

Abstract
Leigh syndrome (LS) is an early-onset progressive neurodegenerative disorder, characterized by a wide clinical and genetic heterogeneity, and is the most frequent disorder of mitochondrial energy production in children. Beside its great variability in clinical, biochemical, and genetic features, LS is pathologically uniformly characterized by multifocal bilateral and symmetric spongiform degeneration of the basal ganglia, brainstem, thalamus, cerebellum, spinal cord, and optic nerves. Isolated complex I deficiency is the most common defect identified in Leigh syndrome. In 2011, the first child with a mutation of NDUFA10 gene, coding for an accessory subunits of complex I, was described. Here, we present an additional description of a child with Leigh syndrome harboring a homozygous mutation in NDUFA10, providing insights in clinical, biochemical, and neuroradiologic features for future earlier recognition.
Keywords: Complex I deficiency, Leigh syndrome, Mitochondrial disorders, NDUFA10 gene, Neuroradiologic features
Introduction
Defects in mitochondrial energy production are the most frequent group of inherited metabolic disorders, with an incidence of at least 1 in 5,000 live births (Skladal et al. 2003). The most common clinical manifestation of mitochondrial disease in children is Leigh syndrome (Leigh 1951) (LS), an early-onset progressive neurodegenerative disorder, characterized by a wide clinical and genetic heterogeneity. Clinical presentation frequently includes psychomotor delay or regression, acute neurological or acidotic episodes, hypotonia, ataxia, spasticity, movement disorders, and corresponding anomalies of the basal ganglia and brainstem on MRI (Tetreault et al. 2015). Up to now, more than 75 disease genes have been linked to LS, involving the oxidative phosphorylation (OXPHOS) system, the pyruvate dehydrogenase complex (PDHc), and multiple other enzymes mostly linked to OXPHOS system or to a broader pathway of energy generation (Lake et al. 2016).
Isolated complex I deficiency is the most common biochemical defect identified in LS, accounting for approximately 25% of all children with OXPHOS deficiencies (Smeitink et al. 2001). Complex I (NADH: ubiquinone oxidoreductase) is the first and the largest multiheteromeric respiratory chain (RC) enzyme complex being composed of 45 protein subunits (7 of which encoded by mtDNA), with a pivotal role in ATP synthesis, transferring electrons from reduced NADH to coenzyme Q10, and pumping protons across the inner mitochondrial membrane. Moreover, its maturation, assembly, and stability rely on a number of ancillary proteins. Up to now, pathogenic mutations have been reported in all the seven mtDNA-encoded subunits and in several nuclear DNA-encoded subunits or assembly factors, including NADH dehydrogenase 1α subcomplex 10 (NDUFA10) (Lake et al. 2016). NDUFA10 is located in the hydrophobic protein fraction and is homologous to deoxyribonucleoside kinases, but it has no enzymatic activity (Elurbe and Huynen 2016). The function of this accessory subunit is still not clear, but it has been suggested to have a role in the transfer of protons across the inner membrane (Hoefs et al. 2011). Until now, only two cases of pathogenic NDUFA10 mutations have been reported (Hoefs et al. 2011; Haack et al. 2012), both presenting with LS and complex I deficiency. In the present paper, we further characterize the clinical, biochemical, and neuroradiologic features of a child with LS due to NDUFA10 mutation.
Case Report: Biochemical and Molecular Analyses
Case Report
We present the case of an Italian boy, the third child of consanguineous parents (third degree cousins), born at 41 weeks of gestation after a normal pregnancy. Family history is remarkable for a paternal cousin who died at the age of 4 months for an unspecified cardiomyopathy, two maternal cousins manifesting an unspecified myopathy, and another cousin affected by an early-onset epileptic encephalopathy. In the proband, birth weight was 3,460 g and length 52 cm. The neonatal period was uneventful, but nystagmus was reported since the first month of life. He reached all his milestones but with a slight delay. At the age of 2 years and 8 months, after a febrile illness, he presented acute-onset ataxia with severe hyposthenia and inability to stand and walk. This condition lasted for approximately a week and was followed by a slow and partial neurological improvement but without complete recovery. A first brain MRI was performed at that age and showed rather symmetrical lesions of the putamina and globi pallidi, characterized by swelling, necrotic changes, and mixed cytotoxic and vasogenic edema. Asymmetric focal lesions of the caudate nuclei were also noted, as well as focal involvement of the left cerebral peduncle (Fig. 1). MR spectroscopy detected a lactate doublet peak.
Fig. 1.
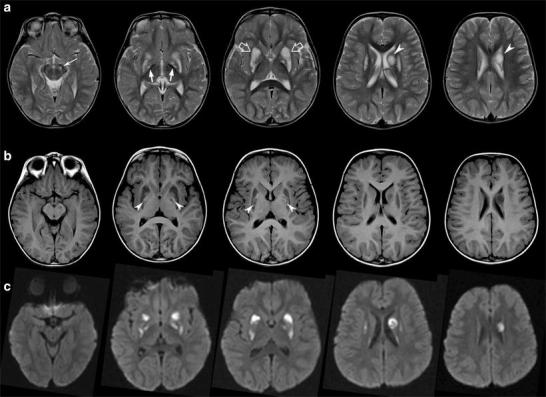
Brain MRI performed at 2 years and 8 months. (a) Axial T2-weighted images demonstrate symmetric swelling and T2 hyperintensity of the putamina (empty arrows) and asymmetric involvement of globi pallidi (arrows), left cerebral peduncle (thin arrows), and left caudate nucleus (arrowheads). Note that the lesion of the left caudate head is round and well demarcated, with moderate mass effect. (b) Hyperintense necrotic foci are present at the level of the medial part of the putamina and globi pallidi on T1-weighted images (arrowheads). On diffusion-weighted images (c), most of the lesions are characterized by restricted diffusion in keeping with cytotoxic edema
The child was then referred to our attention for further evaluation. On clinical examination, we observed a generalized hypotonia with muscular hypotrophy especially in the lower limbs; tendon reflexes were slightly increased, and oscillatory nystagmus was present. Gait ataxia and clumsiness were also observed. The psychomotor evaluation showed a moderate delay in his development. Blood and urine tests were unremarkable with the exception of levels of plasma lactate slightly above normal range (39.2 mg/dl, normal range 8–22 mg/dl). The clinical, neuroimaging, and biochemical features strongly suggested a LS, and a defect of PDHc or OXPHOS pathway was considered. At first, on the basis of his clinical presentation and mostly of intermittent ataxia, molecular analysis on PDHc genes was performed and thiamine-biotin therapy was started.
In the next year, the child presented a slight improvement in neurological development with a partial recovery of gait and language. However, severe difficulties in oculomotor abilities persisted with extrapyramidal signs such as dystonic features, nystagmus, and ataxia. Parents reported intermittent relapses, usually associated with febrile illness, with only partial recovery after each episode, resulting in a progressive neurologic deterioration. Cardiologic evaluations were always unremarkable. A second brain MRI, performed at the age of 3 years and 6 months, showed the chronic evolution of the putaminal lesions and the presence of new symmetric lesions of the caudate heads and Meynert basal nuclei and asymmetric lesions at the level of the right globus pallidus and cerebral peduncle. MR spectroscopy, performed on the right caudate nucleus head, revealed a lactate doublet peak (Fig. 2).
Fig. 2.
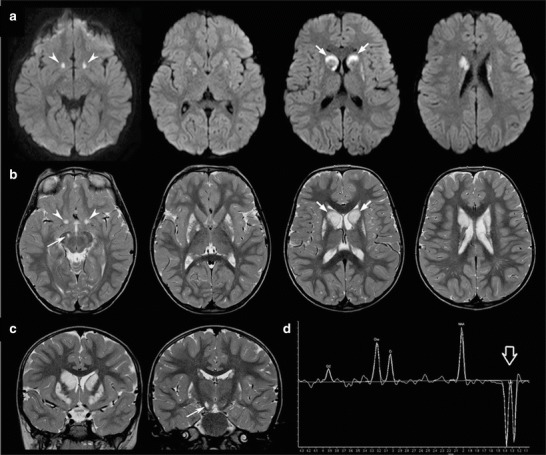
Follow-up brain MRI and MR spectroscopy performed at 3 years and 6 months. Axial diffusion-weighted images (a) and T2-weighted images (axial, b and coronal, c) reveal new acute lesions characterized by T2 hyperintensity and restricted diffusion due to cytotoxic edema at the level of the basal nuclei of Meynert (arrowheads), right cerebral peduncle (thin arrow), and caudate nuclei (arrows). Note the peculiar involvement of the heads of caudate nuclei, with prevalent restricted diffusion in the anterior peripheral portions and increased diffusion in the central portions. The putamina are markedly reduced in size with prevalent increased diffusion, in keeping with chronic evolution of the previous lesions. (d) The MR spectroscopy performed at the level of the right basal ganglia demonstrates slightly reduced NAA peak and markedly elevated lactate peak (empty arrow)
Biochemical and Molecular Analyses
Routine morphology, histochemical stains for oxidative metabolism, and spectrophotometric determination of mitochondrial RC complexes in muscle homogenate were assayed according to standard methods.
Deltoid muscle biopsy was performed under the hypothesis of a mitochondrial disease: histological examinations showed only a slight reduction of cytochrome oxidase activity. Biochemical analyses of the respiratory chain revealed a reduction of complexes I (1.12 mmol/min/g tissue; normal range 1.56–2.60), I + II (0.08 mmol/min/g tissue, normal range 0.11–0.25), and II + III (0.02 mmol/min/g tissue, normal range 0.05–0.08) (details available on request).
Genomic blood and muscle DNA from the patient were purified by standard methods and analyzed by Sanger sequencing to exclude pathogenic variants in mitochondrial DNA (mtDNA) and PDHA1 gene. Then, we used a customized targeted resequencing panel able to investigate the coding regions of 168 genes linked to mitochondrial disorders applying standard methodologies as outlined elsewhere (Mignarri et al. 2016). The identified pathogenic variants in NDUFA10 (NM_004544.3) were confirmed by Sanger sequencing and were tested for segregation in the family.
Molecular analysis identified the homozygous mutation c.296G>A in exon 3 of the NDUFA10 gene (Fig. 3a), causing a substitution of the extremely conserved throughout evolution glycine 99 for glutamate (p.G99E) (Fig. 3b). Both parents carried the heterozygous c.296G>A variant which was predictably damaging when examined in silico using Polyphen2. The mutation has been already reported in a child presenting with severe complex I deficiency (Haack et al. 2012).
Fig. 3.
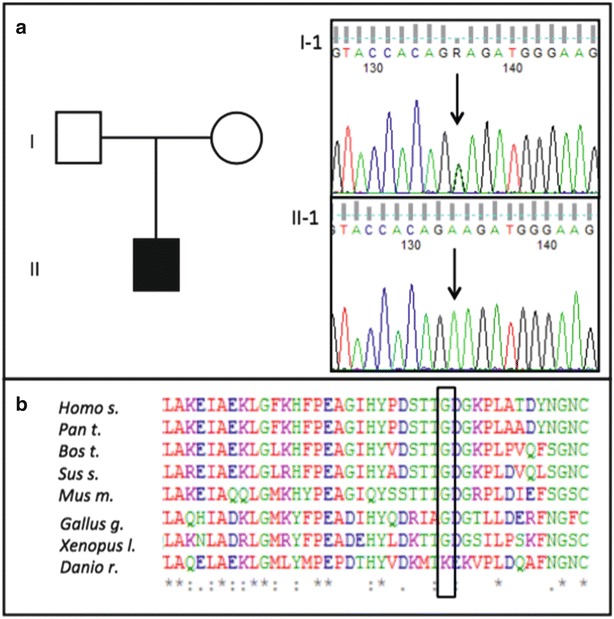
Molecular analysis of NDUFA10 gene. Electropherograms present the nucleotide change in fibroblasts of the patient (II-1); arrows point out the nucleotide substitution c.296G>A in exon 3 (a). Bottom figure show the high conservation of the altered amino acid (p.G99E) throughout evolution (b)
Discussion
The case described here further supports the role of NDUFA10 mutations as a rare cause of LS with reduced complex I activity, providing new data on the related clinical, biochemical, and neuroradiologic features and contributing to the genotype-phenotype correlation in LS.
LS is the most frequent disorder of mitochondrial energy production in children, as the result of a heterogeneous group of defects in the OXPHOS or in the PDHc systems. Therefore, it is unsurprising that a wide variety of multisystemic symptoms may be reported, including cardiac, hepatic, gastrointestinal, and renal dysfunction (Lopez et al. 2006; Monlleo-Neila et al. 2013). The age of presentation is typically set within 2 years of age (Lake et al. 2016), but there is a wide range of disease onset. As in the case here presented, affected children generally have an acute presentation of symptoms, often during an infection, after an initial period of normal development. Progression is typically episodic, with relapses commonly related to febrile illnesses; often, there may be some initial recovery, but never back to the baseline (Lake et al. 2016; Rahman et al. 1996; Sofou et al. 2014). The overall survival is variable, but most patients die after few years from diagnosis. Poor prognosis predictors include age at disease onset less than 6 months, failure to thrive, seizures resistant to pharmacologic treatments, brainstem lesions on neuroimaging, and intensive care unit admission (Lopez et al. 2006; Monlleo-Neila et al. 2013; Rahman et al. 1996; Sofou et al. 2014; Fassone et al. 2011; Benit et al. 2003; Ruhoy and Saneto 2014).
Hoefs et al. firstly described a patient harboring two heterozygous NDUFA10 mutations, one disrupting the start codon in exon 1 (c. c.1A>G) and the other resulting in an amino acid substitution in exon 3 (p.Gln142Arg). The disease was characterized by an early onset, with hypotonia and developmental delay, and by an aggressive course with severe lactic acidosis, progressive respiratory failure, convulsions, hypertrophic cardiomyopathy, and exitus at 23 months of age (Hoefs et al. 2011). Conversely, in the present patient harboring a homozygous mutation c.296G>A in exon 3 of the NDUFA10 gene, the clinical course was less severe with later age of onset, longer survival, and absence of hypertrophic cardiomyopathy. In particular, the developmental delay was very mild, and the prevailing neurological features were the episodes of ataxia with initial intercritical recovery. Interestingly, hypertrophic cardiomyopathy, a potential life-threating finding in many patients with complex I deficiency (Fassone et al. 2011; Benit et al. 2003; Fassone and Rhaman 2012), was absent also in the patient reported by Haack et al., who harbored the same homozygous NDUFA10 mutations in exon 3, suggesting the hypothesis of different effects of specific NDUFA10 mutations in brain and cardiac muscle (Haack et al. 2012).
Of note, the relapsing-remitting course of the disease, and the first positive response to thiamine, initially led us to consider a PDH complex defect as the most likely diagnostic hypothesis (Giribaldi et al. 2012). However, this diagnosis was dismissed by the results of the analysis of RC enzyme activity revealing a defect in the OXPHOS system, subsequently confirmed by molecular assays. In particular, we noticed a clear reduction in complex I activity, associated to a milder decrease in complex I + III and II + III activities. These results are similar to those described by Hoefs et al., underlying the importance of screening for the presence of disease-causing NDUFA10 mutations not only in patients with isolated complex I deficiencies but also in patients with a reduced complex I deficiency combined with a reduction in complex II or III activity. From a histopathologic point of view, similarly to Hoefs et al., we did not found major abnormalities in the patient’s muscle biopsy. Indeed, ragged red fibers, a typical sign of many mitochondrial disorders, are rare in LS, and muscle biopsy histology may be completely normal (Sofou et al. 2014; Gerards et al. 2016; Finsterer 2008).
Besides its wide clinical, biochemical, and genetic heterogeneity, LS is uniformly characterized by the presence of multifocal bilateral and symmetric spongiform degeneration of the basal ganglia, thalamus, cerebellum, brainstem, spinal cord, and optic nerves, variably associated to demyelination and gliosis (Rahman et al. 1996; Ruhoy and Saneto 2014). These pathological changes, also described in the autopsy of the child with NDUFA10 mutation reported by Hoefs et al., are well related to the abnormalities commonly identified on brain MRI.
MR imaging in LS usually reveals symmetrical hyperintense lesions of the basal ganglia and brainstem on T2-weighted images. Cerebellar dentate nuclei are typically abnormal, while spinal cord, hemispheric white matter, and cerebral cortex changes are less frequently found (Lake et al. 2016; Gerards et al. 2016; Lee et al. 2009; Arii and Tanabe 2000). Acutely affected areas may show restricted diffusion, and MR spectroscopy typically reveals elevated lactate, most prominent within the lesions, due to the impairment of the mitochondrial function with secondary energetic failure and cytotoxic edema (Ruhoy and Saneto 2014; Saneto et al. 2008). Interestingly, beside the classical putaminal lesions, the present patient showed a rather unusual asymmetric involvement of the caudate nuclei, globi pallidi, and cerebral peduncles. In particular, peculiar asynchronous round lesions were noted at the level of the caudate heads, characterized by mass effect and mixed restricted and increased diffusion, consistent with the presence of both cytotoxic and vasogenic edema (Fig. 1). Of note, asymmetric involvement of the basal ganglia and brainstem may be found in other mitochondrial diseases. For instance, mutations in MTFMT, encoding a mitochondrial methionyl-tRNA formyl-transferase, may cause variably asymmetric Leigh encephalopathy and cystic or multifocal leukoencephalopathy (Haack et al. 2014; Baertling et al. 2016). Brain MRI images are not provided in the two reported patients with NDUFA10 mutations, but the basal ganglia involvement is described as symmetric in both cases. Further studies are therefore needed to establish if NDUFA10 mutations may cause a specific and recognizable neuroradiologic pattern. In literature, several attempts have been made to correlate the brain MR findings to a specific mutation in LS. For instance, the subthalamic nuclei and brainstem are consistently abnormal in patients with SURF1 mutation (Rossi et al. 2003). On the other hand, abnormalities in the subthalamic nuclei are identified also in patients with other genetic defects in LS, demonstrating the complexity and wide variability of neuroimaging features in these conditions. Indeed, despite its typical features, MRI imaging alone may lack specificity in LS detection, as other mitochondrial conditions and non-mitochondrial diseases can show similar basal ganglia alterations on MRI. Therefore, LS diagnosis is currently based on a combined set of clinical, biochemical, and neuroimaging findings (Rahman et al. 1996; Baertling et al. 2014).
In conclusion, our description adds further evidence on the pathogenic role of NDUFA10 in LS, expanding the spectrum of clinical and brain MRI manifestations. Our data suggest that even in patients apparently sharing the same genotype, the variability of single mutations may lead to different clinical, biochemical, and neuroradiologic phenotype.
Short Running Title (Synopsis)
New Clinical and Neuroradiologic Features of NDUFA10 Mutation.
Authors Contribution
We confirm that all listed authors have provided a significant contribution in the manuscript preparation, in intellectual revision, and in patient’s care.
Conflict of Interest
Francesca Minoia, Marta Bertamino, Paolo Picco, Mariasavina Severino, Andrea Rossi, Chiara Fiorillo, Carlo Minetti, Claudia Nesti, Filippo Maria Santorelli, and Maja Di Rocco declare that they have no conflict of interest related to the present manuscript.
Details of Funding
This study received no specific funding.
Details of Ethical Approval
Specific ethics committee was not requested for anonymous data report.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Contributor Information
Francesca Minoia, Email: francescaminoia@gaslini.org.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Arii J, Tanabe Y. Leigh syndrome: serial MR imaging and clinical follow-up. AJNR Am J Neuroradiol. 2000;21:1502–1509. [PMC free article] [PubMed] [Google Scholar]
- Baertling F, Rodenburg RJ, Schaper J, et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014;85:257–265. doi: 10.1136/jnnp-2012-304426. [DOI] [PubMed] [Google Scholar]
- Baertling F, Klee D, Haack TB, et al. The many faces of paediatric mitochondrial disease on neuroimaging. Childs Nerv Syst. 2016;32:2077–2083. doi: 10.1007/s00381-016-3190-3. [DOI] [PubMed] [Google Scholar]
- Benit P, Beugnot R, Chretien D, et al. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003;21:582–586. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- Elurbe DM, Huynen MA. The origin of the supernumerary subunits and assembly factors of complex I: a treasure trove of pathway evolution. Biochim Biophys Acta. 2016;1857(7):971–979. doi: 10.1016/j.bbabio.2016.03.027. [DOI] [PubMed] [Google Scholar]
- Fassone E, Rhaman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159. [DOI] [PubMed] [Google Scholar]
- Fassone E, Taanman JW, Hargreaves IP, et al. Mutations in the mithocondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J Med Genet. 2011;48:691–697. doi: 10.1136/jmedgenet-2011-100340. [DOI] [PubMed] [Google Scholar]
- Finsterer J. Leigh and Leigh-Like syndrome in children and adults. Pediatr Neurol. 2008;39:223–235. doi: 10.1016/j.pediatrneurol.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Gerards M, Sallevelt SC, Smeets HJ. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab. 2016;117:300–312. doi: 10.1016/j.ymgme.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Giribaldi G, Doria-LAmba L, Biancheri R, et al. Intermittent-relapsing pyruvate dehydrogenase complex deficiency: a case with clinical, biochemical, and neuroradiological reversibility. Dev Med Child Neurol. 2012;54:472–476. doi: 10.1111/j.1469-8749.2011.04151.x. [DOI] [PubMed] [Google Scholar]
- Haack TB, Madignier F, Herzer M, et al. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J Med Genet. 2012;49:83–89. doi: 10.1136/jmedgenet-2011-100577. [DOI] [PubMed] [Google Scholar]
- Haack TB, Gorza M, Danhauser K, et al. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. 2014;111:342–352. doi: 10.1016/j.ymgme.2013.12.010. [DOI] [PubMed] [Google Scholar]
- Hoefs SJ, van Spronsen FJ, Lenssen EW, et al. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur J Hum Genet. 2011;19:270–274. doi: 10.1038/ejhg.2010.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake NJ, Compton AG, Rahman S, Thorburn DR. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol. 2016;79:190–203. doi: 10.1002/ana.24551. [DOI] [PubMed] [Google Scholar]
- Lee H-F, Tsai C-R, Chi C-S, et al. Leigh syndrome: clinical and neuroimaging follow-up. Pediatr Neurol. 2009;40:88–93. doi: 10.1016/j.pediatrneurol.2008.09.020. [DOI] [PubMed] [Google Scholar]
- Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez LC, Schuelke M, Quinzii CM, et al. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125–1129. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignarri A, Rubegni A, Tessa A, et al. Mitochondrial dysfunction in hereditary spastic paraparesis with mutations in DDHD1/SPG28. J Neurol Sci. 2016;362:287–291. doi: 10.1016/j.jns.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Monlleo-Neila L, Toro MD, Bornstein B, et al. Leigh syndrome and the mitochondrial m.13513G>A mutation: expanding the clinical spectrum. J Child Neurol. 2013;28:1531–1534. doi: 10.1177/0883073812460580. [DOI] [PubMed] [Google Scholar]
- Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- Rossi A, Biancheri R, Bruno C, Di Rocco M, Calvi A, Pessagno A, Tortori-Donati P. Leigh syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR Am J Neuroradiol. 2003;24:1188–1191. [PMC free article] [PubMed] [Google Scholar]
- Ruhoy IS, Saneto RP. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014;7:221–234. doi: 10.2147/TACG.S46176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial diseases. Mitochondrion. 2008;8:396–413. doi: 10.1016/j.mito.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–1912. doi: 10.1093/brain/awg170. [DOI] [PubMed] [Google Scholar]
- Smeitink J, van der Huevel L, Di Mauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. 2001;2:342–352. doi: 10.1038/35072063. [DOI] [PubMed] [Google Scholar]
- Sofou K, De Coo IF, Isohanni P, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9:52. doi: 10.1186/1750-1172-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetreault M, Fahiminiya S, Antonicka H, et al. Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum Genet. 2015;134:981–991. doi: 10.1007/s00439-015-1577-y. [DOI] [PubMed] [Google Scholar]