

Abstract
Combined liver/kidney transplant is the preferred transplant option for most patients with primary hyperoxaluria type 1 (PH1) since orthotopic liver transplantation replaces the deficient liver-specific AGT enzyme, thus restoring normal metabolic oxalate production. However, primary hyperoxaluria type 2 (PH2) is caused by deficient glyoxylate reductase / hydroxypyruvate reductase (GRHPR), and this enzyme is widely distributed throughout the body. Though the relative abundance and activity of GRHPR in various tissues is not clear, some evidence suggests that the majority of enzyme activity may indeed reside within the liver. Thus the effectiveness of liver transplantation in correcting this metabolic disorder has not been demonstrated. Here we report a case of 44-year old man with PH2, frequent stone events, and end-stage renal disease who received a combined liver/ kidney transplant. Although requiring confirmation in additional cases, the normalization of plasma oxalate, urine oxalate and urine glycerate levels observed in this patient within a month of the transplant that remain reduced at the most recent follow-up at 13 months suggests correction of the GRHPR deficiency in PH2 can be achieved by liver transplantation.
Introduction
The primary hyperoxalurias are rare inborn errors of glyoxylate metabolism that result in endogenous overproduction and increased renal excretion of oxalate. Complications include nephrocalcinosis, recurrent kidney stones, chronic kidney disease and systemic oxalosis. Primary hyperoxaluria type 1 (PH1) is the most common form of PH and is caused by mutations in the AGXT gene which encodes the liver-specific enzyme alanine/glyoxylate aminotransferase (AGT). Orthotopic liver transplantation provides complete correction of the metabolic deficiency such that combined liver/kidney transplant is the preferred transplant option for most patients with PH1 and end-stage renal disease. In contrast, primary hyperoxaluria type 2 (PH2) generally has a milder clinical course with better preservation of kidney function than PH1 in both native kidneys and renal allografts1. PH2 is caused by mutations in the GRHPR gene and the resulting deficiency of the glyoxylate reductase/hydroxypyruvate reductase (GRHPR) enzyme. In the setting of decreased GRHPR enzyme activity, glyoxylate is aberrantly converted into oxalate resulting in hyperoxaluria. Unlike AGT, GRHPR expression is not limited to the liver; rather it has been detected in multiple tissues throughout the body2, 3. However, the relative amounts of oxalate produced in the liver versus extrahepatic tissues are not well defined. Thus it has been unclear whether liver transplantation can restore oxalate production to normal in patients with PH2. For this reason, along with the milder disease expression, combined kidney and liver transplantation has not been performed in PH2. Certain PH2 patients, however, demonstrate more severe disease, and could potentially benefit from replacement of the deficient enzyme, if indeed liver transplantation substantially reduces oxalate production.
Case description
A 42 year old Caucasian man with PH2 presented to our center with worsening renal allograft function. His medical history was significant for a congenital solitary kidney and recurrent episodes of kidney stones beginning at 6 years of age. As a child he did not receive any diagnostic work-up or stone prophylaxis treatment. His next symptomatic stone event occurred at the age of 37 years when he was diagnosed with a large obstructive stone complicated by acute renal failure with a serum creatinine greater than 4 mg/dL. Despite stone removal, renal function worsened over the next few months. He ultimately received a preemptive living-related kidney transplant from his brother at an outside facility. He did not receive hemodialysis in the post-transplant period. Sudden worsening of kidney function 6 months after transplant prompted a renal allograft biopsy that revealed extensive deposition of calcium oxalate crystals. Twenty-four hour urine oxalate and glycerate excretions were elevated, as was the plasma oxalate level. The patient’s post-transplant course was complicated by frequent and debilitating stone events and progressively worsening renal allograft function.
Upon presentation to our center 5 years after living related kidney transplant, the patient’s serum creatinine was 4.2 mg/dL and iothalamate clearance was 19 ml/min/1.73 m2. A renal allograft biopsy again demonstrated extensive deposition of calcium oxalate crystals without evidence of rejection (Figure 1). Twenty-four hour urine oxalate excretion was markedly increased (180 mg; normal <40.5 mg; Table 1). A random urinary metabolic screen revealed increased urine glycerate (173 mg/g creatinine; normal <25 mg/g creatinine) and normal urine glycolate (5 mg/g creatinine; normal <50 mg/g creatinine). Plasma oxalate was also elevated (22 µmol/L; normal < 1.8 µmol/L). These findings suggested a diagnosis of PH2, which was confirmed after subsequent genetic testing revealed a homozygous mutation of the GRHPR gene (c.139C>T: p.R47X). Additional genetic testing of family members revealed both parents to be heterozygous carriers of the mutation, while it was confirmed that his brother (kidney donor for first kidney transplant) was unaffected. In addition, his brother’s 24 hour urine oxalate measured at this time was normal (16.7 mg /24 hours).
Figure 1.
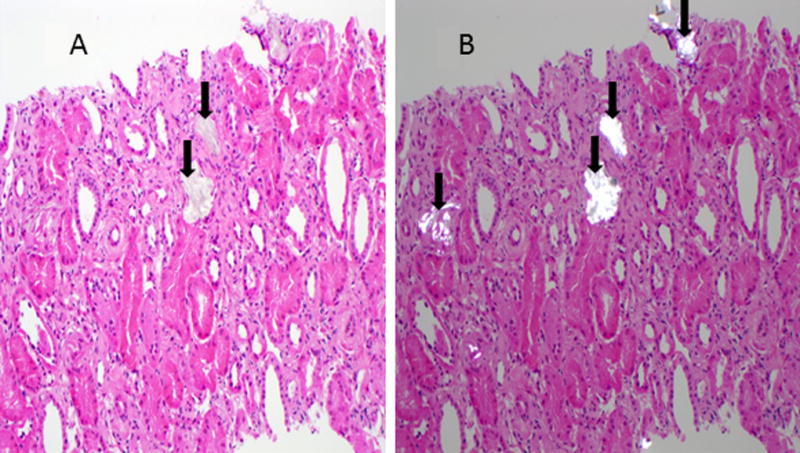
Biopsy of kidney- alone transplant. Calcium oxalate crystals (arrows) are apparent in H&E section (Panel A) and are birefringent under polarizing light (Panel B).
Table 1.
Urine oxalate, creatinine, glycerate levels, pre and post combined liver-kidney transplant
| Pre/ post- transplant day |
-316 (pre- transplant) |
-58 (pre- transplant) |
‘0’ Transplant Day |
21 (post- transplant) |
38 (post- transplant) |
151 (post- transplant) |
384 (post- transplant) |
|---|---|---|---|---|---|---|---|
| 24 hour urine oxalate (mg) normal<40.5 | 136 | 180 | 12 | 37 | 34 | 46 | |
| 24 hour urine creatinine (mg) | 1794 | 2030 | 578 | 1193 | 1378 | 2204 | |
| Random urine glycerate (mg/dL) | 16.781 | 0.304 | 0.436 | 0.308 | |||
| Urine glycerate/ creatinine (mg/g) normal< 25 | 173 | 4 | 4 | 4 | |||
| Plasma oxalate (µmol/L) normal <1.8 | 26.8 | 23.8 | 3.0 | 2.1 | <1.0 | ||
| Serum creatinine (mg/dl) normal <1.4 | 3.5 | 3.8 | 3.9 | 1.6 | 1.6 | 1.4 | 1.3 |
The patient was advised to maintain high fluid intake with a goal urine output of 3.5 to 4 L/day and started on sodium citrate/citric acid solution (500mg-334mg/5ml) 30 ml twice daily. Despite adhering to these treatments he continued to pass frequent kidney stones (3/week on average). A renal stone protocol CT scan revealed multiple non obstructive calyceal tip stones in both the renal allograft and the solitary atrophic native kidney. Analysis confirmed a recently passed stone was composed of 100% calcium oxalate monohydrate. No other systemic manifestations of oxalosis were identified. Specifically, no evidence of oxalate was found on retinal exam, and he had no history of peripheral neuropathy or cardiac disease. The patient was minimally sensitized, despite his history of prior renal transplant, with a calculated PRA of 0%. After careful evaluation by a multidisciplinary team, and full discussion with the patient, he was listed for combined liver/kidney transplant due to his relatively rapid loss of his first renal allograft from oxalate nephropathy and history of frequent and debilitating stone events.
A year after approval, the patient received a preemptive, negative crossmatch combined liver/kidney transplant. He received thymoglobulin induction followed by maintenance immunosuppression with tacrolimus, mycophenolate mofetil and prednisone (tapered off at 4 months per protocol). Surgery was complicated by hepatic artery thrombosis requiring surgical thrombectomy and revision of hepatic artery anastomosis 2 hours later. He also developed a cystic duct leak requiring reoperation. His post-transplant course was further complicated by delayed graft function requiring continuous renal replacement therapy for 3 days followed by 6 days of daily intermittent hemodialysis. Plasma oxalate level was monitored daily after transplant, and decreased from a pre-transplant level of 23.8 µmol/L to 2 µmol/L by post- transplant day 29 (Figure 2). Twenty-four hour urine oxalate excretion decreased from 180 mg before transplant to 37 mg by post-transplant day 38 (Table 1) and urine glycerate level decreased from 173 mg/g creatinine (pre-transplant) to 4 mg/g creatinine (post-transplant) (Table 1). Renal function also improved with serum creatinine falling from 4.1 mg/dL pre-transplant to 1.6 mg /dl by post-transplant day 29. Liver allograft function normalized completely by post-transplant day 20.
Figure 2.
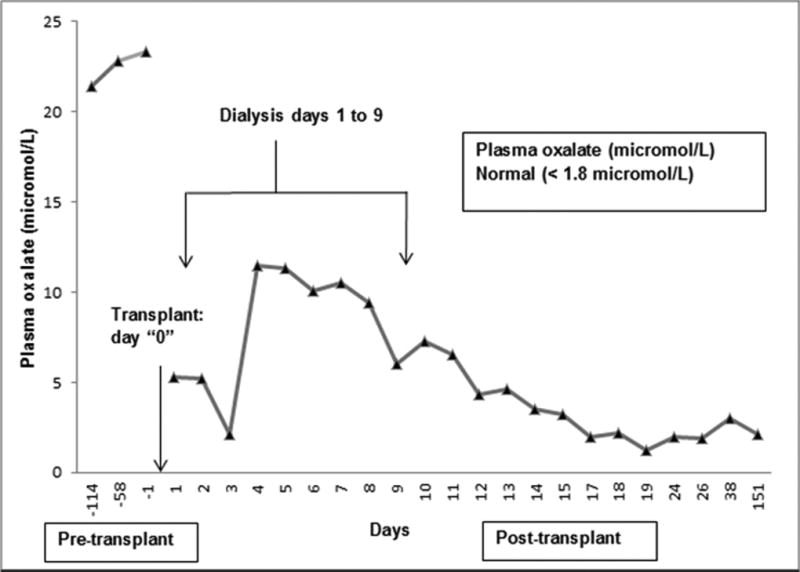
Change in plasma oxalate level pre and post combined Liver-Kidney transplant (Normal plasma oxalate level <1.8 µmol/L).
Four months after transplant, a protocol renal allograft biopsy showed only mild tubular atrophy and interstitial fibrosis (20% of sampled cortex) without evidence of calcium oxalate crystals or rejection. Serum creatinine at that time was 1.4 mg/dL with a measured iothalamate clearance of 78 ml/min/1.73m2. Plasma oxalate level was 2.1µmol/L (normal <1.8 µmol/L), and 24 hour urine oxalate and random urine glycerate were both normal at 34 mg/24 hours and 4 mg/g creatinine, respectively (Table 1). At the last follow-up 384 days post-transplant his plasma oxalate (<1 µmol/L), urine oxalate (46 mg/24 hours) and glycerate (4 mg/g creatinine) remain markedly reduced. Serum creatinine was stable at 1.3 mg/dl and a 1 year protocol renal allograft biopsy did not show any calcium oxalate crystals or rejection. The 60% increase in 24 urine creatinine excretion between 4 and 13 months is consistent with either over collection of urine, regain of muscle mass with post-transplant recovery or combination of both.
Discussion
PH2 is an autosomal recessive disorder caused by mutations in GRHPR that result in endogenous overproduction of oxalate. So far, more than 30 different disease-causing mutations of the GRHPR gene have been described 4. Genetic testing in our patient confirmed a homozygous deleterious mutation of the GRHPR gene (c.139C>T: p.R47X). In PH2, mutations in the GRHPR gene and the consequent absence or deficiency of GRHPR enzyme activity result in increased urinary excretion of oxalate which in turn leads to nephrocalcinosis and recurrent calcium oxalate kidney stones.
PH2 is generally believed to have a milder clinical course than PH1 with a lower risk of ESRD5, 6. The classic presentation of PH2 is recurrent calcium oxalate kidney stones and associated complications, including urinary tract infection and ureteral obstruction7. Our patient had his first episode of nephrolithiasis requiring urologic stone removal at 6 years of age. He then had relatively few stone episodes until he developed an obstructive stone requiring urologic removal and ESRD at the age of 37 years. His history of having a congenital solitary kidney and /or delayed diagnosis of PH2 might have increased the risk of ESRD in light of the observation that many PH2 patients do reasonably well with aggressive medical management targeted to decrease the supersaturation of calcium oxalate in the urine. Typical therapeutic measures include sufficient fluid intake to achieve a urine volume of at least 3 L/day/1.73m2 and treatment with crystallization inhibitors (orthophosphate or citrate). Unlike PH1, there is no role for pyridoxine use in PH2 patients4, 6.
Since oxalate is largely cleared by the kidneys, plasma oxalate levels rise as GFR declines. When the plasma oxalate level reaches 35–50 µmol/L, the supersaturation threshold for calcium oxalate crystallization in the blood is exceeded and crystals can deposit in bone, myocardium, vessels, skin, and retina. Although less common in PH2 (compared to PH1), cases of systemic oxalosis have been described when PH2 patients developed advanced CKD or ESRD8, 9. Plasma oxalate must be closely monitored in all PH patients with declining GFR, with initiation of renal replacement therapy when the plasma oxalate level approaches the supersaturation threshold. After transplant, hemodialysis is often required in cases of delayed graft function to keep the plasma oxalate level below supersaturation threshold until renal allograft function improves. Typically, kidney transplant alone is the preferred option for PH2 patients who develop ESRD1, 10 although loss of renal allograft due to recurrence of oxalate nephropathy, as occurred in our patient, has been reported11.
GRHPR mRNA has been detected in multiple body tissues outside of the liver. Thus it has been unclear that liver transplantation would provide sufficient metabolic correction for the benefit to outweigh the risks. Further there is evidence of renal GRHPR activity12, raising the question whether a solitary liver transplant would entirely prevent hyperoxaluria in PH2 patients. However, Cregeen et al. reported that the greatest concentration of GRHPR mRNA was in liver, with significantly lesser amounts in kidneys and leucocytes13. Similarly, Giafi and Rumsby performed tissue distribution analysis and demonstrated that the liver was the predominant site of the GRHPR enzyme3.
The factors that influenced our decision to proceed with combined liver and kidney transplant in this case included the documented consistently high oxalate excretion, which is predictive of poor renal outcome in all forms of PH14. The patient had frequently recurrent painful stone events following kidney alone transplant despite good compliance with the medical management plan. Further he developed oxalate nephropathy in the first allograft which caused loss of function requiring repeat kidney transplantation. His subsequent post-transplant course and laboratory results suggest successful replacement of GRHPR enzyme that resulted in normalization of plasma oxalate concentration, and urine oxalate and glycerate excretion. However, liver transplantation is never without risk as the perioperative complications in this case attest. Thus, the role of combined liver/kidney transplant in patients with PH2 and ESRD requires further study, and careful consideration of the risks and benefits on an individualized basis.
Acknowledgments
We thank and acknowledge the help of Julie B. Olson RN in getting the family members of the proband getting tested for the GRHPR mutation and obtaining urinary oxalate data of the PH2 carriers in our registry. This study was supported by the Rare Kidney Stone Consortium (U54KD083908), a member of the NIH Rare Diseases Clinical Research Network (RDCRN), funded by the NIDDK and the National Center for Advancing Translational Sciences (NCATS), the Mayo Hyperoxaluria Center, and the Mayo Foundation.
Abbreviations
- PH 1 and PH2
Primary Hyperoxaluria type 1 and 2
- AGT
Alanine glyoxylate aminotransferase
- GR/HPR
Glyoxylate reductase hydroxy pyruvate reductase
- CKD
Chronic Kidney Disease
- ESRD
End stage renal disease
- Pox
Plasma oxalate
- UOx
Urine oxalate
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Bergstralh EJ, Monico CG, Lieske JC, et al. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10(11):2493–2501. doi: 10.1111/j.1600-6143.2010.03271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams HE, Smith LH., Jr Hyperoxaluria in L-glyceric aciduria: possible pathogenic mechanism. Science. 1971;171(3969):390–391. doi: 10.1126/science.171.3969.390. [DOI] [PubMed] [Google Scholar]
- 3.Giafi CF, Rumsby G. Kinetic analysis and tissue distribution of human D-glycerate dehydrogenase/glyoxylate reductase and its relevance to the diagnosis of primary hyperoxaluria type 2. Ann Clin Biochem. 1998;35(Pt 1):104–109. doi: 10.1177/000456329803500114. [DOI] [PubMed] [Google Scholar]
- 4.Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med. 2013;369(7):649–658. doi: 10.1056/NEJMra1301564. [DOI] [PubMed] [Google Scholar]
- 5.Johnson SA, Rumsby G, Cregeen D, Hulton SA. Primary hyperoxaluria type 2 in children. Pediatr Nephrol. 2002;17(8):597–601. doi: 10.1007/s00467-002-0858-6. [DOI] [PubMed] [Google Scholar]
- 6.Milliner DS, Wilson DM, Smith LH. Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int. 2001;59(1):31–36. doi: 10.1046/j.1523-1755.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- 7.Kemper MJ, Muller-Wiefel DE. Nephrocalcinosis in a patient with primary hyperoxaluria type 2. Pediatr Nephrol. 1996;10(4):442–444. doi: 10.1007/s004670050135. [DOI] [PubMed] [Google Scholar]
- 8.Wachter R, Schulze MR, Schmeisser A, Fischer R, Strasser RH. Images in cardiovascular medicine. Cardiomyopathy resulting from primary hyperoxaluria type II. Circulation. 2006;113(3):e39–40. doi: 10.1161/CIRCULATIONAHA.104.517136. [DOI] [PubMed] [Google Scholar]
- 9.Wichmann G, Passauer J, Fischer R, Weise M, Gross P. A young patient with end-stage renal disease, dyspnoea, weakness, peripheral neuropathy and an unsuspected underlying disease. Nephrol Dial Transplant. 2003;18(8):1670–1672. doi: 10.1093/ndt/gfg208. [DOI] [PubMed] [Google Scholar]
- 10.Filler G, Hoppe B. Combined liver-kidney transplantation for hyperoxaluria type II? Pediatr Transplant. 2014;18(3):237–239. doi: 10.1111/petr.12243. [DOI] [PubMed] [Google Scholar]
- 11.Naderi G, Latif A, Tabassomi F, Esfahani ST. Failure of isolated kidney transplantation in a pediatric patient with primary hyperoxaluria type 2. Pediatr Transplant. 2014;18(3):E69–73. doi: 10.1111/petr.12240. [DOI] [PubMed] [Google Scholar]
- 12.Knight J, Holmes RP, Cramer SD, Takayama T, Salido E. Hydroxyproline metabolism in mouse models of primary hyperoxaluria. Am J Physiol Renal Physiol. 2012;302(6):F688–693. doi: 10.1152/ajprenal.00473.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cregeen DP, Williams EL, Hulton S, Rumsby G. Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat. 2003;22(6):497. doi: 10.1002/humu.9200. [DOI] [PubMed] [Google Scholar]
- 14.Zhao F, Bergstralh EJ, Mehta RA, et al. Predictors of Incident ESRD among Patients with Primary Hyperoxaluria Presenting Prior to Kidney Failure. Clin J Am Soc Nephrol. 2016;11(1):119–126. doi: 10.2215/CJN.02810315. [DOI] [PMC free article] [PubMed] [Google Scholar]