranes, had the same growth-retarding effect on cells whether or not they expressed FliP (Figure S5, panel C).
Experiments with the somewhat larger ion guanidinium also showed increased sensitivity of cells expressing FliP, in both the ΔfliP and ΔflhDC backgrounds (Figure S6). In contrast to the situation with the divalent ions, however, guanidium sensitization was no stronger with the F226C protein than with wild type (not shown). Sensitivity to the much larger ion choline, by contrast, was not increased upon expression of FliP, in either background. We hypothesized that a highly conserved, methionine-rich segment of FliP between TM3 and TM4 might form a seal at the inner end of the pore (Figure 1), in which case conductance to a larger ion might be increased by deletion of one of the Met residues. A FliP variant lacking one of the three consecutive Met residues (ΔMet-FliP) was constructed and shown to have essentially normal function in motility assays (Figure S7). When expressed in the ΔfliP background, this ΔMet FliP variant sensitized the cells to choline (Figure 4, upper two panels). Unlike the Cd2+ and guanidium effects, the sensitization to choline was not observed in the ΔflhDC background or in strains with deletions of fliQ, flhA, or flhB (Figure 4), and thus appears to require the other components of the apparatus. Together, these observations indicate that the ΔMet variant of FliP can be incorporated into the export apparatus and confers increased conductance to choline. Loss of the Met residue did not increase sensitivity to other ions; the already significant sensitization to Cd2+ and guanidinium was not increased in the ΔMet mutant (Figure S8).
Figure 4.
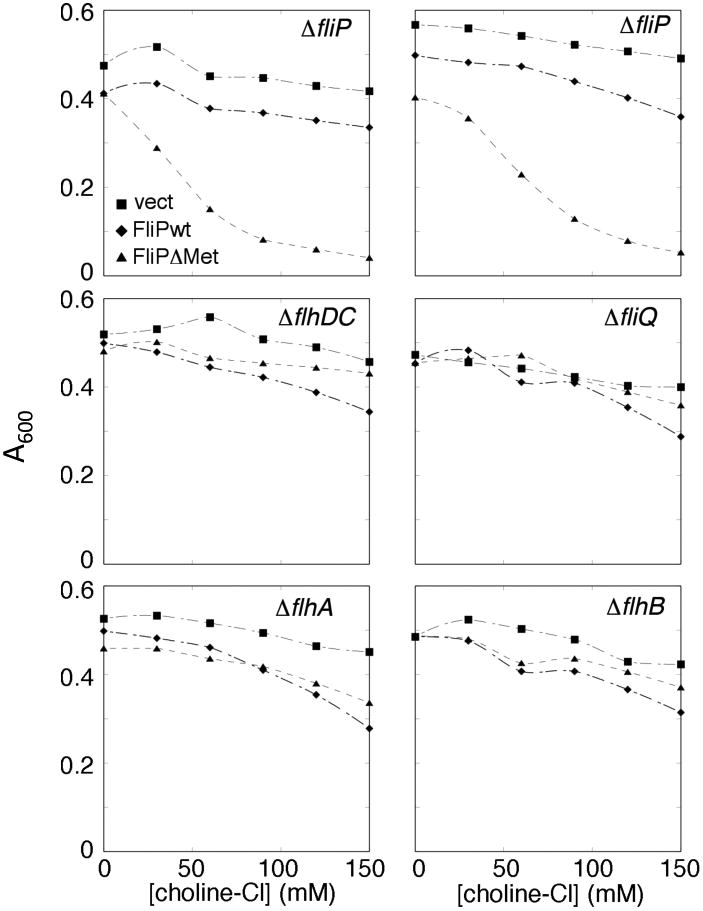
Sensitization of cells to choline upon expression of a FliP variant deleted for one of the conserved Met residues in segment 209-211, but not wild-type FliP. FliP expression was induced with 10 μM salicylate. The upper panels show two independent experiments carried out in ΔfliP cells, where expression of FliP from the plasmid allows assembly of the full export apparatus. The other panels show representative single experiments carried out in the indicated deletion backgrounds.
If the sensitization to choline is due to its movement through the protein-export pore, then the effect should be diminished when the pore is blocked by substrate trapped in the secretion channel. A pore-blocking substrate was engineered by fusing the fliK gene to the gene encoding the very stable designed protein TOP7 [36], and inserted into the chromosomal ara locus. On induction with arabinose, the FliK-Top7 protein is targeted to the export apparatus and becomes trapped in the secretion channel. Jamming of the export apparatus by the pore-blocking FliK-Top7 protein prevented secretion of wild-type FliK and abolished motility (Figure S9). We hypothesized that blocking the secretion pore by an actively secreting FliK-Top7 protein might prevent sensitization to choline (whose conductance requires the other proteins of the apparatus in addition to FliP), but probably not to Cd 2+ (because the Cd 2+ effect requires only FliP, to which FliK should not be targeted). In cells expressing the ΔMet-FliP variant, sensitization to choline was almost fully reversed on induction of the FliK-TOP7 blocking construct with arabinose (Figure 5). This did not occur when arabinose was added to control cells lacking the blocking construct (Figure 5), or when the experiment used Cd2+ instead of choline (Figure S9).
Figure 5.
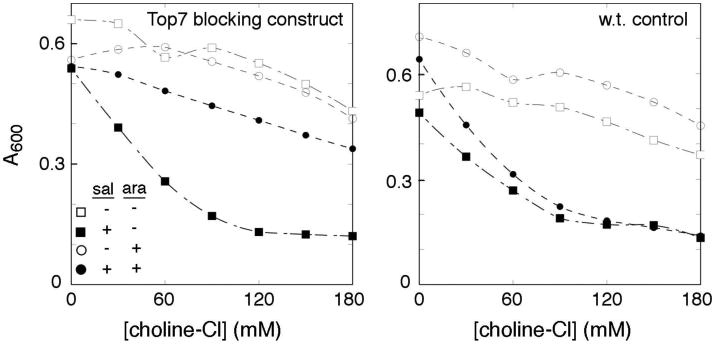
Reversal of the choline-induced growth defect upon expression of the pore-blocking construct FliK-TOP7. The experimental strain (left-hand panel) contained the FliK-TOP7 construct in the chromosomal ara locus; the control strain (right-hand panel) lacked the pore-blocking construct but was otherwise the same. Expression of ΔMet-FliP was induced by 10 μM salicylate; induction of FliK-TOP7 was with 0.01% arabinose. A representative experiment is shown.
As a further means of probing the effects of FliP on membrane permeability, we monitored solute movements across the membrane through the accompanying changes in cellular refractility. Changes in absorbance have been used previously to monitor water efflux following osmotic upshift and the subsequent adaptation events that allow water to flow back into the cell [37]. Here, we used absorbance measurements to monitor trans-membrane solute movement in cells expressing either wild-type FliP or the ΔMet variant. Cells were equilibrated in medium of moderate osmotic strength (PBS), then diluted two-fold into the same medium supplemented with various salts at a concentration of 0.5 M (1.0 Osm/L). Water movements were monitored by the resulting changes in OD600. The initial transfer into high-salt media causes rapid water efflux that increases refractility of the cells and causes a rapid increase in OD600 (typically by about 10-15%) that is largely complete in the dead time of a manual-mixing experiment (a few seconds). In a subsequent slower phase, optical density decreases as water re-enters the cell, at a rate that is mainly dictated by re-entry of the osmotic agent. The experiment was carried out with chloride salts of Na+, K+, NH4+, CH3NH3+, guanidinium, and choline. For all the ions, including the large ion choline, a fairly rapid inward flow was observed when cells expressed the ΔMet-FliP variant (Figure 6). When wild-type FliP was used, fluxes were much smaller for all ions except guanidinium, which was conducted about as well by wild-type FliP. A triple mutant with the three conserved Met residues replaced by alanine also displayed high conductance to guanidinium; a triple mutant with Phe residues in place of the methionines did not (Figure S10). Function evidently depends on having side-chains of appropriate size and/or flexibility in this region: both the Ala and Phe triple-mutants were immotile (Figure S10).
Figure 6.
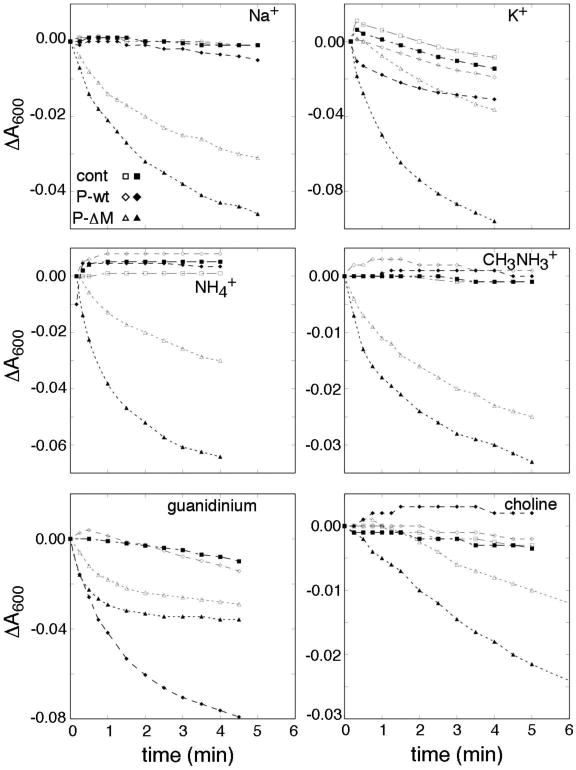
Inward fluxes in presence of various ions, in cells expressing the Δ-Met FliP variant. Cations are indicated in the figure panels. The counter-ion was Cl- in all cases. Induced cultures contained 10 μM salicylate. Outward water flux was induced by dilution into PBS containing the indicated salts at 0.5 M, as described in Materials and Methods. Measurements began a few (6-8) seconds after mixing, and monitored the slower return toward the initial absorbance level. Experiments were carried out at room temperature (ca. 20° C). Open symbols indicate non-induced cultures, and filled symbols cultures with FliP expression induced with 10 μM salicylate; ‘cont’ indicates the vector-only controls not expressing FliP.
Osmotic-upshift experiments were also carried out in the ΔflhDC strain that expresses no other flagellar proteins. In this background, FliP-dependent conductance was observed only with guanidinium; the flow was slower than in the ΔfliP background and was somewhat greater for wild-type FliP than for the delta-Met variant (Figure S11). Thus, FliP suffices to conduct guanidinium but additional components of the apparatus appear necessary to conduct the other ions examined in this experiment.
As a further test of the proposal that solutes flow through the flagellum, experiments were next carried out in a ΔflgG mutant strain. FlgG forms the distal part of the basal-body rod, and ΔfliG cells assemble basal bodies that contain an intact export apparatus but lack structures beyond the inner rod [38, 39]. Fluxes were examined with choline chloride as the osmotic agent, and with the ΔMet-FliP variant expressed in either the ΔflgG or wild-type background. (A background level of wild-type FliP, expressed from the chromosome, was thus also present.) The initial absorbance increase associated with water outflow was of similar magnitude in the two strains. A reproducible difference was observed in the time-course of the subsequent recovery phase, with water re-entry into the wild-type cells showing a lag in wild-type relative to that in the ΔflgG strain (Figure 7). This is as expected if choline must flow through the flagellar filament in the wild-type cells but through only the rod in the ΔflgG mutant. Consistent with this proposal, inflow in the wild type was made faster (becoming similar to that in the ΔflgG mutant) when the flagellar filaments were sheared just prior to the experiment (Figure 7, panel B). As expected, shearing had no effect in the ΔflgG strain (Figure 7, panel C).
Figure 7.
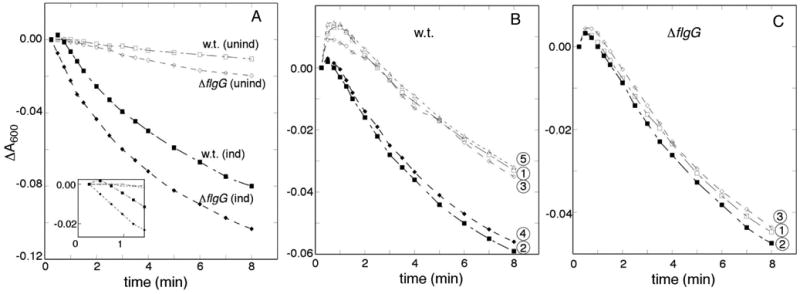
(A) Comparison of choline flux in cells that assemble normal flagella (w.t.) vs. cells that can assemble only the lower portion of the basal body (ΔflgG). The ΔMet variant of FliP was expressed from a plasmid, with induction by 10 μM salicylate where indicated. An initial delay in inward flow is seen in w.t. cells but not in cells with truncated basal bodies. (Inset shows an enlargement of the early time-course.) (B) Effect of shearing (filled symbols) on choline influx in w.t. cells. Flagellar filaments were sheared by passing samples twice rapidly through a 1-inch, 23G needle. Traces are numbered according to the order in which measurements were made (each involving an aliquot of cells that was either sheared or not prior to osmotic upshift). (C) Absence of a shearing effect with cells of the ΔflgG strain that does not make flagellar filaments. Numbers again indicate the sequence of the measurements.
Discussion
Several lines of evidence presented here indicate that FliP is the major component forming the cargo-conducting pore of the flagellar secretion apparatus. Recent work on SpaP, a homolog of FliP, endorses this view, and further establishes that several copies of SpaP, most likely five, can assemble into a ring with a cavity at its center [33, 40]. The present results on FliP suggest that FliP suffices to form a pore conductive to guanidium and can sensitize cells to certain cations, but does not, by itself, support large fluxes of most ions.
Chemical-accessibility experiments and mutational effects point to the close involvement of segments TM3 and TM4 in forming the pore. These segments exhibit strong sequence conservation that includes two invariant charged residues, Asp197 and Lys222, near the middles of the segments. Although these residues are not strictly required for transport function [31], their conservation, and the significant functional impairment that results from their mutation, point to an important role in transport. Because a protein-conducting pore must in any case be of fairly large diameter, it is not clear why the interior of the pore would need to contain these particular residues; a variety of polar groups could presumably serve to form a large, hydrated conduit. Based on the strict conservation and chemical properties (charge and proton-binding ability) of these residues, we suggest that they might have an active role in the transport of protein cargo. Specifically, we suggest that they could contribute to gradient-energized active transport by enforcing the following mechanistic constraints, or “rules” on cargo transit: 1) acidic groups of the cargo will most often pass through the channel in deprotonated (negatively charged) form, and 2) basic groups of the cargo will also most often pass through in deprotonated (but in this case electrically neutral) form. The first condition would ensure that the transit of each acidic group is actively driven by the membrane electric potential, while the second would couple transport to the pH gradient by ensuring that passage of each basic group is linked with what is effectively the movement of a proton from the outside of the cell to the inside (because the dissociating proton will leave to the inside and the new one is acquired from the outside).
If the TM3 and TM4 segments are helical, the part of the pore just ‘above’ the invariant charges should contain the side-chains of residues Phe193 and Phe226. These large, hydrophobic groups might be important in modulating the conductance of the pore, possibly in minimizing leakage of solutes past the cargo. Alternatively, or in addition, they might help shape electric fields in the channel to facilitate cargo movement (possibly contributing to the active transport process hypothesized above). Whatever their precise function, the strict conservation of these residues implies that they contribute something important to the transport mechanism in both the injectisome and flagellar systems.
A final feature of note is the Met-rich segment predicted to lie at the inner mouth of the channel. Among hydrophobic residues, the side-chain of Met is most flexible. Several such side-chains (15-18 in all, in a channel formed from five or six FliP subunits) might together form a deformable gasket at the inner end of the channel, accommodating the passage of a fast-moving, irregularly shaped cargo subunit while slowing or preventing the leakage of valuable metabolites and the energy-wasting influx of protons and other ions. The increased conductance that is observed on deletion of one of the Met residues, or mutation to residues of decreased bulk, is consistent with such a barrier function.
The structure of the FliP pore remains undetermined and will be an important goal of future studies. Other important questions concern the stoichiometry, positions, topologies and functions of FliQ and FliR.
Materials and Methods
Strains and plasmids
Strains and plasmids are listed in Table 1. Most experiments used derivatives of Salmonella enterica Serovar typhimurium strain LT2 (a gift from J. Roth). In-frame deletions were constructed using lambda-RED mediated recombineering [41]. Plasmids were based on the IPTG-inducible vector pRR48 or the salicylate-inducible vector pKG116, gifts from J. S. Parkinson. A KmR variant of pKG116 was constructed by insertion of the Km-resistance gene into the CmR gene.
Table 1. Strains and plasmids.
| Strain | Relevant genotype or property | Source or reference |
|---|---|---|
| LT2 | Wild-type Salmonella enterica serovar typhimurium | K. T. Hughes |
| TH2230 | ΔflgG-L2157 | K. T. Hughes |
| TH10549 | ΔfliP6709 | [31] |
| TH10550 | ΔfliQ6710 | [31] |
| TH12642 | ΔflhA7452 | [31] |
| TH12644 | ΔflhB7454 | [31] |
| TH13564 | ΔaraBAD7609::fliK+ | K. T. Hughes |
| TH13569 | ΔaraBAD7609::fliK-top7 | K. T. Hughes |
| TH15712 | ΔflhDC7902::FRT | This study |
| EM671 | ΔfliHIJ7367 ΔflgBC6557 ΔaraBAD1005::FRT | This study |
| EM808 | ΔaraBAD1005::FRT | This study |
| EM2225 | fliP22354::G157-3×FLAG | This study |
| EM3035 | fliP22424::G157- 3×FLAG MMM209AAA | This study |
| EM3748 | fliP22989::G157- 3×FLAG MMM209FFF | This study |
| Plasmid | ||
| pKG116 | Salicylate-inducible vector; Cmr | J. S. Parkinson |
| pDB1002 | Kmr variant of pKG116 (vector control) | This study |
| pDB1007 | fliP E178A in Kmr variant of pKG116 | This study |
| pDB1019 | fliP ΔMet in Kmr variant of pKG116 | This study |
| pMS10 | fliP in pKG116 | [31] |
| pMS11 | fliQ in pKG116 | [31] |
| pMS12 | fliR in pKG116 | [31] |
| pMS22 | fliP in Kmr variant of pKG116 | This study |
| pMS122 | flhA in pKG116 | [31] |
| pMS123 | flhB in pKG116 | [31] |
| pMS64 | fliP D197A in pKG116 | This study |
| pMS141 | fliP D197P in pKG116 | This study |
| pMS142 | fliP D197N in pKG116 | This study |
| pMS144 | fliP D197W in pKG116 | This study |
| pMS89 | N-terminal 3xHA tag fused to fliP codons 22-245, cloned in pKG116 | This study |
Media and Reagents
TB contained, per liter, 10 g tryptone and 5 g NaCl. Chloramphenical (Cm) and Kanamycin (Km) were used at 50 μg/ml. Motility plates contained TB, 0.27% Bacto-agar, appropriate antibiotics, and salicylate at the concentrations indicated in the figures. PBS buffer contained, per liter, 8 g NaCl, 0.2 g KCl, 1.22 g Na2HPO4, and 0.24 g KH2PO4.
PEG modification
To ensure that FliP is not shielded by other components of the apparatus, PEG-accessibility experiments were carried out in a ΔflhDC background that expresses no flagellar genes from the chromosome. Epitope-tagged FliP was made by fusing three repeats of the haemagglutinin (HA) antigen to codons 22-285 of fliP (the part following the signal sequence). Cells were cultured overnight at 32° C in TB plus antibiotic, then diluted 100-fold into fresh media containing antibiotic and 10 mM sodium salicylate to induce expression of the plasmid-encoded FliP. Following 6 h of growth at 32° C, optical density at 600 nm was measured, and equal amounts of cells as judged by OD600 were centrifuged and re-suspended in 2 ml spheroplast buffer (10 mM EDTA, 0.5 M sucrose, 50 mM Tris-pH 8.0, 200 μg/ml lysozyme) then left on ice for 1 hour. Dithiothreitol (DTT) was added to a concentration of 0.5 mM, and cells were incubated at room temperature for 40 minutes. Aliquots of 1 ml were transferred to 1.5-ml microfuge tubes, then spun and resuspended two times in spheroplast buffer to remove the DTT. Experimental samples received m-polyethylene glycol (PEG) maleimide (Laysan Bio Inc., average MW 1000) to a final concentration of 0.2 mM, added from a 20 mM stock in DMSO, then were incubated at room temperature for 1 hour. Cells were washed by centrifugation two more times, then sonicated on ice (Branson model 450, 30 pulses, power level 3, 50% duty cycle). The lysed cells were pelleted and re-suspended in 150 μl 2x SDS-PAGE reducing buffer. Electrophoresis used 15% polyacrylymide gels run overnight at 4 °C at relatively low voltage (80 volts). Proteins were transferred to nitrocellulose membrane using a semi-dry transfer apparatus (BioRad) at 25 volts for 50 minutes. Membranes were incubated in 5% milk for 30 minutes, washed several times with 1x TBS, then incubated in the primary antibody (anti-HA, 1:1000 dilution) overnight. The membrane was washed with TBS 3 times, and then incubated in the secondary antibody (1:10,000 dilution) for 1.5 hours. The membrane was washed with TBS three more times then scanned with a LI-COR Odyssey scanner to visualize FliP and its PEG-modified adduct.
Growth inhibition assays
Cells were cultured overnight at 32° C in TB medium containing appropriate antibiotics. Cultures were diluted 50-fold into the same media, with or without salicylate (10 μM) to induce expression of plasmid-borne fliP genes, then cultured for 3.5 h at 32° C. These mid-log cultures were then diluted 50-fold into the same media (again with or without salicylate) containing the growth-inhibiting agent at the concentrations indicated in the figures. OD600 was measured after an additional 3.5 h of growth at 32° C. In the experiment measuring effects of the pore-blocking FliK-Top7 construct, the construct was expressed from the chromosomal ara locus. Arabinose was omitted from the overnight cultures but was added to the induced cultures (to .01%) during both of the 3.5-hr periods of re-growth.
Growth curves
Growth curves (Figures 3D and S5) were carried out in TB medium at 37° C, using 1:100 dilutions of saturated cultures grown at 32° C. Where indicated, cultures were induced with 1.5 μM salicylate. Following an initial growth of about 2 h, cultures were split into equal volumes, and the stressing agent (Cd2+, Mn2+, or ethanol) added to one of the cultures, to the concentration indicated in the legends.
Soft-agar motility assay
Cells were cultured overnight at 32° C in TB medium with the appropriate antibiotics, then 3 μL of the saturated culture was spotted onto motility plates. Plates were incubated at 32° C and photographed at the times indicated in the figures. For the assay of inter-subunit complementation, motility plates were directly inoculated with 3 independent pickates from a fresh transformation plate.
Water movement following osmotic upshift
Cells were cultured overnight at 32° C in TB with appropriate antibiotics, in the presence or absence of 10 μM salicylate to induce expression of plasmid-borne fliP. Cultures were diluted 100-fold into the same media (again with or without salicylate) and cultured for 3.5 h at 32° C. Cells were collected by centrifugation and resuspended in an equal volume of PBS, then re-pelleted and suspended in half the original volume of PBS. Some experiments were carried out using the same medium but with KCl omitted and KH2PO4 replaced with NaH2PO4, to test for the potential involvement of K+ uptake systems; equivalent results were obtained in this nominally K+-free PBS. Samples were diluted two-fold into PBS, OD600 measured, then adjusted to equal densities using PBS. To examine responses to osmotic upshift, samples were diluted 2-fold into PBS containing 1 M salt (to give a final salt concentration of 0.5 M), mixed by pipetting, and placed in the spectrophotometer for measurement of OD600 through a time-course of 8-10 minutes.
Supplementary Material
Acknowledgments
We thank Lynette Scott and Lauren Williams for assistance with growth-impairment assays and Takanori Hirano and Mayukh Sarkar for strains and plasmids. Supported by NIH grants R01-GM64664 (to D.F.B.) and GM087260Z (to D.F.B. and K.T.H), Deutsche Forschungsgemeinschaft (DFG) research grant no. ER 778/2-1 (to M.E.), and a fellowship from the Alexander von Humboldt foundation (to T.T.R.).
Footnotes
The authors declare no conflict of interest.
References
- 1.Berg HC, Anderson RA. Bacteria swim by rotating their flagellar filaments. Nature. 1973;245:380–2. doi: 10.1038/245380a0. [DOI] [PubMed] [Google Scholar]
- 2.Larsen SH, Adler J, Gargus JJ, Hogg RW. Chemomechanical coupling without ATP: the source of energy for motility and chemotaxis in bacteria. Proc Natl Acad Sci USA. 1974;71:1239–43. doi: 10.1073/pnas.71.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hirota N, Imae Y. Na+-driven flagellar motors of an alkalophilic Bacillus strain YN-1. J Biol Chem. 1983;258:10577–81. [PubMed] [Google Scholar]
- 4.DePamphilis ML, Adler J. Purification of intact flagella from Escherichia coli and Bacillus subtilis. J Bacteriol. 1971;105:376–83. doi: 10.1128/jb.105.1.376-383.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morimoto YV, Minamino T. Structure and function of the bi-directional bacterial flagellar motor. Biomolecules. 2014;4:217–34. doi: 10.3390/biom4010217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macnab RM. How bacteria assemble flagella. Ann Rev Microbiol. 2003;57 doi: 10.1146/annurev.micro.57.030502.090832. (epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 7.Macnab RM. Type III flagellar protein export and flagellar assembly. Biochim Biophys Acta. 2004;1694:207–17. doi: 10.1016/j.bbamcr.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Brown J, Faulds-Pain A, Aldridge P. The coordination of flagellar gene expression and the flagellar assembly pathway. In: Jarrell K, editor. Pili and Flagella: Current Research and Future Trends. Norfolk: Caister Scientific Press; 2009. p. 238. [Google Scholar]
- 9.Aizawa SI. What is essential for flagellar assembly? In: Jarrell K, editor. Pili and Flagella: Current Research and Future Trends. Norfolk: Caister Academic Press; 2009. p. 238. [Google Scholar]
- 10.Chevance FFV, Hughes KT. Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol. 2008;6:455–65. doi: 10.1038/nrmicro1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu X, Matsumura P. The FlhD/FlhC complex, a transcriptional activator of the Escherichia coli flagellar class II operons. J Bacteriol. 1994;176:7345–51. doi: 10.1128/jb.176.23.7345-7351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science. 1993;262:1277–80. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- 13.Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4:811–25. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 14.Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Reviews. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macnab RM. The bacterial flagellum: reversible rotary propellor and type III export apparatus. J Bacteriol. 1999;181:7149–53. doi: 10.1128/jb.181.23.7149-7153.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blocker A, Komoriya K, Aizawa S. Type III secretion systems and bacterial flagella: insights into their function from structural similarities. Proc Natl Acad Sci U S A. 2003;100:3027–30. doi: 10.1073/pnas.0535335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iino T. Assembly of Salmonella flagellin in vitro and in vivo. J Supramol Struct. 1974;2:372–84. doi: 10.1002/jss.400020226. [DOI] [PubMed] [Google Scholar]
- 18.Renault TT, Abraham AO, Bergmiller T, Paradis G, Rainville S, Charpentier E, et al. Bacterial flagella grow through an injection-diffusion mechanism. eLife. 2017;6:e23135. doi: 10.7554/eLife.23136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schlumberger MC, Müller AJ, Ehrbar K, Winnen B, Duss I, Stecher B, et al. Real-time imaging of type III secretion: Salmonella SipA injection into host cells. Proc Natl Acad Sci USA. 2005;102:12548–53. doi: 10.1073/pnas.0503407102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aizawa SI, Vonderviszt F, Ishima R, Akasaka K. Termini of Salmonella flagellin are disordered and become organized upon polymerization into flagellar filament. J Mol Biol. 1990;211:673–7. doi: 10.1016/0022-2836(90)90064-S. [DOI] [PubMed] [Google Scholar]
- 21.Parsot C, Hamiaux C, Page AL. The various and varying roles of specific chaperones in type III secretion systems. Curr Opin Microbiol. 2003;6:7–14. doi: 10.1016/s1369-5274(02)00002-4. [DOI] [PubMed] [Google Scholar]
- 22.Aldridge PD, Karlinsey JE, Aldridge C, Birchall C, Thompson D, Yagasaki J, et al. The flagellar-specific transcription factor, σ28, is the Type III secretion chaperone for the flagellar-specific anti-σ28 factor FlgM. Genes Dev. 2006;20:2315–26. doi: 10.1101/gad.380406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morimoto YV, Ito M, Hiraoka KD, Che YS, Bai F, Kami-Ike N, et al. Assembly and stoichiometry of FliF and FlhA in Salmonella flagellar basal body. Mol Microbiol. 2014;91:1214–26. doi: 10.1111/mmi.12529. [DOI] [PubMed] [Google Scholar]
- 24.Barker CS, Meshcheryakova IV, Kostyukova AS, Samatey FA. FliO regulation of FliP in the formation of the Salmonella enterica flagellum. PLoS Genetics. 2010;6 doi: 10.1371/journal.pgen.1001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minamino T, Macnab RM. Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol. 1999;181:1388–94. doi: 10.1128/jb.181.5.1388-1394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilharm G, Lehmann V, Krauss K, Lehnert B, Richter S, Ruckdeschel K, et al. Yersinia enterocolitica type III secretion depends on the proton motive force but not on the flagellar motor components MotA and MotB. Infect and Immun. 2004;72:4004–9. doi: 10.1128/IAI.72.7.4004-4009.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul K, Erhardt M, Hirano T, Blair DF, Hughes KT. Energy source of flagellar type III secretion. Nature. 2008;451:489–92. doi: 10.1038/nature06497. [DOI] [PubMed] [Google Scholar]
- 28.Minamino T, Namba K. Distinct roles of the FliI ATPase and proton motive force in bacterial flagellar protein export. Nature. 2008;451:485–8. doi: 10.1038/nature06449. [DOI] [PubMed] [Google Scholar]
- 29.Morimoto YV, Kami-Ike N, Miyata T, Kawamoto A, Kato T, Namba K, et al. High-resolution pH imaging of living bacterial cells to detect local pH differences. mBio. 2016;7:e01911–16. doi: 10.1128/mBio.01911-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minamino T, Morimoto YV, Hara N, Namba K. An energy transduction mechanism used in bacterial flagellar type III protein export. Nat Commun. 2011;2 doi: 10.1038/ncomms1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erhardt M, Wheatley P, Kim EA, Hirano T, Zhang Y, Sarkar MK, et al. Mechanism of type-III protein secretion: Regulation of FlhA conformation by a functionally critical charged-residue cluster. Mol Microbiol. 2017;104:234–49. doi: 10.1111/mmi.13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minamino T, Morimoto YV, Hara N, Aldridge PD, Namba K. The bacterial flagellar type III export gate complex is a dual fuel engine that can use both H+ and Na+ for flagellar protein export. PLOS Pathogens. 2016;12:e1005495. doi: 10.1371/journal.ppat.1005495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dietsche T, Mebrhatu MT, Brunner MJ, Abrusci P, Yan J, Franz-Wachtel M, et al. Structural and functional characterization of the bacterial type III secretion export apparatus. PLOS Pathogens. 2016 doi: 10.1371/journal.ppat.1006071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan F, Ohnishi K, Francis NR, Macnab RM. The FliP and FiR proteins of Salmonella typhimurium, putative components of the type III flagellar export apparatus, are loated in the flagellar basal body. Mol Microbiol. 1997;26:1035–46. doi: 10.1046/j.1365-2958.1997.6412010.x. [DOI] [PubMed] [Google Scholar]
- 35.Fukumura T, Furukawa Y, Kawaguchi T, Saijo-Hamano Y, Namba K, Imada K, et al. Crystallization and preliminary x-ray analysis of the periplasmic domain of FliP, an integral membrane component of the bacterial flagellar type III protein-export apparatus. Acta Crystallogr F Struct Biol Commun. 2014;70 doi: 10.1107/S2053230X14014678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–8. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 37.Mallo RC, Ashby MT. AqpZ-mediated water permeability in Escherichia coli measured by stopped-flow spectroscopy. J Bacteriol. 2006;188:820–2. doi: 10.1128/JB.188.2.820-822.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jones CJ, Macnab RM. Flagellar assembly in Salmonella typhimurium: analysis with temperature-sensitive mutants. J Bacteriol. 1990;172:1327–39. doi: 10.1128/jb.172.3.1327-1339.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Homma M, Kutsukake K, Hasebe M, Iino T, Macnab RM. FlgB, FlgC, FlgF and FlgG. A family of structurally related proteins in the flagellar basal body of Salmonella typhimurium. J Mol Biol. 1990;211:465–77. doi: 10.1016/0022-2836(90)90365-S. [DOI] [PubMed] [Google Scholar]
- 40.Zilkenat S, Franz-Wachtel M, Stierhof YD, Galán JE, Macek B, Wagner S. Determination of the stoichiometry of the complete bacterial tpe III secretion needle complex using a combined quantitative proteomic approach. Mol Cell Proteomics. 2016;15:1598–609. doi: 10.1074/mcp.M115.056598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karlinsey JE. lambda-Red genetic engineering in Salmonella enterica serovar Typhimurium. Methods Enzymol. 2007;421:199–209. doi: 10.1016/S0076-6879(06)21016-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.