

Abstract
Objective
Obesity is a major risk factor for multiple diseases and is in part heritable, yet the majority of causative genetic variants that drive excessive adiposity remain unknown. Here, we used outbred heterogeneous stock (HS) rats in controlled environmental conditions to fine-map novel genetic modifiers of adiposity.
Methods
Body weight and visceral fat pad weights were measured in male HS rats that were also genotyped genome-wide. Quantitative trait loci (QTL) were identified by genome-wide association of imputed single nucleotide polymorphism (SNP) genotypes using a linear mixed effect model that accounts for unequal relatedness between the HS rats. Candidate genes were assessed by protein modeling and mediation analysis of expression for coding and noncoding variants, respectively.
Results
HS rats exhibited large variation in adiposity traits, which were highly heritable and correlated with metabolic health. Fine-mapping of fat pad weight and body weight revealed three QTL and prioritized five candidate genes. Fat pad weight was associated with missense SNPs in Adcy3 and Prlhr and altered expression of Krtcap3 and Slc30a3, whereas Grid2 was identified as a candidate within the body weight locus.
Conclusions
These data demonstrate the power of HS rats for identification of known and novel heritable mediators of obesity traits.
Keywords: Quantitative Trait Loci, adenylate cyclase, rat models, adipose tissue, body weight
Introduction
Obesity and overweight are major risk factors for multiple cardiovascular and metabolic diseases [1]. Of particular importance is visceral, or abdominal, adipose tissue, which is strongly predictive of metabolic health [2]. Multiple environmental (e.g., lifestyle) and genetic factors contribute to obesity with genetics accounting for up to 70% of the population variance for human body mass index (BMI) and obesity [3] and visceral adiposity [4]. To date, human genome-wide association studies have identified many genes for anthropomorphic traits [5-8], but these genes explain only a small proportion of the heritable variation [5], indicating many genes are yet unidentified. Identification of additional genes is particularly important because there has been a steady increase in prevalence of overweight and obesity since the 1970's [1], with over one third of adults and almost one fifth of all children in the United States being classified as obese [9].
One strategy for identifying the heritable modifiers of obesity is to control for exogenous environmental factors using experimental genetic mapping strategies such as the outbred heterogeneous stock (HS) rats. HS rats descend from eight inbred founder strains and have been out-bred for over 70 generations, such that the fine recombination block structure allows genetic mapping to identify regions that are only a few Mb [10]. In previous work, we used HS rats to fine-map a single region on rat chromosome 1 previously identified for glucose and insulin traits [11, 12], and identified Tpcn2 as a likely causal gene at this locus [13]. Here, we demonstrate that HS rats vary for adiposity traits including body weight and visceral fat pad weight, and that these measures correlate with metabolic health. We then detect and fine-map QTL for these traits genome-wide and identify five likely causal genes within these loci.
Methods and Procedures
Animals
Heterogeneous stock colony
The NMcwi:HS colony, hereafter referred to as HS, was initiated by the NIH in 1984 using the following eight inbred founder strains: ACI/N, BN/SsN, BUF/N, F344/N, M520/N, MR/N, WKY/N and WN/N [14]. This colony has been maintained at the Medical College of Wisconsin since 2006 and has been through over 70 generations of breeding. Rats were given ad libitum access to Teklad 5010 diet (Harlan Laboratories). Additional housing conditions are detailed in Supplementary Methods.
Founding inbred sub-strains
Other than M520/N (now maintained at MCW), phenotyping of the founders was conducted in the following sub-strains (abbreviated names to be used throughout manuscript in parentheses): ACI/Eur or ACI/Seg (ACI), BN/SsnHsd (BN), BUF/NHsd (BUF), F344/NHsd (F344), and WKY/NHsd (WKY). We tested 8-19 male rats per inbred strain.
Phenotyping protocol
We measured body weight at 16 weeks of age in 989 male HS rats. Rats underwent an intra-peritoneal glucose tolerance test (IPGTT) as described previously [11, 12]. We used the Ascensia Elite system for reading blood glucose values (Bayer, Elkhart, IN). Plasma insulin levels were determined using an ultrasensitive ELISA kit (Alpco Diagnostics, Salem, NH). The following metabolic measures were calculated: area under the curve for glucose (glucose_AUC) and insulin (insulin_AUC) during the IPGTT, the quantitative insulin sensitivity check (QUICKI) as a measure of insulin sensitivity, and the insulinogenic index (IGI) as a measure of beta cell sensitivity to glucose [12].
Inbreds and 743 HS rats were euthanized after an overnight fast at 17 weeks of age. Body weight and two measures of body length (from nose to base of the tail and from nose to end of tail) were collected, allowing us to calculate two measures of body mass index: BMI_Tail_Base and BMI_Tail_End. BMI was calculated as: (body weight/body length2)×10. Rats were euthanized by decapitation and trunk blood was collected. Fasting cholesterol and triglycerides were determined from fasting serum on an ACE Alera autoanalyzer using an enzymatic method for detection. Several tissues were dissected and weighed including retroperitoneal and epididymal visceral fat pads, hereafter referred to as RetroFat and EpiFat, respectively. Liver and adipose tissues were snap-frozen in liquid nitrogen for subsequent expression analysis. All protocols were approved by the IACUC committee at MCW. Phenotyping data have been deposited in RGD (www.rgd.mcw.edu).
Genotyping
We extracted DNA from tail tissue from HS and the original eight inbred founder strains (tissue obtained from the NIH) using either the Qiagen DNeasy kit (Valencia, CA) or a phenol-chloroform extraction. Founder and HS rats were genotyped using the Affymetrix GeneChip Targeted Genotyping technology on a custom 10K SNP array panel as previously described [15], with marker locations based on rat genome assembly 6.0. 147 samples were genotyped by the Vanderbilt Microarray Shared Resource center at Vanderbilt University in Tennessee (currently VANTAGE: http://www.vmsr.edu) and the remaining 842 by HudsonAlpha Institute (http://hudsonalpha.org). From the 10,846 SNPs on the array, 8,218 were informative and produced reliable genotypes in the HS rats. From these final informative markers, the average SNP spacing was 284 Kb, with an average heterozygosity of 25.68%. Principle Component Analysis was used to confirm there were no systematic genotyping differences between the two centers (Figure S1).
RNAseq
RNA was extracted from liver of 398 HS rats using Trizol. Illumina kits were used to create library preps and RNA-seq was run on the Illumina HiSeq 2500. RSEM and Bowtie were used to align reads and compute transcript level expression abundance (Supplementary Methods).
Statistical Analysis
Estimating heritability of adiposity traits
Narrow-sense heritability was estimated for each transformed phenotype using a Bayesian linear mixed model (LMM) implemented in INLA [16, 17]. The LMM included fixed effects representing time food deprived, order of tissue harvest, and dissector (notably, dissector significantly affected EpiFat and BMI_Tail_Base), and a random “polygenic” effect, which represented the effect of overall relatedness (calculated using [18]). Heritability, h2, was defined as the proportion of variance attributed to polygenic effects vs residual noise (Supplemental Methods).
Genome-wide association
QTL were identified by genome-wide association of imputed allele dosages of genotyped SNPs. A hidden Markov model [19] was used to infer each HS rat's haplotype mosaic and thereby obtain robust estimates of each SNP's genotype. Association tests were then performed, SNP-by-SNP, on each trait using a frequentist version of the LMM described for estimating heritability but with an added SNP effect term. Tests of the SNP effect yielded p-values that are here reported as negative log to the base 10, or “logP”. Genome-wide significance thresholds for logP scores were estimated by parametric bootstrap samples from the fitted null [11, 20]. Linkage Disequilibrium (LD) intervals for the detected QTL were defined by including neighboring markers that met a set level of LD, measured with the squared correlation coefficient r2; we used r2=0.5 to define intervals based on the plots of the SNP associations overlaid with LD information (Supplemental Methods).
Fine-mapping and haplotype effect estimation at detected QTL
SNP variants within the LD interval were prioritized used the multi-SNP method LLARRMA-dawg [21], which calculates for each SNP a resample model inclusion probability (RMIP): SNPs with high RMIPs represent strong, independent signals, and the existence of multiple SNPs with a high RMIP is consistent with the presence of multiple independent signals. To characterize each QTL signal, we used the Diploffect model [22], which estimates the relative contributions of alternate founder haplotypes (Supplemental Methods).
Candidate gene identification
Two parallel approaches were used: 1) bioinformatic analysis and protein modeling of known sequence variants; and, 2) mediation analysis of expression levels. For (1), we used HS founder sequence (www.rgd.mcw.edu; genome build Rn6) to identify highly conserved, non-synonymous coding variants within each QTL that were predicted to be damaging by Polyphen (http://genetics.bwh.harvard.edu/pph/) and/or SIFT, focusing on variants in founder strains that showed haplotype effects at the locus. Variants were confirmed using Sanger sequencing and then analyzed in the Sequence-to-Structure-to-Function analysis as previously described [23]. Briefly, proteins were assessed with codon selection analysis of multiple species open reading frames, inspected for linear motif impact near variants of interest, and modeled with I-TASSER [24] and YASARA [25]. Models were then assessed for likely impact on protein folding and/or function based on model confidence, phylogenetic sequence alignment, conservation, and whether or not the variant altered structural packing, molecular dynamic simulations, binding partners, linear motifs or post-translational modifications. For (2), transcript abundance levels of genes within HS liver were evaluated as potential causal mediators of the physiological QTL through mediation analysis [26] (Supplementary Methods).
Results
HS founder strains exhibit large variation in adiposity traits
All phenotypes were rank-inverse normal transformed except EpiFat, which instead was log transformed based on the Box-Cox procedure. All traits differed significantly between the inbred founder strains: body weight (F5, 76 = 15.492, p = 1.74e-10), BMI_Tail_End (F5, 73 = 25.024, p = 1.34e-14), BMI_Tail_Base (F5, 73 = 9.683, p = 4.02e-7), EpiFat (F5, 78 = 69.541, p < 2.2e-16) and RetroFat (F5, 78 = 38.157, p < 2.2e-16; Figure 1). The BUF inbred strain had significantly more EpiFat mass (Tukey-Kramer p ≤ 0.05) and BMI_Tail_End (Tukey-Kramer p < 0.05) relative to all other strains. BUF also had significantly more RetroFat mass compared to all strains (Tukey-Kramer p < 0.001) except F344 (Tukey-Kramer p = 0.06175), higher body weight relative to ACI, BN, and M520 (Tukey-Kramer p < 0.01), and higher BMI_Tail_Base than ACI and M520 (Tukey-Kramer p < 0.05). ACI, BN and M520 were the lightest strains, with BN and M520 showing significantly lighter EpiFat (Tukey-Kramer p ≤ 1e-5) and RetroFat (Tukey-Kramer p ≤ 0.001) relative to other strains.
Figure 1.
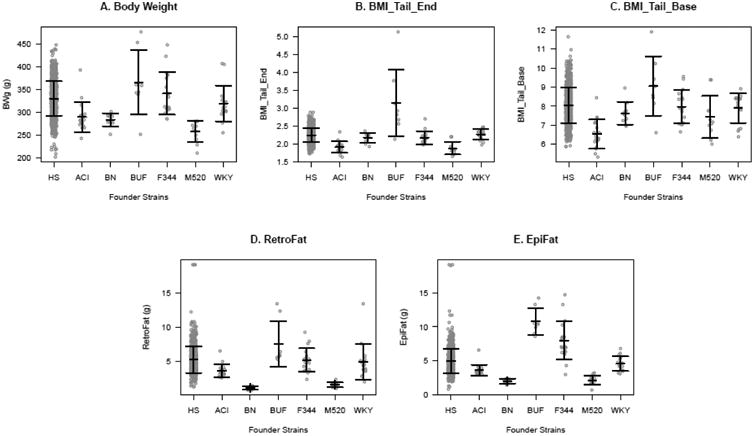
Adiposity traits in inbred founders and HS rats. Mean + SD are shown. BMI is body mass index from nose to end of tail (BMI_Tail_End) and from nose to base of the tail (BMI_Tail_Base). EpiFat and RetroFat are epididymal and retroperitoneal fat pad weight, respectively. Gray circles represent individual animals from 8-19 individuals from 6 of the founder strains, and the HS rats (989 in body weight; 741 in RetroFat, EpiFat, and BMI_Tail_End; and 740 in BMI_Tail_Base). See text for statistical differences between founder strains.
Adiposity traits are highly correlated with measures of metabolic health in HS rats
Variation between the founder strains is represented within the HS colony (Figure 1). Adiposity measures were highly correlated with several measures of metabolic health (Table 1, Figure 2). EpiFat significantly correlated with every measure of metabolic health and RetroFat correlated with all but fasting glucose. Body weight significantly correlated with all measures except fasting triglycerides. BMI_Tail_End significantly correlated with fasting total cholesterol, fasting tricglycerides, glucose AUC, insulin AUC, and IGI, whereas BMI_Tail_Base did not significantly correlate with any of the measures of metabolic health.
Table 1. Correlations between adiposity traits and measures of metabolic health in HS rats.
| Body Weight | BMI Tail End | BMI Tail Base | EpiFat | RetroFat | |
|---|---|---|---|---|---|
| Fasting Glucose | 0.1453 (0.0012) | 0.0952 (0.76) | 0.0886 (1) | 0.1931 (3.24e-05) | 0.1132 (0.1009) |
| Fasting Insulin | 0.1936 (3.48-07) | 0.1153 (0.091) | -0.0248 (1) | 0.4314 (1.35e-31) | 0.3516 (4.75e-27) |
| Fasting Total Cholesterol | 0.2644 (7.10e-11) | 0.2428 (5.75e-09) | 0.1172 (0. 39) | 0.2535 (6.85e-10) | 0.3529 (1.02e-20) |
| Fasting Triglycerides | 0.1291 (0.12) | 0.2426 (6.07e-09) | 0.0950 (1) | 0.3096 (1.75e-15) | 0.3499 (2.55e-20) |
| Glucose AUC | 0.1378 (0.0040) | 0.1259 (0.0214) | 0.0652 (1) | 0.1663 (0.0016) | 0.1718 (1.71e-05) |
| Insulin AUC | 0.2937 (5.67e-18) | 0.2238 (7.99e-10) | 0.0312 (1) | 0.4670 (2.71e-37) | 0.4397 (8.79e-44) |
| QUICKI | -0.2042 (3.91e-08) | -0.1198 (0.0523) | 0.0211 (1) | -0.4338 (5.24e-32) | -0.3544 (1.57e-27) |
| IGI | 0.1629 (0.0001) | 0.1220 (0.0430) | 0.0001 (1) | 0.2475 (5.46e-09) | 0.2038 (5.46e-08) |
Spearman's rank correlation with Bonferonni-adjusted p-values in parentheses (bold if <0.05). To mitigate potentially confounding effects of experimental covariates, correlations are performed on phenotypic residuals (ie, on the residuals after regressing out covariate effects from the rank-inverse normal transformed phenotype).
Figure 2.
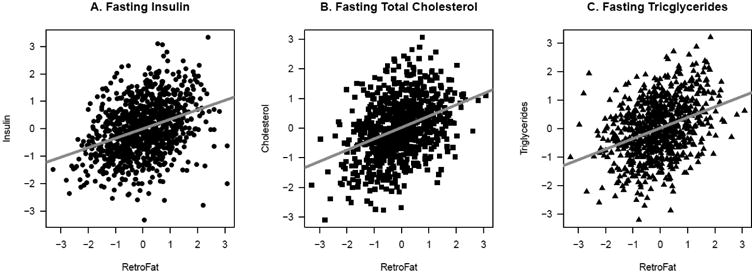
Significant correlations between RetroFat (retroperitoneal fat pad weight) and A) fasting insulin (p = 4.75e-27), B) fasting total cholesterol (p = 1.02e-20) and C) fasting triglycerides (p = 2.55e-20) in HS rats. Plots show the residuals of rank-inverse normal transformed phenotypes with nuisance factors regressed out to restrict correlation estimates to that between RetroFat and these metabolic traits. Significant correlations were also found between RetroFat and several other measures of metabolic health (see Table 1).
Adiposity traits are highly heritable
Adiposity traits were highly heritable in HS rats: body weight (posterior mode of h2 = 75.3%; 95% highest posterior density interval = 67.0-81.7%), EpiFat (54.1%; 40.1-66.0%), RetroFat (53.9%; 39.7-66.7%), BMI_Tail_End (45.0%; 32.3%-57.2%) and BMITailBase (25.4%; 13.6- 41.8%).
RetroFat QTL on chromosomes 1 and 6
Two 90% significant QTL were identified for RetroFat, a QTL on rat chromosome 6: 22.79-28.93 Mb (6.14 Mb, -logP = 4.73) and a QTL on chromosome 1: 280.63 – 281.82 Mb (1.19 Mb, -logP = 4.69; Figure 3ABC, 6ABC). The LLARRMA-dawg multi-SNP fine-mapping analysis narrowed the most likely region of the broader chromosome 6 QTL to 1.46 Mb region (27.17 –28.63 Mb; Figure S2) narrowing the number of the genes from 130 to 30 (Table S1, Figure S3). Estimating founder haplotype effects at the chromosome 6 QTL gave an effect size (posterior median) of 11.05% and showed that at this locus, decreased fat pad weight is associated with the WKY haplotype (Figure 3D). For the chromosome 1 QTL the effect size was 13.33%, with increased fat pad weight associated with BUF, MR and WKY haplotypes (Figure 6D).
Figure 3.
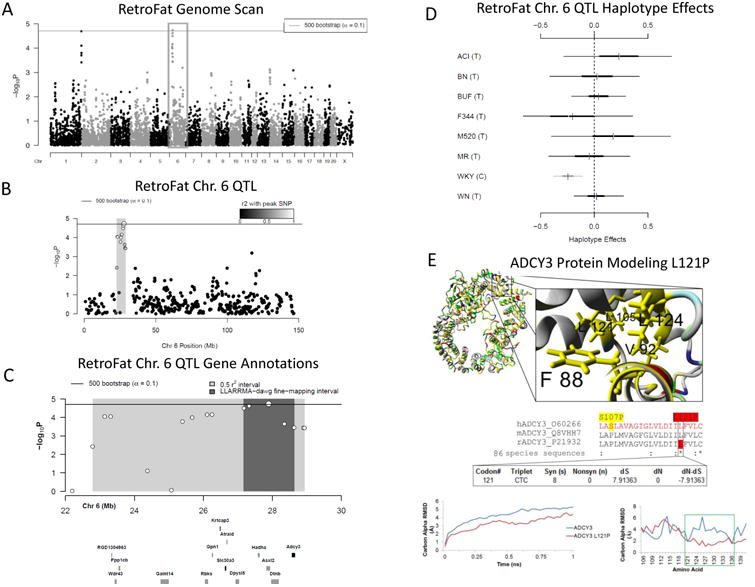
A) Genome scan of RetroFat. X-axis is position on chromosome and y-axis is the −logP level of association. Genome-wide significance thresholds were calculated using parametric bootstraps from the null model (α = 0.1, logP = 4.70). B) The grey region highlights the 6.14 LD support interval of the chromosome 6 QTL showing neighboring markers that are correlated with the peak marker, representing genomic regions likely to contain the causal variant underlying the statistical signal. C) Annotation of genes that fall within the support interval. The entire 6.14 region is shaded in grey, with the fine-mapped 1.46 Mb region shaded in dark grey. Only genes that have a cis eQTL are shown. All 130 genes within the region are listed in Supplementary Table S1. D) Additive haplotype effects were estimated using the Diploffect model, which takes into account uncertainty in haplotype state. SNP allele information is overlaid on the haplotype effects, and are distinguished by black or gray. The WKY haplotype, the only haplotype with the C allele at the chromosome 6 locus, has a significantly negative effect on phenotype. E) Protein modeling for ADCY3. Variant L121P of ADCY3 is found with the conserved hydrophobic core of the transmembrane helices. A zoomed in view is shown to the right. The middle panel shows sequence alignments of amino acids. ADCY3 amino acid 121 is also 100% conserved (red) as a leucine in 86 analyzed vertebrate species. A human SNP is known at amino acid 107 (yellow). Using the DNA information from the 86 nucleotide sequences for ADCY3, there is also evidence of selective pressure in the DNA sequence to conserve the amino acid. Bottom panel shows molecular dynamic simulations for ADCY3. Simulations performed on the protein dimer for wild type (WT blue) or the mutant (ADCY3 L121P, red) suggests that the models' average movement over time is altered. Altered movement is seen in the simulations for ADCY3 with fluctuation of amino acids found near amino acid 121 when mutated.
Figure 6.
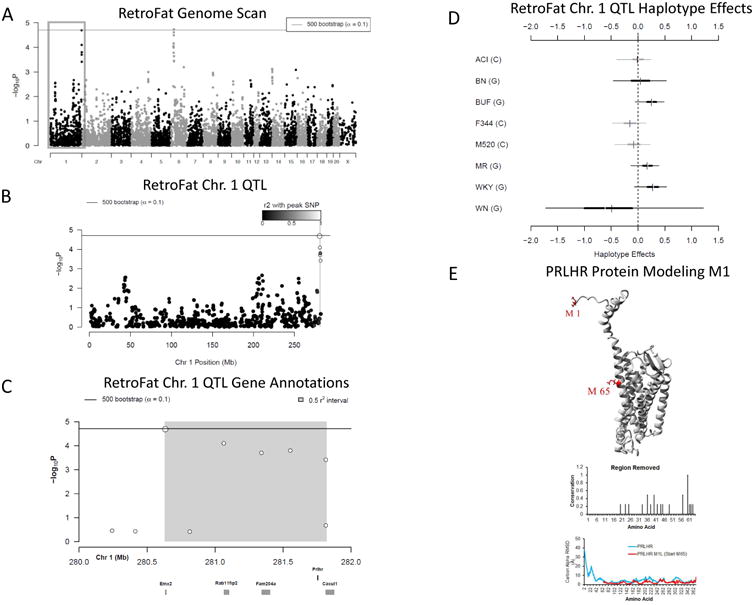
A) Genome scan of RetroFat as described in Figure 3A. B) The grey region highlights the 1.19 Mb LD support interval for the chromosome 1 locus representing neighboring markers that are correlated with the peak marker, representing genomic regions likely to contain the causal variant underlying the statistical signal. C) Annotation of the five characterized genes that fall within the support interval. D) Additive founder haplotype effects for the chromosome 1 RetroFat locus. Additive haplotype effects were estimated using the Diploffect model, which takes into account uncertainty in haplotype state. SNP allele information is also overlaid on the haplotype effects. The C allele is shared by ACI, F344, and M520, that possesses a variant with a negative effect on RetroFat, whereas BUF, MR and WKY haplotypes result in increased RetroFat at this locus. E) Protein modeling for PRLHR. Variant M1I of PRLHR is found within the methionine start site. The next start site is at position 65 leading to removal of the conserved N-terminal region and half of transmembrane helix 1. 16 amino acids removed are under selective pressure (middle panel) and the deletion of the first 64 amino acids causes a destabilization of the entire proteins dynamics as seen by the molecular dynamic simulations (bottom panel).
Identification of Adcy3, Krtcap3, Slc30a3 within the chromosome 6 Retrofat QTL
Within the chromosome 6 RetroFat QTL, bioinformatic analysis revealed only one gene, Adcy3, that had a highly conserved, potentially damaging, non-synonymous variant in the WKY rat, the founder haplotype associated with decreased Retrofat at this locus. Adcy3 also falls within the fine-mapped support interval of the QTL (Figure 3C). The WKY founder strain harbors a C at position 28,572,363 bp within Adcy3 while all other strains harbor a T, resulting in a leucine-to-proline substitution at amino acid 121. Based on DNA information from 86 nucleotide sequences for ADCY3, this variant is highly conserved with evidence for selective pressure. Protein modeling indicated that amino acid 121 is located within the first transmembrane region, with a proline likely causing a bend in the helix and thus altered transmembrane packing (Figure 3E).
Since fine-mapping supported multiple independent signals at the QTL, we investigated potential mediators among the cis-expressed genes. Mediation analysis identified six potential mediators, all driven by the WKY haplotype (Figure 4; Tables S4 and S5), suggesting that multiple genes may influence RetroFat at this locus. Two in particular were strongly supported: Krtcap3 as a full mediator and Slc30a3 as a partial mediator, remaining significant after controlling for Krtcap3 Under the proposed model (Figure 5), the WKY haplotype increases expression of Krtcap3, which is itself negatively correlated with RetroFat (Figure S4), and thus the causal path is consistent with the negative WKY effect on RetroFat at the locus; meanwhile, WKY decreases expression of Slc30a3, which is also negatively correlated RetroFat, suggesting Slc30a3 is a suppressor of the QTL/Krtcap3 effect.
Figure 4.
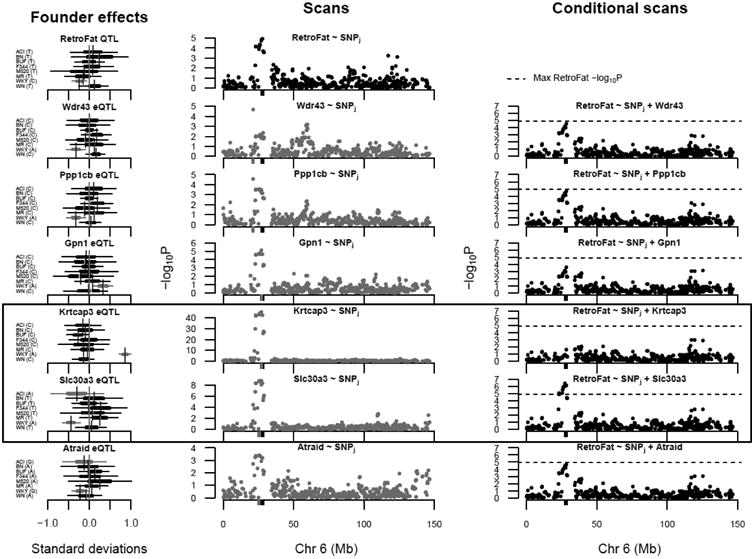
Mediation analysis identified the expression levels of six genes (Wdr43, Ppp1cb, Gpn1, Krtcap3, Slc30a3, and Atraid;Table S5) in the RetroFat chromosome 6 QTL interval as potential mediators of the QTL effect on the phenotype. [Middle column] Comparisons of the RetroFat chromosome 6 association scan with association scans for the potential mediators reveals them to likely have co-localizing cis eQTL with the RetroFat QTL. [Left column] The haplotype effects on RetroFat at the QTL and on the mediators at the eQTL reveals that in this region, the WKY haplotype is largely driving the differences in RetroFat and mediator gene expression, suggesting a possible connection between RetroFat and local gene expression. [Right column] RetroFat chromosome 6 association scans, conditioned on candidate gene expression, is consistent with the mediation analysis finding that Krtcap3 is a strong candidate as full mediator of the effect of QTL on RetroFat. When Krtcap3 expression is included in the model, the QTL is largely removed. Slc30a3, as a potential suppressor of the QTL effect on RetroFat, actually increases the significance seen at the QTL.
Figure 5.

Model demonstrating role of Adcy3, Krtcap3 and Slc30a3 on RetroFat. WKY haplotype increases expression of Krtcap3, which is itself negatively correlated with RetroFat (Figure S4), and thus the causal path is consistent with the negative WKY effect on RetroFat at the locus. In contrast, WKY decreases expression of Slc30a3, which is also negatively correlated RetroFat, suggesting Slc30a3 is a suppressor of the QTL/Krtcap3 effect. Finally, the non-synonymous variant with Adcy3 causes amino acid change L121P leading to lower RetroFat.
Identification of Prlhr within the chromosome 1 RetroFat QTL
Within the chromosome 1 RetroFat QTL, there are 15 genes, ten of which are uncharacterized (Figure 6BC, Table S2). One gene, Prlhr, contains a non-synonymous variant in the BUF and WKY founder strains, two founder haplotypes associated with an increase fat pad weight at this locus. The variant falls within the start codon, changing methionine to isoleucine. The next methionine falls at amino acid 65, such that the conserved N-terminal region and half of transmembrane helix 1 would be deleted with the variant (Figure 4E, https://youtu.be/vRTIkITXRbw). A molecular dynamic simulation of the PRLHR protein with and without the first 64 amino acids showed strong changes to the entire GPCR transmembrane region. Prlhr is expressed mainly in adrenal and brain such that expression levels could not be determined in liver tissue. None of the liver-expressed genes local to the QTL map as cis-eQTL, thus Prlhr remains the strongest candidate within this region.
Body weight QTL on chromosome 4 and identification of Grid2
A 95% significant QTL for body weight was also detected on rat chromosome 4: 91.35Mb to 94.7Mb (3.35 Mb, -logP = 5.32) (Figure 7ABC). At this locus, whose effect size was 12.33%, decreases in body weight were associated with ACI, BUF, F344 and MR haplotypes, and increases with BN (Figure 7D).
Figure 7.
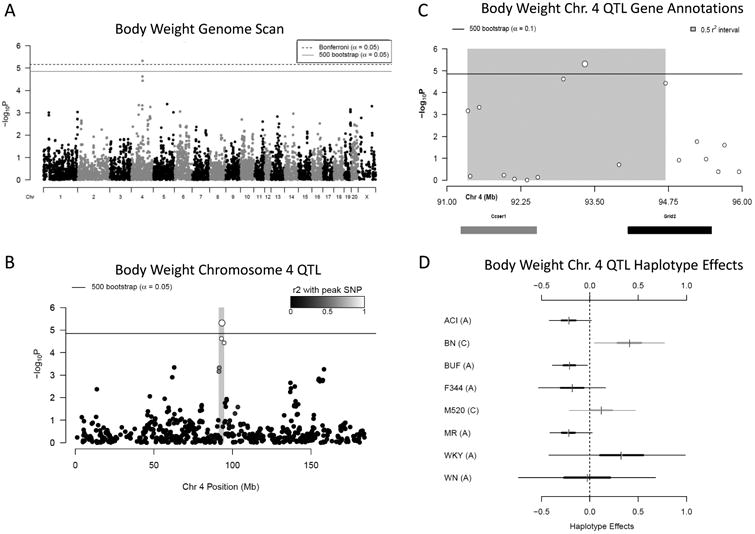
A) Genome scan of body weight. X-axis is position on chromosome and y-axis is the −logP level of association. Genome-wide significance thresholds were calculated using parametric bootstraps from the null model (significant: α = 0.05, logP = 4.86) and conservative α = 0.05 Bonferroni thresholds (logP = 5.16). B) Linkage disequilibrium support interval in grey is 3.35 Mb. C) Annotation of the two characterized genes that fall within the support interval. D) Additive haplotype effects for chromosome 4 body weight QTL. The C allele at the marker could represent shared haplotype descent between BN and M520, both which have an increasing effect on body weight at this locus. ACI, BUF, F344 and MR haplotypes have a decreasing effect of body weight at this locus, all of which share the A allele. The WKY and WN also have an A allele and the WKY haplotype has an increasing effect on body weight, while the WN haplotype appears not to effect body weight, although the credible interval of both is fairly large and not well represented in the data at this locus.
Within the body weight QTL, there are only 11 genes, nine of which are pseudogenes or uncharacterized LOC proteins, leaving only Ccser1 and Grid2 (Figure 7C, Table S3). None of the genes at this locus contained highly conserved potentially damaging non-synonymous variants. Both Ccser1 and LOC108350839 are expressed in liver, but neither's expression was significantly associated with the body weight QTL, ruling these out as candidate mediators. The brain-specific Grid2 is the only gene that has previously been linked to body weight [27] and Grid1 was recently associated with BMI in human GWAS [5], implicating Grid2 as the most likely candidate at this locus.
Discussion
This is the first study to map adiposity traits genome-wide using HS rats and demonstrates their utility for uncovering genes and variants likely to impact human adiposity. We identified QTL for RetroFat on rat chromosomes 1 and 6 and a QTL for body weight on chromosome 4. Using various fine-mapping procedures, we identified three likely candidate genes within the chromosome 6 RetroFat locus: a protein-coding variant within Adcy3, and transcriptional regulation of Krtcap3 and Slc30a3 that mediate between the QTL and RetroFat. Within the chromosome 1 RetroFat QTL, we identified a variant within Prlhr that increases fat pad weight. Lastly, Grid2 was identified as the most likely candidate gene within the body weight locus. It is of interest that several of these candidate genes play a role in neural regulation of energy metabolism and/or feeding behavior.
As expected, both the HS founders and the HS population varied for adiposity traits, with BUF showing the highest adiposity and ACI, BN and M520 showing the lowest. As seen in humans [3, 4], these traits were highly heritable. Also similar to humans [2], increased body weight, particularly visceral fat pad weight (RetroFat and EpiFat), was significantly associated with several measures of metabolic health in the HS rats, indicating that genes underlying QTL for adiposity traits are likely to contribute to overall metabolic health in HS rats.
Despite high heritability for adiposity traits in this model, we found only three QTL for the five traits that were studied. The remaining heritability could be attributable to loci of small effect and/or complex genetic architecture that lie below the limit of detection in this study, and this accords with the fact that the QTL we identified were each of relatively large effect (12.33%, 13.33% and 11.05% respectively). The identified QTL also had relatively small LD support intervals (3.35 Mb, 1.19 Mb and 6.14 Mb, respectively), significantly decreasing the number of potential candidate genes within each QTL relative to traditional QTL studies using F2 intercross or backcross animals; the map density, however, was too low for high resolution mapping of the genes within the interval. We expect that increasing both the number of animals used as well as the density of genotyping would serve to uncover additional loci.
The chromosome 6 RetroFat locus encompassed 6.14 Mb, contained 130 genes and was driven by the WKY haplotype. Using a fine-mapping procedure that allowed for the presence of multiple signals, we identified a 1.46 Mb plausible region for the QTL. Adcy3 was the only gene in this region to contain a highly conserved, non-synonymous variant in the WKY founder strain that is predicted to be damaging: the leucine-to-proline switch at amino acid 121 would likely induce a bend in the helix leading to altered membrane interactions and binding. In addition, we found that multiple genes within the locus map as eQTL. Subsequent mediation analysis supported roles for Krtcap3 and Slc30a3, with Krtcap3 expression presenting as a full mediator of the QTL and Slc30a3 expression as a partial/suppressor mediator. Although little is known about Krtcap3, Slc30a3 is a zinc transporter that plays a role in glucose transport and metabolism [28], and Adcy3 is an enzyme that catalyzes the cAMP second messenger system and is likely involved in energy homeostasis [29]. The POMC/RBJ/ADCY3 region has previously been identified in multiple human GWAS for BMI and obesity [8, 30-32]. Interestingly, a non-synonymous amino acid change (Ser107Pro) in the human Adcy3 gene [8], which falls within the same transmembrane helix as the rat variant, has been identified as the causal variant in height-adjusted childhood BMI [31], indicating the same likely causal variant between rat and human. Adcy3 knock-out and haplo-insufficient mice become obese with age, exhibiting increased food intake and decreased locomotion [33, 34]. In addition, gain of function in Adcy3 protects against diet-induced obesity [35], further supporting a causal role for this gene.
The chromosome 1 RetroFat locus encompassed 1.19 Mb, contained 15 genes, with BUF, MR, and WKY haplotypes increasing RetroFat. Prlhr, containing a non-synonymous variant in both the BUF and WKY strains, stood out as the most likely candidate gene: the variant fell within the methionine start site and leads to removal of the conserved N-terminal region and half of transmembrane helix 1, likely having a large impact on protein function. This variant is found in several other rat strains including FHH, GK, LEW and SD. Prlhr is known to play a role in feeding behavior, with ICV administration in the hypothalamus leading to decreased food intake [36], and Prlhr knock-out mice exhibiting increased food intake, body weight and fat pad weight [37]. Interestingly, this specific variant did not alter feeding behavior in outbred Sprague-Dawley rats [38], indicating that the effect of the variant on fat pad weight may be independent of food intake, although additional studies are needed to confirm this.
The body weight locus encompassed 3.35 Mb and contained 11 genes, none of which contained highly conserved non-synonymous variants predicted to be damaging between the two haplotype effect groups: ACI, BUR, F344, MR versus BN. Only one gene in the region, Grid2, has previously been linked to obesity, jointly with tobacco use, in a family-based study [27], making it the most likely candidate gene. Interestingly, Grid1 was associated with BMI in a recent human GWAS [5], further supporting a potentially causal role for Grid2 within the rat body weight locus. Grid2 encodes the glutamate ionotropic receptor delta type subunit 2 and is known to play a role in synapse formation, particularly within the cerebellum [39]. Synaptic formation and plasticity are increasingly being recognized as playing a role in metabolism and energy balance [40]. Additional work, including assessing Grid2 expression levels in brain, is needed to confirm or eliminate Grid2 as the causal gene at this locus.
In summary, we have used HS rats to identify QTL for adiposity traits, leading to identification of five candidate genes and two likely causal variants. Some genes have previously been identified in human GWAS or linkage studies (Adcy3, Grid2) or implicated in rodent models of obesity (Adcy3, Prlhr), while two genes are novel (Krtcap3, Slc30a3). The Adcy3 variant falls within the same transmembrane helix as that found in humans indicating direct human relevance of this work. It is also of interest that Adcy3, Prlhr and Grid2 have previously been found to impact feeding behavior and/or neural regulation of metabolism. This work demonstrates the power of HS rats for genetic fine-mapping and identification of underlying candidate genes and variants that will likely be relevant to human adiposity.
Supplementary Material
Figure S1: Principal component analysis for the first ten principal components of genotypes between two genotyping centers. Those genotyped at Hudson Alpha are plotted in red and those genotyped at Vanderbilt are plotted in blue. For all plots, red and blue points fall within the same general region indicating that there are no systematic differences in genotype between the two centers.
Figure S2: Fine-mapping of the chromosome 6 locus using LLARRMA-dawg reduced the LD support interval from 6.14 Mb to 1.46 Mb. LLARRMA-dawg jointly models and selects SNPs in a region, and returns probabilities corresponding to how often a SNP was included over many re-samples of the data (RMIP). Multiple SNPs with high RMIP suggests the potential for multiple independent signals beneath the QTL peak.
Figure S3: The SNP association (7 markers) present in the LLARRMA-dawg fine-mapping interval (Figure S2), including the annotations of the 30 genes local to the region. The candidate gene Adcy3 is in bold, and possesses a non-synonymous WKY variant that is predicted to alter protein function (Figure 3E).
Figure S4: Scatterplot of RetroFat and mediator expression levels, with data points colored by the peak SNP minor allele dosages at the QTL. RetroFat and expression levels are rank-inverse normal transformed.
Table S1: Genes in RetroFat chromosome 6 QTL interval.
Table S2: Genes in RetroFat chromosome 1 QTL interval.
Table S3: Genes in body weight chromosome 4 QTL interval.
Table S4: Genes in RetroFat chromosome 6 QTL interval that support X→M relationship.
Table S5: Genes in RetroFat chromosome 6 QTL interval that mediation analysis supports as candidate mediators of the effect of QTL on RetroFat.
What is already known about this subject?
Traits associated with adiposity such as body mass index and visceral adiposity are highly heritable, with genetics accounting for ∼70% of the population variance.
Although many genes have been identified for adiposity traits, a small proportion of the heritable variance has been identified, indicating a critical need to identify the remaining genes.
Outbred rodent models enable rapid fine-mapping of genetic loci involved in complex traits to only a few Megabases, and may prove useful for uncovering some of the missing heritability for adiposity traits.
What this study adds.
Similar to humans, we show that visceral adiposity is highly correlated with traits of metabolic health in outbred heterogeneous stock (HS) rats, demonstrating that this is an excellent model to study the underlying genetics of adiposity.
Using HS rats, we identify two quantitative trait loci (QTL) for visceral fat pad weight and one for body weight. Linkage disequilibrium-based intervals for these loci are relatively small: 6.14, 1.19, and 3.35 Mb, respectively.
We identify candidate genes within each QTL, and likely causal variants within visceral fat pad loci. Genes identified include Adcy3, Krtcap3, Slc30a3, Prlhr and Grid 2. Likely causal variants are identified in Adcy3 and Prlhr, genes identified in the QTL for visceral fat pad weight.
Acknowledgments
We would like to thank Jennifer Phillips for running the cholesterol and triglyceride assays. We would like to thank Mary Shimoyama and Jeffrey DePons for help in transferring founder sequence to Rn6 in RGD.
This work was funded by R01 DK-088975, R01 DK-106386 (LSW) and funding from the Individualized Medicine Institute at MCW (LSW). Partial support for GK and YK was provided by R01 GM-104125 (WV). DMG and KWB were supported by R01 GM 070683. SWT and MJF were supported by R01 CA193343 (MJF). Protein modeling analysis was supported by the NIH office of the director award K01ES025435 (JWP).
Footnotes
Conflicts of interest statement: The authors have no conflicts of interest.
References
- 1.Wang YC, et al. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378(9793):815–25. doi: 10.1016/S0140-6736(11)60814-3. [DOI] [PubMed] [Google Scholar]
- 2.Emdin CA, et al. Genetic Association of Waist-to-Hip Ratio With Cardiometabolic Traits, Type 2 Diabetes, and Coronary Heart Disease. JAMA. 2017;317(6):626–634. doi: 10.1001/jama.2016.21042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. Jama. 1986;256(1):51–4. [PubMed] [Google Scholar]
- 4.Katzmarzyk PT, et al. Familial resemblance in fatness and fat distribution. Am J Hum Biol. 2000;12(3):395–404. doi: 10.1002/(SICI)1520-6300(200005/06)12:3<395::AID-AJHB10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 5.Locke AE, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun. 2016;7:10495. doi: 10.1038/ncomms10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng MCY, et al. Discovery and fine-mapping of adiposity loci using high density imputation of genome-wide association studies in individuals of African ancestry: African Ancestry Anthropometry Genetics Consortium. PLoS Genet. 2017;13(4):e1006719. doi: 10.1371/journal.pgen.1006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speliotes EK, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flegal KM, et al. Trends in Obesity Among Adults in the United States, 2005 to 2014. JAMA. 2016;315(21):2284–91. doi: 10.1001/jama.2016.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Solberg Woods LC. QTL mapping in outbred populations: successes and challenges. Physiol Genomics. 2014;46(3):81–90. doi: 10.1152/physiolgenomics.00127.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Solberg Woods LC, et al. Fine-mapping a locus for glucose tolerance using heterogeneous stock rats. Physiol Genomics. 2010;41(1):102–8. doi: 10.1152/physiolgenomics.00178.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solberg Woods LC, et al. Fine-mapping diabetes-related traits, including insulin resistance, in heterogeneous stock rats. Physiol Genomics. 2012;44(21):1013–26. doi: 10.1152/physiolgenomics.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsaih SW, et al. Identification of a novel gene for diabetic traits in rats, mice, and humans. Genetics. 2014;198(1):17–29. doi: 10.1534/genetics.114.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen C, Spuhler K. Development of the National Institutes of Health genetically heterogeneous rat stock. Alcohol Clin Exp Res. 1984;8(5):477–9. doi: 10.1111/j.1530-0277.1984.tb05706.x. [DOI] [PubMed] [Google Scholar]
- 15.Saar K, et al. SNP and haplotype mapping for genetic analysis in the rat. Nat Genet. 2008;40(5):560–6. doi: 10.1038/ng.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holand AM, et al. Animal models and integrated nested Laplace approximations. G3 (Bethesda) 2013;3(8):1241–51. doi: 10.1534/g3.113.006700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rue H, Martino S, Chopin N. Approximate Bayesian inference for latent Gaussian models by using integrated nested Laplace approximations. Journal of the Royal Statistical Society: Series B. 2009;71(2):319–392. [Google Scholar]
- 18.Gatti DM, et al. Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 2014;4(9):1623–33. doi: 10.1534/g3.114.013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broman KW. qtl2geno: Treatment of Marker Genotypes for QTL Experiments. R package version 0.4-21. 2016 http://kbroman.org/qtl2.
- 20.Valdar W, et al. Mapping in structured populations by resample model averaging. Genetics. 2009;182(4):1263–77. doi: 10.1534/genetics.109.100727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabourin J, Nobel AB, Valdar W. Fine-mapping additive and dominant SNP effects using group-LASSO and fractional resample model averaging. Genet Epidemiol. 2015;39(2):77–88. doi: 10.1002/gepi.21869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Wang W, Valdar W. Bayesian modeling of haplotype effects in multiparent populations. Genetics. 2014;198(1):139–56. doi: 10.1534/genetics.114.166249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prokop JW, et al. Molecular modeling in the age of clinical genomics, the enterprise of the next generation. J Mol Model. 2017;23(3):75. doi: 10.1007/s00894-017-3258-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krieger E, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins. 2009;77 Suppl 9:114–22. doi: 10.1002/prot.22570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baron RA, Kenny DA. The Moderator-Mediator Variable Distinction in Social Psychological Research: Conceptual, Strategic, and Statistical Considerations. Journal of Personality and Social Psychology. 1986;51(6):1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- 27.Nikpay M, et al. Genetic mapping of habitual substance use, obesity-related traits, responses to mental and physical stress, and heart rate and blood pressure measurements reveals shared genes that are overrepresented in the neural synapse. Hypertens Res. 2012;35(6):585–91. doi: 10.1038/hr.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smidt K, et al. SLC30A3 responds to glucose- and zinc variations in beta-cells and is critical for insulin production and in vivo glucose-metabolism during beta-cell stress. PLoS One. 2009;4(5):e5684. doi: 10.1371/journal.pone.0005684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu L, et al. Adenylate cyclase 3: a new target for anti-obesity drug development. Obes Rev. 2016;17(9):907–14. doi: 10.1111/obr.12430. [DOI] [PubMed] [Google Scholar]
- 30.Nordman S, et al. Genetic variation of the adenylyl cyclase 3 (AC3) locus and its influence on type 2 diabetes and obesity susceptibility in Swedish men. Int J Obes (Lond) 2008;32(3):407–12. doi: 10.1038/sj.ijo.0803742. [DOI] [PubMed] [Google Scholar]
- 31.Stergiakouli E, et al. Genome-wide association study of height-adjusted BMI in childhood identifies functional variant in ADCY3. Obesity (Silver Spring) 2014;22(10):2252–9. doi: 10.1002/oby.20840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wen W, et al. Meta-analysis identifies common variants associated with body mass index in east Asians. Nat Genet. 2012;44(3):307–11. doi: 10.1038/ng.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong T, et al. Adenylyl cyclase 3 haploinsufficiency confers susceptibility to diet-induced obesity and insulin resistance in mice. Sci Rep. 2016;6:34179. doi: 10.1038/srep34179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z, et al. Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS One. 2009;4(9):e6979. doi: 10.1371/journal.pone.0006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pitman JL, et al. A gain-of-function mutation in adenylate cyclase 3 protects mice from diet-induced obesity. PLoS ONE. 2014;9(10):e110226. doi: 10.1371/journal.pone.0110226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence CB, et al. Alternative role for prolactin-releasing peptide in the regulation of food intake. Nat Neurosci. 2000;3(7):645–6. doi: 10.1038/76597. [DOI] [PubMed] [Google Scholar]
- 37.Gu W, et al. The prolactin-releasing peptide receptor (GPR10) regulates body weight homeostasis in mice. J Mol Neurosci. 2004;22(1-2):93–103. doi: 10.1385/JMN:22:1-2:93. [DOI] [PubMed] [Google Scholar]
- 38.Ellacott KL, et al. Characterization of a naturally-occurring polymorphism in the UHR-1 gene encoding the putative rat prolactin-releasing peptide receptor. Peptides. 2005;26(4):675–81. doi: 10.1016/j.peptides.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 39.Hirai H, et al. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat Neurosci. 2003;6(8):869–76. doi: 10.1038/nn1086. [DOI] [PubMed] [Google Scholar]
- 40.Dietrich MO, Horvath TL. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends Neurosci. 2013;36(2):65–73. doi: 10.1016/j.tins.2012.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Principal component analysis for the first ten principal components of genotypes between two genotyping centers. Those genotyped at Hudson Alpha are plotted in red and those genotyped at Vanderbilt are plotted in blue. For all plots, red and blue points fall within the same general region indicating that there are no systematic differences in genotype between the two centers.
Figure S2: Fine-mapping of the chromosome 6 locus using LLARRMA-dawg reduced the LD support interval from 6.14 Mb to 1.46 Mb. LLARRMA-dawg jointly models and selects SNPs in a region, and returns probabilities corresponding to how often a SNP was included over many re-samples of the data (RMIP). Multiple SNPs with high RMIP suggests the potential for multiple independent signals beneath the QTL peak.
Figure S3: The SNP association (7 markers) present in the LLARRMA-dawg fine-mapping interval (Figure S2), including the annotations of the 30 genes local to the region. The candidate gene Adcy3 is in bold, and possesses a non-synonymous WKY variant that is predicted to alter protein function (Figure 3E).
Figure S4: Scatterplot of RetroFat and mediator expression levels, with data points colored by the peak SNP minor allele dosages at the QTL. RetroFat and expression levels are rank-inverse normal transformed.
Table S1: Genes in RetroFat chromosome 6 QTL interval.
Table S2: Genes in RetroFat chromosome 1 QTL interval.
Table S3: Genes in body weight chromosome 4 QTL interval.
Table S4: Genes in RetroFat chromosome 6 QTL interval that support X→M relationship.
Table S5: Genes in RetroFat chromosome 6 QTL interval that mediation analysis supports as candidate mediators of the effect of QTL on RetroFat.