

Abstract
Objective
To characterize the clinical features and neuropathology associated with recessive VAC14 mutations.
Methods
Whole‐exome sequencing was used to identify the genetic etiology of a rapidly progressive neurological disease presenting in early childhood in two deceased siblings with distinct neuropathological features on post mortem examination.
Results
We identified compound heterozygous variants in VAC14 in two deceased siblings with early childhood onset of severe, progressive dystonia, and neurodegeneration. Their clinical phenotype is consistent with the VAC14–related childhood‐onset, striatonigral degeneration recently described in two unrelated children. Post mortem examination demonstrated prominent vacuolation associated with degenerating neurons in the caudate nucleus, putamen, and globus pallidus, similar to previously reported ex vivo vacuoles seen in the late‐endosome/lysosome of VAC14‐deficient neurons. We identified upregulation of ubiquitinated granules within the cell cytoplasm and lysosomal‐associated membrane protein (LAMP2) around the vacuole edge to suggest a process of vacuolation of lysosomal structures associated with active autophagocytic‐associated neuronal degeneration.
Interpretation
Our findings reveal a distinct clinicopathological phenotype associated with recessive VAC14 mutations.
Introduction
VAC14 is a scaffold protein that complexes with FIG4 and PIKFYVE to regulate biosynthesis of phosphatidylinositol 3,5‐bisphosphate (PI(3,5)P2), a signaling lipid that is important for membrane trafficking of endolysosomal vesicles.1 Deficiency of PI(3,5)P2 leads to the formation of large intracellular endolysosomal vacuoles, identifiable by the presence of late endosomal marker proteins bound to the membranes of the vacuoles.2, 3 In mice, partial or complete loss of VAC14 causes accumulation of vacuoles within cells and neurodegeneration.4 Compound heterozygous variants in VAC14 were recently described as the cause of childhood‐onset striatonigral degeneration in two unrelated males (MIM:617054),5 however, neuropathologic data were not reported. Here we report the clinical features and post mortem findings in two siblings with childhood onset neurodegenerative disease and compound heterozygous variants in VAC14.
Methods
The Royal Children's Hospital Human Research Ethics Committee approved the study. Informed consent was obtained from the patients' father. Clinical details were obtained from the deceased patients' medical records. Exome sequencing of genomic DNA isolated from fibroblasts from both patients was carried out at the Australian Genome Research Facility. Exons were captured using the SureSelect Human All Exon, V5 + UTRs (Agilent) and sequencing, linkage and variant analysis was performed as previously described.6
Results
Clinical data
The affected brothers were born to healthy unrelated parents of Anglo‐Celtic (paternal) and Anglo‐Polish (maternal) descent. The older had an unremarkable perinatal course and normal development for the first 3 years. At three and a half years he presented with clumsy walking, frequent falls, and intention tremor. Within 6 months he was unable to walk due to dystonia, predominantly affecting his left leg. Dystonia progressed to involve all four limbs and he developed dysarthria and impaired truncal balance. One year after symptom onset he developed painful muscle spasms, joint contractures, and urinary incontinence. He was treated simultaneously with trihexyphenidyl and a cervical cord stimulator with some improvement. His dystonia progressed leading to severe weight loss and death at age 5 years.
The second sibling was born 2 years later. He had normal motor and cognitive development until age 2 years when he presented with abnormal movement of his left leg, slowed walking, and slowed speech. This was followed by progressive loss of motor function with severe muscle spasms and dystonia with relative preservation of intellectual capacity. A low‐normal level of free gaba‐aminobutyric acid (GABA) detected in the CSF led to a trial of vigabatrin, which dramatically reduced his muscle spasms, transiently enabling him to stand and walk. The benefit was not sustained and did not slow the progression of his motor impairment and dystonia. At age 4 years he had acute neurological deterioration with vomiting in the setting of fever and died. Both children had normal growth with head circumferences +1–2 SD from the mean and no dysmorphic features. Neither child had seizures.
Both children had urine, plasma, and CSF analysis for evidence of metabolic disease, which were normal. Electroencephalograms for both children were normal. MRI brain was performed for the first child and was reported as normal. The images were not available for review. MRI was not performed for the second child. Electromyography and nerve conduction studies in the second child were normal.
Genetic studies
Thirty two regions achieved a LOD score of 0.6, the maximum score possible using a fully penetrant, recessive genetic model (Fig. 1). Seventy‐one candidate variants were identified after filtration, which included compound heterozygous variants in VAC14. VAC14 is relatively intolerant to functional genetic variation with an RVIS score of 8.57%.7 Variant 1, NM_018052 c.1271 G>T, p.Trp424Leu is a very rare missense mutation with only two heterozygous alleles reported in the gnomAD database in the European (non‐Finnish) population. This variant was reported by Lenk et al.5 as pathogenic when carried in trans with a splice‐site variant. The substitution affects a highly conserved amino acid located within one of the heat repeat domains. Variant 2 in the siblings, NM_018052 c.1096 + 1 G>C is novel and disrupts the consensus splice‐donor site of intron 9. It is absent from population databases and affects a highly conserved nucleotide, predicted by in silico tools to cause a frameshift and premature stop codon, leading to truncation of the transcript by more than 50%, therefore leaving it subject to nonsense‐mediated decay (NMD). These variants were confirmed by Sanger sequencing of gDNA (Fig. 1). The c.1096 + 1 G>C variant was paternally inherited. The children's mother is deceased and therefore could not be tested. The effect of the splice‐site variant was assessed using RNA isolated from patient fibroblasts. Sequencing of the cDNA region spanning the c.1271 G>T, p.Trp424Leu mutation revealed only the mutant maternally inherited transcript. The paternally inherited allele encoding the reference G at position c.1271 was not detectable, consistent with loss of the c.1096 + 1 G>C allele in the cDNA due to nonsense‐mediated decay of the mRNA (Fig. 2).
Figure 1.
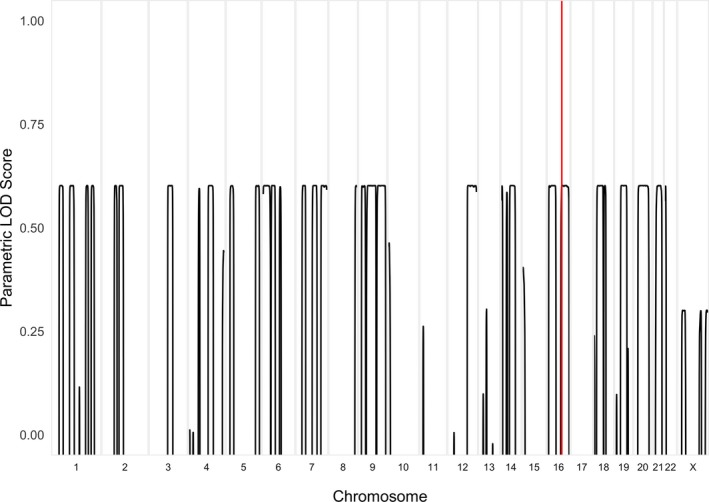
LOD plot. Linkage analysis was performed using SNP genotype calls based on the HapMap database. A recessive genetic model was applied with a 100% penetrance for homozygous carriers and 0% for heterozygous or homozygous reference samples, with an estimated population prevalence of 0.001%. This analysis identified several linkage regions genome‐wide with a near maximal LOD score (~0.6). The two VAC14 mutations were within the chromosome 16 linkage region, highlighted in red.
Figure 2.
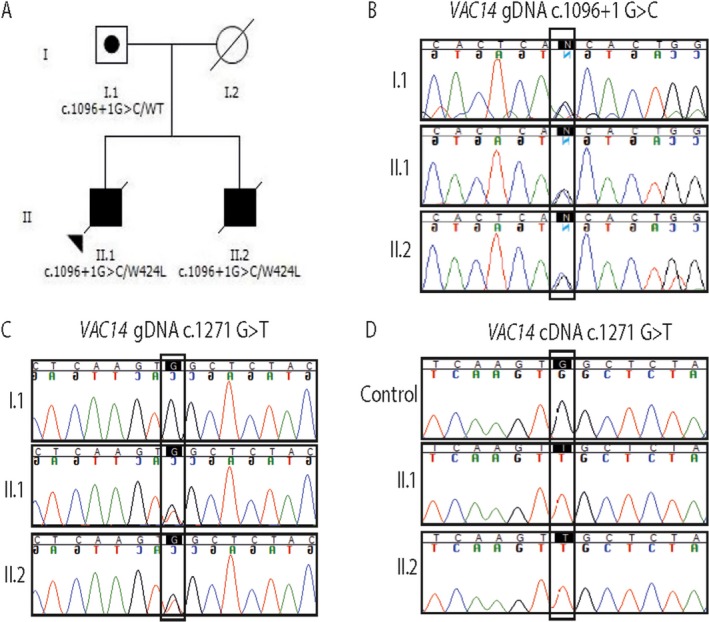
(A) Pedigree with segregation of two allelic VAC14 variants. (B) Sanger sequencing of the paternally inherited c.1096 + 1 G>C variant. (C–D) Comparison of genomic DNA and complementary DNA sequencing of the c.1271 G>T variant; Both alleles are detected in the patients' genomic sequence (C), however, sequencing of the cDNA detected only the allele carrying the c.1271 G>T variant, consistent with loss of the c.1096 + 1 G>C allele due to nonsense‐mediated decay of the mRNA (D).
Neuropathology
Histology of the brain showed very prominent vacuolation in the neuropil in the caudate nucleus, putamen, and globus pallidus associated with degenerating neurons (Fig. 3A). The vacuolation could be seen to extend from the cell cytoplasm of degenerating neurons showing eosinophilic granular cell cytoplasm (Fig. 3B), immunoreactive with ubiquitin (DAKO) and LAMP2 (Chemicon) (Fig. 3C–D). A thin LAMP2 immunoreactive membrane lining the vacuoles was also noted (Fig. 3D). There was no spheroidal accumulation of neurofilament protein (DAKO) (not shown). There was a background of minor gliosis but no inflammatory infiltrate. There was no iron pigment but focal areas of calcification were seen. There was no demyelination on luxol fast blue stains and there were no PAS‐positive bodies accumulating. Minor changes were seen in the pons where similar vacuolation was seen in the tegmentum. The substantia nigra was well preserved with no obvious neuronal loss and very sparse eosinophilic bodies identified. Elsewhere in the cortex, cerebellum, and brain stem appeared normal with the exception of occasional eosinophilic bodies in the amygdala, anterior horn, and thalamus. No peripheral nervous system tissue was available for analysis.
Figure 3.
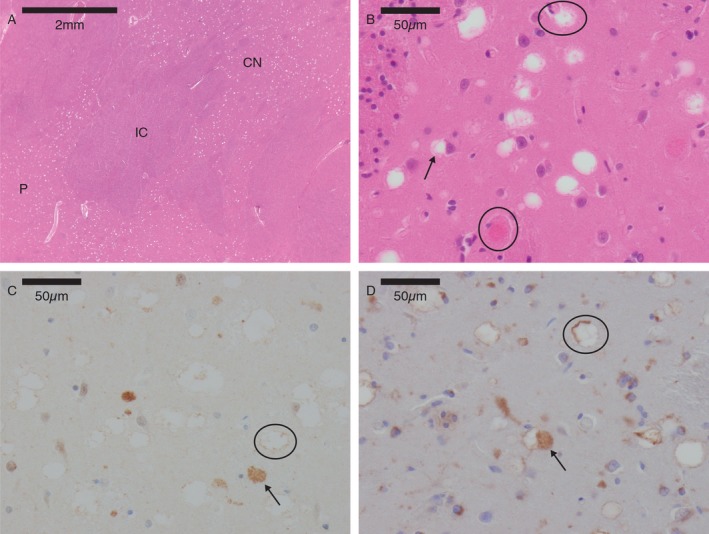
Neuropathology. (A) Marked vacuolation in the caudate nucleus (CN) and putamen (P), with unremarkable internal capsule (IC). Hematoxylin and Eosin × 4. (B) Vacuoles present in the neuropil could sometimes be seen to extend from the cell body (arrow). Degenerating granular eosinophilic neurons were seen (circles). Putamen, Hematoxylin, and Eosin × 400. (C) Vacuoles showed a fine ubiquitin immunoreactive rim (circle). Granular dense ubiquitin was present in degenerating neurons (arrow). Putamen, ubiquitin immunoperoxidase × 400. (D) Vacuoles within the putamen were lined by LAMP2 immunoreactive membrane (circle). LAMP 2 granular bodies were present in cytoplasmic bodies (arrow). Putamen, LAMP2 immunoperoxidase × 400.
Discussion
Here we describe for the first time the neuropathological features of VAC14‐associated childhood‐onset basal ganglia degeneration. VAC14 is a dimeric protein involved in intracellular vesicle transport through the endolysosome pathway. It forms a trimolecular complex with FIG4 and PIKFYVE that regulates biosynthesis of PI(3,5)P2 in the endosomal membrane. The membrane content of PI(3,5)P2 is regulated by a dynamic process mediating the generation and fusion of intracellular transport vesicles. The tight regulation of PI(3,5)P2 levels is achieved by the combined action of the lipid kinase PIKFYVE, which synthesizes PI(3,5)P2 from PI3P, and the PI(3,5)P2 phosphatase FIG4 that can remove the 5‐phosphate, as well as the scaffold protein VAC14, which stabilizes the protein complex and may activate PIKFYVE.1, 3 Impaired regulation of PI(3,5)P2 levels can result from mutation of any of these three components of the VAC14 complex.8 The resultant effect on the endolysosomal pathway manifests as vacuolation of the cell, which has been observed in patient fibroblasts9 and brain tissue from mouse models10, 11 and our patients.
The functions of PI(3,5)P2 appear to be critical for neuronal survival. A VAC14‐null mouse model demonstrates extensive neurodegeneration and early lethality with neuronal cell bodies in the CNS that are completely vacuolated.4 A hypomorphic mouse model homozygous for a missense mutation in VAC14 lives up to 3 weeks and demonstrates smaller areas of spongiform degeneration in the brain involving the thalamus, brainstem, and cerebellar nucleus, which more closely resembles the pathological changes in our patients.3 It has also been demonstrated that neurodegeneration in the FIG4 null mouse can be rescued by the activity of a neuron‐specific transgene.11 In our patients, the cellular vacuolation is seen in the neuritic processes rather than the cytoplasm of the CNS neurons and neurodegeneration is mainly limited to the basal ganglia. The reason for this localization is not known as VAC14 is ubiquitously expressed and not GABA specific.
The presence of large apparently empty cytoplasmic vacuoles lined by LAMP2 suggests a vacuole arising within a dilated lysosomal structure. A lack of material centrally is in keeping with lipid dissolution during the process of tissue fixation. These findings reflect the previously reported ex vivo vacuoles seen in the late‐endosome/lysosome of VAC14 deficient neurons.12 Upregulation of ubiquitinated, LAMP‐positive granules in degenerating neurons suggests that the process of massive vacuolation of lysosomal structures is associated with active autophagocytic associated neuronal degeneration.
The patients described here are compound heterozygotes carrying one loss‐of‐function allele and the missense allele p.Trp424Leu. One of the previously reported patients also carries the p.Trp424Leu allele in combination with a different protein truncating mutation. Both families include Polish ancestry, suggesting that this missense allele may be enriched in that population.
The two previously reported cases of recessive VAC14‐related childhood‐onset neurodegeneration share several clinical features with our patients. All four affected males had normal motor and cognitive development prior to onset of disease in early childhood, between 1.5 and 3 years of age. Symptom onset was marked by spasticity and dystonia with relative preservation of cognitive function. The disease course is rapidly progressive, leading to premature death as early as 2 years after onset. The slightly low GABA levels in the CSF fluid and positive response to the GABA‐ergic anticonvulsant vigabatrin in one child may reflect destruction of the basal ganglia and loss of GABA‐ergic neurons. Vigabatrin may therefore be considered for temporary relief of symptoms in other cases of VAC14‐related neurodegerative disease.
The patients reported by Lenk et al.5 have signal abnormalities in the striatum and substantia nigra on susceptibility and diffusion‐weighted images. Our patient had MRI prior to the availability of these sequences and it was reported as normal. Although our patient's MRI is not available for comparison, neither of our patients showed histological abnormalities in the substantia nigra. Evidence of nigral involvement is therefore not an essential feature of this disease.
Conclusion
VAC14 is essential for vesicular trafficking in the endolysosomal pathway. Deficiency of this protein leads to vacuolation of the neuropilin in the brain, particularly the basal ganglia. Biallelic VAC14 mutations cause a distinctive phenotype of childhood‐onset progressive dystonia that is symptomatic of the underlying basal ganglia degeneration.
Author Contributions
Conception and design of the study: CS, CM, RL, MD, DA, PL. Acquisition and analysis of data: CS, PD, CM, MB, MFF, SS, CWC, PL. Drafting a significant portion of the manuscript or figures: CS, PD, CM, CWC, PL, MM.
Conflicts of Interests
PL and MB receive salary support and project funding from grants from the National Health and Medical Research Council of Australia.
Acknowledgments
We thank the family for participating in this study. CS was supported by NHMRC Postgraduate Scholarship (ID: APP1133266) and the Royal Children's Hospital/Murdoch Childrens Research Institute Flora Suttie Neurogenetics Fellowship made possible by the Thyne‐Reid Foundation and the Macquarie Foundation. MB was supported by NHMRC Program Grant (ID: 1054618) and NHMRC Senior Research Fellowship (ID: 1102971). PJL was supported by an NHMRC Career Development Fellowship (GNT1032364). This work was supported by the Victorian Government's Operational Infrastructure Support Program and Australian Government National Health and Medical Research Council Independent Research Institute Infrastructure Support Scheme (NHMRC IRIISS). MHM acknowledges support from the USPHS (GM24872). RJL is supported by a Melbourne Children's Clinician Scientist Fellowship. We thank Bronwyn Christiansen from Anatomical Pathology at the Royal Children's Hospital, Melbourne for her assistance with access to the pathology specimens. We thank Michael Wilson from Bruce Lefroy Centre for Genetic Health Research, MCRI, for his assistance in formatting figures for the manuscript.
[Correction added on 21 December 2017 after first online publication: Reference 3 and 4 have been renumbered]
Funding Statement
This work was funded by NHMRC Postgraduate Scholarship grant APP1133266; Royal Children's Hospital/Murdoch Childrens Research Institute grant ; Thyne‐Reid Foundation grant ; Macquarie Foundation grant ; NHMRC Program Grant grant 1054618; NHMRC Senior Research Fellowship grant 1102971; NHMRC Career Development Fellowship grant GNT1032364; Victorian Government's Operational Infrastructure Support Program grant ; Australian Government National Health grant ; Medical Research Council Independent Research Institute Infrastructure grant ; USPHS grant GM24872; Melbourne Children's Clinician Scientist Fellowship grant .
References
- 1. Sbrissa D, Ikonomov OC, Fu Z, et al. Core protein machinery for mammalian phosphatidylinositol 3,5‐bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve‐PIKfyve complex. J Biol Chem 2007;282:23878–23891. [DOI] [PubMed] [Google Scholar]
- 2. Schulze U, Vollenbroker B, Braun DA, et al. The Vac14‐interaction network is linked to regulators of the endolysosomal and autophagic pathway. Mol Cell Proteomics 2014;13:1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin N, Chow CY, Liu L, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J 2008;27:3221–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Y, Zolov SN, Chow CY, et al. Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5‐bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci USA 2007;104:17518–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lenk GM, Szymanska K, Debska‐Vielhaber G, et al. Biallelic mutations of VAC14 in pediatric‐onset neurological disease. Am J Hum Genet 2016;99:188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marsh AP, Heron D, Edwards TJ, et al. Mutations in DCC cause isolated agenesis of the corpus callosum with incomplete penetrance. Nat Genet 2017;49:511–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petrovski S, Wang Q, Heinzen EL, et al. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lenk GM, Meisler MH. Mouse models of PI(3,5)P2 deficiency with impaired lysosome function. Methods Enzymol 2014;534:245–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X, Chow CY, Sahenk Z, et al. Mutation of FIG 4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain 2008;131:1990–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Campeau PM, Lenk GM, Lu JT, et al. Yunis‐Varon syndrome is caused by mutations in FIG 4, encoding a phosphoinositide phosphatase. Am J Hum Genet 2013;92:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferguson CJ, Lenk GM, Jones JM, et al. Neuronal expression of Fig 4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet 2012;21:3525–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y, McCartney AJ, Zolov SN, et al. Modulation of synaptic function by VAC14, a protein that regulates the phosphoinositides PI(3,5)P(2) and PI(5)P. EMBO J 2012;31:3442–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]