

Abstract
Background and Purpose
Multicatalytic endopeptidase complex‐like‐1 (β2i), low molecular mass polypeptide (LMP) 2 (β1i) and LMP7 (β5i) are the proteolytically active subunits of the immunoproteasome, a special type of proteasome mainly expressed in haematopoietic cells. Targeting LMP7 has been shown to be therapeutically effective in preclinical models of autoimmune diseases. In this study, we investigated the selectivity and biological activity of LU‐005i, a recently described inhibitor of the immunoproteasome.
Experimental Approach
The specificity of LU‐005i and other immunoproteasome‐selective inhibitors was characterized using fluorogenic peptide substrates. The effect of proteasome inhibition on cytokine release was investigated in endotoxin‐stimulated mouse splenocytes or human peripheral blood mononuclear cells (PBMCs). The effect of proteasome inhibition on inflammatory bowel disease in the dextran sulfate sodium (DSS)‐induced colitis model was assessed by measuring weight loss and colon length.
Key Results
LU‐005i is the first human and mouse immunoproteasome‐selective inhibitor that targets all three proteolytically active immunoproteasome subunits. LU‐005i inhibited cytokine secretion from endotoxin‐stimulated mouse splenocytes or human PBMCs. Furthermore, differentiation of naïve T helper cells to T helper 17 cells was impaired in the presence of LU‐005i. Additionally, LU‐005i ameliorated DSS‐induced colitis.
Conclusion and Implications
This study with a novel pan‐immunoproteasome inhibitor substantiates that the immunoproteasome is a promising drug target for the treatment of inflammatory diseases and that exclusive inhibition of LMP7 is not necessary for therapeutic effectiveness. Our results will promote the design of new generations of immunoproteasome inhibitors with optimal therapeutic efficacy for clinical use in the treatment of autoimmunity and cancer.
Abbreviations
- CD
cluster of differentiation
- CP
constitutive proteasome
- DAI
disease activity index
- DSS
dextran sulfate sodium
- EAE
experimental autoimmune encephalomyelitis
- IP
immunoproteasome
- LMP
low molecular mass polypeptide
- MECL‐1
multicatalytic endopeptidase complex‐like‐1
- MHC
major histocompatibility complex
- NEPHGE
non‐equilibrium pH gradient gel electrophoresis
- PBMCs
peripheral blood mononuclear cells
- TCR
T cell receptor
- Th
T helper cell
- UTY
ubiquitously transcribed tetratricopeptide repeat gene, Y‐linked
Introduction
The proteasome is the main protease in charge of generating ligands for major histocompatibility complex (MHC) I presentation (Groettrup et al., 1996; Basler et al., 2009). It has a barrel‐shaped structure consisting of four rings, each with seven subunits. The inner two rings consist of β‐subunits and bear the catalytically active subunits proteasome subunit beta 6 (β1c), proteasome subunit beta 7 (β2c) and proteasome subunit beta 5 (β5c) (Huber et al., 2012). In haematopoietic cells and in cells stimulated with IFN‐γ or TNF‐α, these proteolytically active subunits are replaced by proteasome subunit beta 9 [β1i; low molecular mass polypeptide (LMP)2], proteasome subunit 10 [β2i; multicatalytic endopeptidase complex‐like (MECL)‐1] and proteasome subunit beta 8 (β5i; LMP7) forming the immunoproteasome (IP). The immunoproteasome is involved in the generation of MHC‐I ligands (Basler et al., 2004; 2011; 2012; 2013; Kincaid et al., 2012), in T cell expansion (Basler et al., 2006; Moebius et al., 2010), T helper cell differentiation (Muchamuel et al., 2009; Kalim et al., 2012; Basler et al., 2014; Vachharajani et al., 2017), in protection from immunopathological damage in the brain (Kremer et al., 2010; Mundt et al., 2016) and in autoimmune diseases (Muchamuel et al., 2009; Basler et al., 2010, 2014, 2015; Zaiss et al., 2011; Ichikawa et al., 2012; Nagayama et al., 2012; Liu et al., 2017).
Immunoproteasome inhibition ameliorated disease symptoms in different animal models of autoimmune diseases, like rheumatoid arthritis, inflammatory bowel disease, Hashimoto's thyroiditis, systemic lupus erythematosus, neuritis (Liu et al., 2017) and multiple sclerosis (summarized in ref. Basler et al., 2015). Two major pathways involved in disease development are affected by LMP7 inhibition, namely, cytokine secretion and T helper cell differentiation. LMP7 inhibition of T cell receptor (TCR)‐activated T cells and endotoxin‐stimulated human peripheral blood mononuclear cells (PBMCs) or mouse splenocytes strongly reduced the secretion of numerous pro‐inflammatory cytokines (Muchamuel et al., 2009; Basler et al., 2010; 2011; 2012; 2014; Ichikawa et al., 2012; Sula Karreci et al., 2016). Additionally, LMP7 inhibition prevents the differentiation of naïve T helper cells to polarized Th17 cells in vitro (Muchamuel et al., 2009; Kalim et al., 2012; Liu et al., 2017). These findings indicate that immunoproteasome is a valid therapeutic target for the treatment of autoimmune diseases. Nevertheless, except for one study (Sula Karreci et al., 2016), all these results were based on studies with a single compound: ONX 0914 (formerly named PR‐957). Therefore, it is essential to confirm previous data with new compounds and to investigate the therapeutic benefit of simultaneously inhibiting all three active site subunits of the immunoproteasome.
Recently, using a structure‐based approach, new immunoproteasome‐selective compounds were identified (de Bruin et al., 2014). In the present study, we characterized the biological activity of a panel of these compounds. We found that an inhibitor selective for all three catalytically active subunits of the immunoproteasome (LU‐005i) was able to inhibit pro‐inflammatory cytokine release from LPS‐stimulated mouse splenocytes and human PBMCs, reduce T helper cell differentiation and ameliorate experimental colitis.
Methods
Animals
C57BL/6 mice (H‐2b) were originally purchased from Charles River, Germany. LMP7 gene‐targeted mice (Fehling et al., 1994) were kindly provided by Dr John J. Monaco (Department of Molecular Genetics, Cincinnati Medical Centre, Cincinnati, OH, USA). Animal experiments were approved by the Review Board of Regierungspräsidium Freiburg in accordance with German Animal Protection Law. The study went through a process of ethical review (Regierungspräsidium Freiburg) prior to its commencement. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All the animals were housed in groups of three to five animals in Eurotype II long clear‐transparent plastic cages with autoclaved dust‐free sawdust bedding. They were fed a pelleted and extruded mouse diet ad libitum and had unrestricted access to drinking water. The light/dark cycle in the room consisted of 12/12 h with artificial light. Mice were kept in a specific pathogen‐free facility, and female mice were used at 8–12 weeks of age, at a weight of 20–25 g. Mice were killed by cervical dislocation or CO2 inhalation.
Cell lines and media
LCL721.174 cells [contain constitutive proteasome (CP)] and LCL721.145 cells (contain immunoproteasome) (Salter and Cresswell, 1986) were kindly provided by Hansjörg Schild (Mainz University). All media were purchased from Invitrogen‐Life Technologies (Karlsruhe, Germany) and contained GlutaMAX, 10% FCS and 100 U·mL−1 penicillin/streptomycin.
Purification of 20S proteasome from mouse organs and fluorogenic assays
The purification of 20S proteasomes from the liver of LMP7−/−/MECL‐1−/− double deficient mice (source of mouse constitutive proteasome), from the liver of lymphocytic choriomeningitis virus (LCMV)‐infected (8 days post infection with 200 pfu LCMV‐WE i.v.) BALB/c mice (source of mouse immunoproteasome), LCL 721.174 cells (source of human constitutive proteasome) or LCL 721.145 cells (source of human immunoproteasome) as well as the analysis of their subunit composition on two‐dimensional non‐equilibrium pH gradient electrophoresis (NEPHGE)‐SDS‐PAGE was performed as described previously (Basler and Groettrup, 2012).
Hydrolytic assays for proteasome activity were performed using fluorogenic substrates, as detailed previously (Basler and Groettrup, 2012). Substrates were dissolved in DMSO (10 mM) and used at 12.5 μM (Ac‐PAL‐AMC; BostonBiochem) or at 100 μM (Suc‐LLVY‐AMC, Bz‐VRG‐AMC, Z‐LLE‐βNA; Bachem). IC50 values were determined using GraphPad Prism Software (version 4.03).
To investigate the cell permeability of LU‐005i, LCL721.174 cells (contain CP) and LCL721.145 cells (contain immunoproteasome) were treated with different inhibitor concentrations in PBS + 25 mM HEPES for 30 min at 37°C. The cell permeable substrate MeoSuc‐GLF‐AMC (Bachem) (in DMSO 10 mM) was added at 40 μM to the cells and incubated at 37°C. The fluorescence intensity in the wells containing the cells was measured at an excitation wavelength of 360 nm and emission wavelength of 465 nm (infinite M200 pro, TECAN).
LacZ assay
The LacZ assay was performed exactly as described previously (Basler and Groettrup, 2013). Briefly, 105 cells of the ubiquitously transcribed tetratricopeptide repeat gene, Y‐linked (UTY)246–254‐specific T cell hybridoma (donated by N. Shastri, University of California, Berkeley, Berkeley, CA, USA) were co‐cultured with 1.5 × 106 stimulator cells in 96‐well plates overnight. The lacZ‐based colour reaction was achieved and measured, as described in detail previously (Basler et al., 2004).
Cytokine secretion assay
Splenocytes (106 per well) from C57BL/6 mice were incubated with proteasome inhibitors or DMSO and stimulated with plate bound anti‐cluster of differentiation (CD) 3 (5 μg·mL−1, clone 145‐2C11, eBioscience) and anti‐CD28 (5 μg·mL−1, clone 37.51, eBioscience) antibodies or stimulated with 3 μg·mL−1 LPS. After 20 h, IFN‐γ or IL‐6 in the supernatant was determined by elisa, according to the manufacturer's protocol (eBioscience). Human PBMCs from healthy volunteers were incubated with proteasome inhibitors or DMSO at 37°C and stimulated with 3 μg·mL−1 LPS (from E. coli 0111:B4) (Sigma). After 20 h, IL‐23 and IL‐6 levels in the supernatant were determined by elisa, according to the manufacturer's protocol (eBioscience).
Proteasome immunoprecipitation
To determine the proteasomal activity in lysates with the fluorogenic substrate Ac‐PAL‐AMC (BostonBiochem), LCL721.174 cells were incubated with 300 nM LU‐001i or DMSO overnight at 37°C. Immunoprecipitation was performed as described previously (Basler and Groettrup, 2012). Briefly, after extensive washing, equal amounts of cells were lysed and incubated with 3 μL of polyclonal rabbit anti‐mouse proteasome antibodies (Schwarz et al., 2000) and 50 μL protein A microbeads (μMACSProtein A microbeads; Miltenyi Biotec) for 30 min on ice. The lysate was applied to the μ column (Miltenyi Biotec), and the column was washed five times. Then, 50 μL fluorogenic peptide substrate (Ac‐PAL‐AMC) was applied to the column at a concentration of 12.5 μM. The μ column was incubated at 37°C for 30 min. A total of 100 μL lysis buffer was added, and the fluorescence intensity in the eluate was measured at an excitation wavelength of 360 nm and emission wavelength of 465 nm (infinite M200 pro, TECAN).
MHC‐I surface expression
Splenocytes from C57BL/6 or LMP7−/− mice were incubated overnight with various concentrations of LU‐005i or LU‐001i or DMSO control. H‐2Kb surface staining was performed as described previously (Basler and Groettrup, 2007). Briefly, splenocytes were incubated with anti‐mouse CD16/32 (clone 2.4G2) to block Fc‐receptors and then stained for H‐2Kb (clone AF6‐88.5, BD Biosciences) for 30 min. After two washes, cells were acquired with the use of the Accuri C6 flow cytometer system (BD Biosciences). Lymphocytes were gated based on forward scatter and side scatter, and the median H‐2Kb fluorescence of gated lymphocytes was calculated.
Propidium iodide staining
Splenocytes from C57BL/6 mice were incubated with various concentrations of LU‐005i or DMSO control for 1 to 3 days. Cells were stained with antibodies (all BioLegend) for CD11c, CD11b, F4/80, CD4, CD8, CD19 and NK1.1. The viability of these cells was assessed with propidium iodide (PI) (eBioscience) using the BD FACSVerse™ flow cytometer (BD Biosciences).
Th17 cell differentiation
Magnetically purified CD4+ T cells (MACS; Miltenyi Biotech) were stimulated (8 × 104 cells per well) with plate‐bound antibodies to CD3 (5 μg·mL−1, clone 145‐2C11, eBioscience) and CD28 (5 mg·mL−1, clone 37.51, eBioscience) in the presence of 2.5 ng·mL−1 TGF‐β (Preprotech), 30 ng·mL−1 IL‐6 (eBioscience) and antibodies to IL‐4 (10 μg·mL−1; eBioscience) and IFN‐γ (10 μg·mL−1; eBioscience) for 3 days. Intracellular IL‐17A expression was measured 4 h after exposure to 50 ng·mL−1 PMA (Sigma) and 500 ng·mL−1 ionomycin (Sigma) in the presence of 10 mg·mL−1 brefeldin A (Sigma) by flow cytometery (Accuri C6).
Induction of colitis
Oral administration of dextran sulfate sodium (DSS) in mice has been widely used to study the mechanisms of colonic inflammation and to evaluate the effect of any candidate drug for inflammatory bowel disease. Additionally, this animal model has been chosen to compare the new data with previous results with different proteasome inhibitors obtained in this model (Basler et al., 2010; Schmidt et al., 2010). Mice were randomly separated into three groups (vehicle, LU‐005i and LU‐001i). Colitis was induced in 8 to 10‐week‐old mice by adding 3% DSS (m.w. 36 000–50 000; MP Biomedicals, Solon, OH, USA) to the drinking water, beginning at day 0 for 5 days; thereafter, mice were given regular drinking water. The body weight was measured daily throughout the experiment. Mice were killed with CO2. A weight loss >25% was defined as a humane endpoint, no animal reached the endpoint. Colitis severity was scored by evaluating clinical disease activity through daily observation of the following parameters: weight loss (0: no weight loss or weight gain; 1: 5–10% weight loss; 2: 10–15% weight loss; 3: 15–20% weight loss; 4: >20% weight loss); stool consistency (0: normal and well formed; 2: very soft and unformed; 4: watery stool); and bleeding stool score (0: normal colour stool; 2: reddish colour stool; 4: bloody stool). The disease activity index (DAI) was calculated based on the combined scores of weight loss, stool consistency and bleeding ranging from 0 to 12.
Real‐time RT‐PCR
Real‐time RT‐PCR was used to quantify cytokine expression levels in mouse colons. Total RNA was extracted from the colon of mice using a NucleoSpinRNA II extraction kit (MACHEREY‐NAGEL, Dueren, Germany). One microgram of total RNA was reverse transcribed using oligonucleotide (dT) primers and the reverse transcription system (Promega, Madison, WI, USA). Quantitative PCR was performed with the Toptical professional thermocyler instrument (Analytic Jena), using the Biozym Blue S'Green qPCR Kit (Biozym), with the following primers: IL‐1β‐specific forward: 5′‐TTGACGGACCCCAAAAGATG‐3′; IL‐1β reverse primer: 5′‐AGAAGGTGCTCATGTCCTCA‐3′; TNF‐α–specific forward: 5′‐ATGAGCACAGAAAGCATGATC‐3′; TNF‐ α reverse primer: 5′‐TACAGGCTTGTCACTCGAATT‐3′; IL‐17–specific forward: 5′‐CTCCAGAAGGCCCTCAGACTAC‐3′; IL‐17 reverse primer: 5′‐GCTTTCCCTCCGCATTGACACAG‐3′; IL‐6‐specific forward: TGGACAGGACTGAAAGACTTG‐3′; IL‐6 reverse primer: 5′‐CCAGCAGGTCAGCAAAGAACTTA‐3′; IFN‐γ–specific forward: 5′‐ATGAAC GCTACACACTGCATC‐3′; and IFN‐γ reverse primer: 5′‐CCATCCTTTTGCCAGTTCCTC‐3′. Mouse hypoxanthineguanine phosphoribosyl transferase was used as a reference gene with the following primers: 5′‐TGGACAGGACTGAAAGACTTG‐3′ (forward) and 5′‐CCA GCA GGT CAG CAA AGA ACT TA‐3′ (reverse). Data were analysed by ΔCT‐method (2‐ΔΔCT) (Schmittgen and Livak, 2008).
Histology
Mouse colons were Swiss‐rolled and fixed in 10% formalin followed by paraffin embedding. Tissue sections of 4 μm were stained with haematoxylin and eosin for pathological evaluation in a blinded manner. Histology scores were assessed as previously defined by Horino et al. (2008) with slight modifications. The score is based on three different parameters: inflammation severity (0–3); extent of inflammation (0–3); and crypt damage (0–4).
Data and statistical analysis
In the DSS‐induced colitis animal experiment, a co‐worker blinded to the experimental protocol randomized animals into groups. For all other experiments randomization is not applicable. DAI was determined in a blinded manner. Histological scoring was performed in a blinded manner by two people who were unaware of the sample group. All other experiments were not subjected to blinding since they result in values not influenced by personal bias. Groups which contained five or more independent replicates were subjected to statistical analysis. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). elisa and flow cytometry experiments were performed in triplicate to ensure the reliability of singe values. Group sizes within an experiment are equal, except for the naïve mice used in Figure 5. This group was used as an illustrative control group, and therefore, the group size was reduced so as not to unnecessarily kill mice. The statistical significance of the differences was determined using either Student's t‐test or one‐way ANOVA with Dunnett's multiple comparisons test. Dunnett's multiple comparisons test was run only if F achieved P < 0.05 and there was no significant variance inhomogeneity as determined with Bartlett's test. All statistical analyses were performed using GraphPad Prism Software (version 4.03) (GraphPad, San Diego, CA, USA). A P value of <0.05 was considered significant. If not indicated otherwise, differences are not significant. The number of mice for the colitis experiment was determined using G*Power 3.0.10 (comparison of two independent groups) (Faul et al., 2007).
Materials
Proteasome inhibitors LU‐005i, LU‐015i, LU‐025i, LU‐035i, LU‐045i, LU‐055i and LU‐001i were synthesized as described previously (de Bruin et al., 2014). ONX 0914 (Muchamuel et al., 2009) (formerly called PR‐957) (Onyx Pharmaceuticals) and other proteasome inhibitors were dissolved at a concentration of 10 mM in DMSO and stored at −80°C (Muchamuel et al., 2009). For proteasome inhibition in mice, LU‐005i and LU‐001i were dissolved in 5% ethanol, 10% PEG 300 (Sigma) in an aqueous solution of 20% (w . v‐1) sulfobutylether‐ß‐cyclodextrin and 10 mM sodium citrate (pH 6) and administered to mice s.c. (15 mg·kg−1).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Specificity of LU‐005i, LU‐015i, LU‐025i, LU‐035i, LU‐045i, LU‐055i and LU‐001i
Using a structure‐based design approach, de Bruin et al. (2014) obtained proteasome inhibitors with considerably improved selectivity for β5i over β5c and β2i over β2c. Out of these inhibitors, compounds LU‐005i, LU‐015i, LU‐025i, LU‐035i, LU‐045i, LU‐055i and LU‐001i were chosen to confirm the selectivity of these inhibitors with purified proteasomes in fluorogenic peptide assays. Therefore, human constitutive proteasomes and human immunoproteasomes were purified from the Epstein–Barr virus‐positive lymphoblastoid cell line LCL721.145 (source of immunoproteasome) and its LMP2‐ and LMP7‐deficient deletion mutant LCL721.174 (source of CP) (Salter and Cresswell, 1986). Purity and proteasome composition of the 20S proteasomes from these cell lines was analysed by NEPHGE‐2D‐gel electrophoresis (Supporting Information Figure S1). Proteasomes derived from LCL721.174 cells exclusively contain constitutive proteasomes, whereas immunoproteasomes derived from LCL721.145 cells contain approximately 10% constitutive subunits. Human CP and immunoproteasome were incubated with different concentrations of LU‐005i, LU‐015i, LU‐025i, LU‐035i, LU‐045i, LU‐055i and LU‐001i and assayed with a fluorogenic substrate specific for the chymotrypsin‐like activity of the proteasome (Suc‐LLVY‐AMC). To compare the selectivity of these proteasome inhibitors, the IC50 values were calculated (Table 1). No inhibition of the chymotrypsin‐like activity for the LMP2‐selective inhibitor LU‐001i (de Bruin et al., 2014) was observed, indicating that LU‐001i did not inhibit β5c or β5i. LU‐015i, LU‐025i, LU‐035i and LU‐055i showed IC50 values for the immunoproteasome of around 10−7 M whereas no inhibition of the CP below 10−6 M was observed. LU‐005i and LU‐045i inhibited the chymotrypsin‐like activity of the immunoproteasome at IC50 values of around 10−7 M and were at least 10 times more selective for the immunoproteasome than for the CP. Although human and murine proteasome subunits share more than 90% sequence identity, small differences in the substrate binding pockets can affect inhibitor binding (Huber et al., 2012). Therefore, we tested whether compounds LU‐005i, LU‐015i, LU‐025i, LU‐035i, LU‐045i, LU‐055i and LU‐001i also inhibit mouse 20S proteasomes. Immunoproteasomes were isolated from livers of BALB/c mice 8 days after infection with LCMV, and constitutive proteasomes were purified from livers of uninfected LMP7−/−/MECL‐1−/− gene‐targeted mice (Khan et al., 2001; Huber et al., 2012). After assessment of their purity by two‐dimensional gel electrophoresis as described previously (Khan et al., 2001; Huber et al., 2012), mouse CPs and mouse immunoproteasomes were incubated with different concentrations of LU‐005i, LU‐015i, LU‐025i, LU‐035i, LU‐045i, LU‐055i and LU‐001i and assayed for the chymotrypsin‐like activity (Table 1). No inhibition of the chymotrypsin‐like activity of mouse CP and immunoproteasome below 10−6 M could be observed for LU‐035i, LU‐055i and LU‐001i. LU‐015i, LU‐025i and LU‐045i showed IC50 values in the μM range for mouse immunoproteasome and were therefore at least 10 times less potent compared to human immunoproteasome. In contrast, IC50 values of LU‐005i for mouse immunoproteasome and CP were comparable to human proteasome. To investigate the impact of the inhibitors on the caspase‐like activity, which is evoked by β1c, human and mouse CPs were incubated with different inhibitor concentrations and assayed with the fluorogenic substrate z‐LLE‐βNA (Table 1). None of the inhibitors tested reduced the caspase‐like activity below 10−6 M. Next, mouse and human immunoproteasome were incubated with proteasome inhibitors, and the trypsin‐like activity was assessed with the fluorogenic substrate Bz‐VGR‐AMC (Table 1). Apart from LU‐005i, none of the inhibitors tested affected the trypsin‐like activity, indicating that these inhibitors do not inhibit MECL‐1, the proteolytically active subunit in the immunoproteasome responsible for the trypsin‐like activity.
Table 1.
IC50 values of the indicated compounds were determined using the hydrolysis of fluorogenic substrates for the chymotrypsin‐like (LLVY), caspase‐like (LLE) or trypsin‐like (VGR) activity of 20S immunoproteasomes or constitutive proteasomes of human or mouse origin
| LU‐005i | LU‐055i | LU‐001i | LU‐015i | LU‐045i | LU‐025i | LU‐035i | ||
|---|---|---|---|---|---|---|---|---|
| Chymotrypsin‐like activity (LLVY) | ||||||||
| Human | IP | 1.6 ± 0.8 10−7 M | 3 ± 2 10−7 M | n.i. | 9.9 ± 5.2 10−8 M | 3.4 ± 1.6 10−7 M | 5.1 ± 1.6 10−7 M | 1.4 ± 0.4 10−7 M |
| CP | 3 ± 1.6 10−6 M | n.i. | n.i. | n.i. | 4.6 ± 0.4 10−6 M | n.i. | n.i. | |
| Mouse | IP | 3.8 ± 0.8 10−7 M | n.i. | n.i. | 1.2 ± 0.4 10−6 M | 1.8 ± 0.6 10−6 M | 2.7 ± 0.9 10−6 M | n.i. |
| CP | 3.8 ± 0.9 10−6 M | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. | |
| Caspase‐like activity (LLE) | ||||||||
| Human | CP | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. |
| Mouse | CP | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. |
| Trypsin‐like activity (VGR) | ||||||||
| Human | IP | 4.7 ± 0.9 10−7 M | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. |
| Mouse | IP | 1.8 ± 0.5 10−7 M | n.i. | n.i. | n.i. | n.i. | n.i. | n.i. |
IC50 values ± SD are indicated. n.i., no inhibition observed below 1 μM.
Since we intended to evaluate the potential of immunoproteasome‐selective inhibitors in the mouse system, we further focused on LU‐005i (Figure 1A). Additionally, LU‐001i, described as an LMP2‐selective inhibitor, was further characterized (Figure 1A). Ac‐PAL‐AMC is a fluorescent substrate exclusively cleaved by LMP2 (Blackburn et al., 2010; Basler et al., 2012). Mouse and human immunoproteasomes were incubated with different concentrations of LU‐005i or LU‐001i and assayed with the fluorogenic substrate specific for LMP2 activity (Table 2). Both compounds were potent LMP2 inhibitors. Lastly, the inhibition of the trypsin‐like activity of human and mouse CP was analysed (Table 3). LU‐001i did not affect the trypsin‐like activity of either mouse or human CP. In contrast to mouse CP, for which LU‐005i showed no inhibition below 10−6 M, LU‐005i affected the trypsin‐like activity of human CP with IC50 values of 3.1 μM. Taken together, these findings indicate that LU‐001i (Figure 1A) is a potent mouse and human LMP2 inhibitor that does not affect other catalytically active proteasome subunits (Tables 1, 2, 3). LU‐005i (Figure 1A) is an effective inhibitor of MECL‐1, LMP2 and LMP7 with at least 10‐fold selectivity over β2c, β1c and β5c and, therefore, can be considered as a general immunoproteasome‐selective inhibitor (Tables 1, 2, 3).
Figure 1.
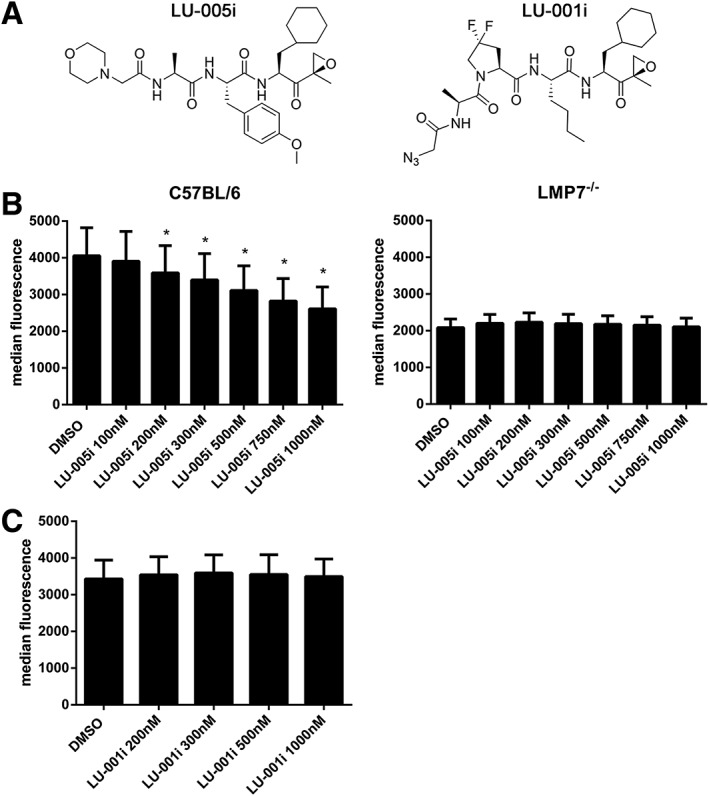
LU‐005i reduces H‐2Kb surface expression on splenocytes. (A) Structure of LU‐005i (left side) or LU‐001i (right side). (B, C) Flow cytometry analysis of H‐2Kb surface expression on splenocytes derived from C57BL/6 (left side) or LMP7−/− (right side) mice treated with the indicated concentrations of LU‐005i (B) or LU‐001i (C) overnight. Pooled data from five independent experiments performed in triplicates are shown as the means of median fluorescent intensity ± SEM. All data were statistically compared to the DMSO treated group. *P < 0.05.
Table 2.
IC50 values of the indicated compounds were determined using the hydrolysis of fluorogenic substrates for LMP2‐activity (PAL‐AMC) of 20S immunoproteasomes of human or mouse origin
| LU‐005i | LU‐001i | ||
|---|---|---|---|
| Human | IP | 5.2 ± 2.9 10−8 M | 1.6 ± 0.2 10−8 M |
| Mouse | IP | 2.0 ± 0.7 10−8 M | 3.9 ± 1.2 10−9 M |
IC50 values ± SD are indicated.
Table 3.
IC50 values of the indicated compounds were determined using the hydrolysis of fluorogenic substrates for the trypsin‐like activity (VGR) of 20S immunoproteasomes or constitutive proteasomes derived from humans or mice
| LU‐005i | LU‐001i | ||
|---|---|---|---|
| Human | IP | 4.7 ± 0.9 10−7 M | n.i. |
| CP | 3.1 ± 0.5 10−6 M | n.i. | |
| Mouse | IP | 1.8 ± 0.5 10−7 M | n.i. |
| CP | n.i. | n.i. |
IC50 values ± SD are indicated. n.i., no inhibition observed below 1 μM.
Next, we tested the cell permeability of LU‐005i and LU‐001i cells. LCL721.174 (contain human CP) or LCL721.145 (contain human IP) cells were incubated with different concentrations of LU‐005i and assayed with the cell permeable substrate MeO‐Suc‐GLF‐AMC (Harding et al., 1995) cleaved by the chymotrypsin‐like activity of the proteasome (Supporting Information Figure S2A). LU‐005i inhibited the cleavage of this substrate in cells containing immunoproteasomes, demonstrating that LU‐005i is cell permeable. Since the substrate to test LMP2‐activity (Ac‐PAL‐AMC) is poorly cell permeable, we tested the cell permeability of LU‐001i with a method based on immunoprecipitation. LCL721.145 (contain IP) cells were incubated with 300 nM LU‐001i. After extensive washing, cells were lysed and the proteasome was immunoprecipitated. The precipitated proteasome was assayed for the cleavage of the LMP2‐specific substrate Ac‐PAL‐AMC (Supporting Information Figure S2B). Whereas the substrate was converted by proteasomes from untreated cells, proteasomes derived from cells incubated with 300 nM LU‐001i had strongly reduced activity, demonstrating the cell permeability of LU‐001i. To determine whether complete inhibition of immunoproteasome induces cell death, mouse splenocytes were incubated with different concentrations of LU‐005i. After 24, 48 and 72 h, CD11c+, CD11b+, NK1.1+, CD19+, CD4+ and CD8+ populations were analysed for cell death by PI staining (Supporting Information Figure S3). Cell death of all populations increased over time independently of LU‐005i treatment. Whereas at 24 h, no significant effect of LU‐005i on cell death could be observed for all populations, a slight increase in the death of CD4+ and CD8+ T cells at 500 and 1000 nM was observed at 48 and 72 h. This is not surprising since the proteasome is essential for cell survival and T cells mainly express immunoproteasome.
LU‐005i reduces the cell surface expression of MHC class I
The cell surface expression of MHC‐I is reduced in LMP7‐deficient mice (Fehling et al., 1994). Therefore, we compared the effect of LU‐005i and LU‐001i on MHC‐I H‐2Kb surface expression in wild‐type or LMP7‐deficient splenocytes (Figure 1B, C). Based on the data obtained in in vitro fluorogenic assays (Tables 1, 2, 3), splenocytes were incubated with different LU‐001i and LU‐005i concentrations ranging from 50 to 1000 nM overnight: LU‐005i reduced the expression of H‐2Kb in a dose‐dependent manner, whereas basal H‐2Kb expression on LMP7−/− splenocytes, which is approximately 50% lower relative to wild‐type levels, was not further affected by LU‐005i treatment (Figure 1B). The LMP2‐selective inhibitor LU‐001i had no influence on the cell surface expression of MHC‐I (Figure 1C).
LU‐005i affects the presentation of an LMP7‐dependent epitope
The generation of the cytotoxic T lymphocyte epitope UTY246–254, derived from the endogenously expressed Y‐chromosome–encoded HY‐Ag, has been shown to be LMP7‐dependent (Palmowski et al., 2006; Basler et al., 2012). Furthermore, the presentation of the UTY246–254 epitope can be reduced by an LMP7‐selective, but not by an LMP2‐selective inhibitor (Muchamuel et al., 2009; Basler et al., 2012). To confirm these results with LU‐005i and LU‐001i, UTY246–254 presentation on the H‐2Db class I molecule of male‐derived splenocytes was determined with a UTY246–254‐specific T cell hybridoma in lacZ assays (Figure 2). As reported previously, the presentation of UTY246–254 was shown to be dependent on LMP7 and could be reduced in a dose‐dependent manner with the immunoproteasome‐selective inhibitor LU‐005i (Figure 2A). The LMP2‐selective inhibitor LU‐001i had no influence on the presentation of the UTY246–254 epitope (Figure 2B).
Figure 2.
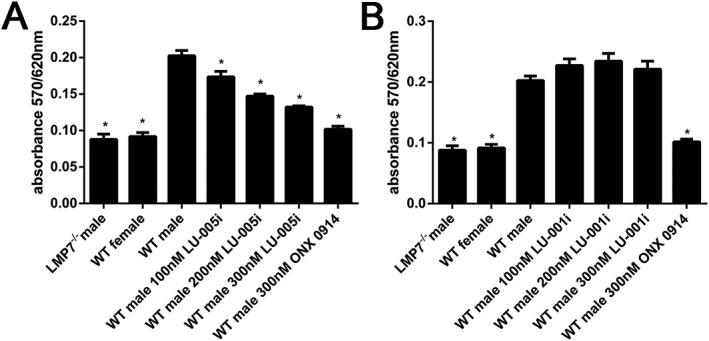
LU‐005i blocks MHC‐I–restricted presentation of an LMP7‐dependent epitope. Presentation of UTY246–254 on MHC‐I by wild‐type (WT) splenocytes after exposure to the indicated concentrations of LU‐005i (A) or LU‐001i (B), as analysed by exposure to H‐2Db‐UTY246–254‐specific, LacZ‐expressing T cell hybridomas. Female, LMP7‐deficient and ONX 0914 (300 nM) treated splenocytes were used as controls. Pooled data from five independent experiments conducted in triplicates are shown as means ± SEM. All data were statistically compared to the WT male group. *P < 0.05.
LU‐005i inhibits cytokine secretion
It has previously been shown that a selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production (Muchamuel et al., 2009; Basler et al., 2010, 2011; 2014). Therefore, splenocytes derived from wild‐type or LMP7‐deficient mice were incubated with different doses of LU‐005i and stimulated with LPS overnight. IL‐6 in the supernatant was detected by elisa (Figure 3A). Whereas IL‐6 secretion of wild‐type splenocytes was reduced in a dose‐dependent manner by LU‐005i, it had no influence on IL‐6 secretion of LMP7‐deficient cells. LU‐001i did not alter IL‐6 secretion of LPS‐stimulated wild‐type splenocytes (Figure 3B). Next, splenocytes derived from wild‐type mice were treated with LU‐005i or LU‐001i and stimulated with plate‐bound anti‐CD3/anti‐CD28 antibodies overnight. IFN‐γ secretion into the supernatant was determined by elisa (Figure 3C). In contrast to LU‐001i, LU‐005i reduced IFN‐γ secretion into the supernatant in a dose‐dependent manner.
Figure 3.
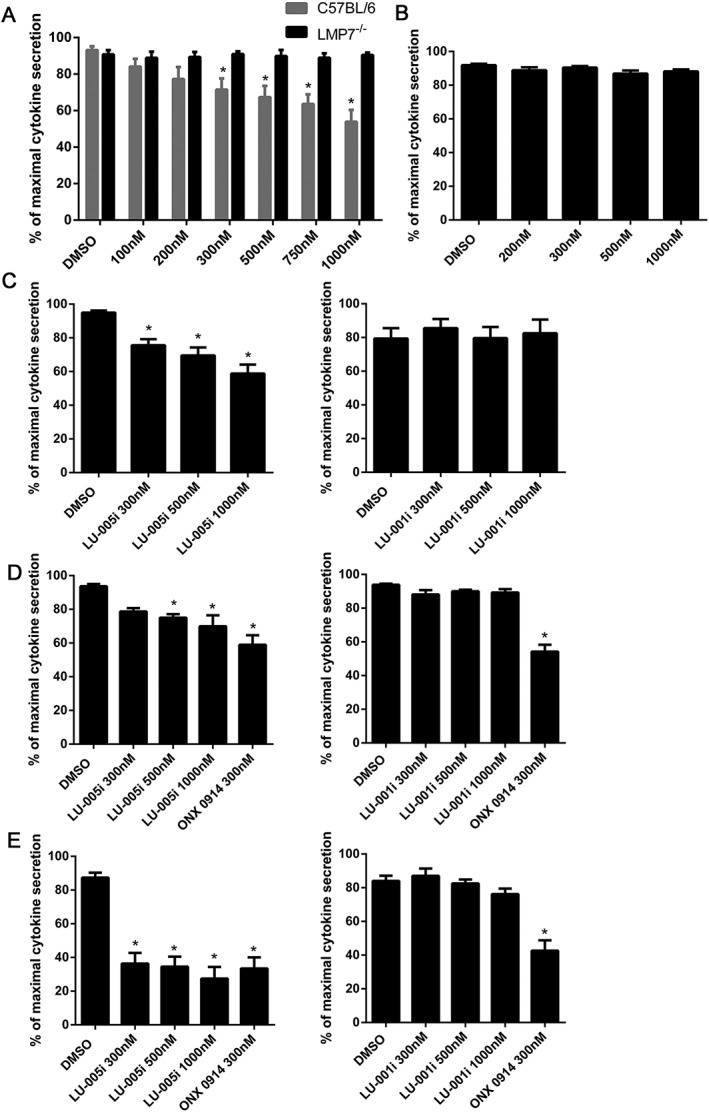
LU‐005i inhibits the production of inflammatory cytokines. (A–C) Splenocytes derived from C57BL/6 (A–C) or LMP7−/− (A) mice were exposed to the indicated concentrations of LU‐005i (A, C) or LU‐001i (B, C) and stimulated with LPS (A, B) or plate‐bound antibodies specific for CD3/CD28 (C) overnight. IL‐6 (A, B) or IFN‐γ (C) in the supernatant was analysed by elisa. Each data point was measured in triplicates. The highest cytokine concentration from each experiment was set to 100% to control for unwanted variations in cytokine secretion between individual experiments. Data are presented as the mean ± SEM of six (A, B) or five (C) independent experiments. (D, E) Human PBMCs were exposed to the indicated concentrations of LU‐005i (left side) or LU‐001i (right side) and stimulated with LPS overnight. IL‐6 (D) or IL‐23 (E) in the supernatant was analysed by elisa. Each data point was measured in triplicates. The highest cytokine concentration from each experiment was set to 100% to control for unwanted variations between individual donors. Data are presented as the mean ± SEM of six (D) or eight (E) independent donors. All data were statistically compared to the DMSO treated group. *P < 0.05.
Next, we evaluated the impact of selective immunoproteasome inhibition on cytokine production of endotoxin‐stimulated human PBMCs. Human PBMCs from healthy volunteers were exposed to LU‐005i or LU‐001i and stimulated with LPS (Figure 3D, E). Compared to 300 nM ONX 0914, LU‐005i only slightly reduced IL‐6 production at 300, 500 and 1000 nM (Figure 3D). In contrast, IL‐23 secretion was fully blocked at 300 nM (Figure 3E). LU‐001i had no effect on cytokine secretion, either of IL‐6 or of IL‐23 (Figure 3D, E).
LU‐005i blocks Th17 differentiation
The importance of the IL‐23/IL‐17 axis in the pathogenesis of autoimmunity is well established. LMP7 inhibition prevents the differentiation of naïve T helper cells to polarized Th1 or Th17 cells in vitro (Muchamuel et al., 2009; Kalim et al., 2012). Therefore, we investigated the effect of LU‐005i on Th17 differentiation. Magnetically‐sorted CD4+ T cells derived from wild‐type or LMP7‐deficient mice were cultured in vitro under Th17 polarizing conditions in the presence of different concentrations of LU‐005i (Figure 4). LU‐005i inhibited the Th17 differentiation in a dose‐dependent manner (Figure 4A). This inhibition was still LMP7‐selective, since LU‐005i did not affect Th17 differentiation in LMP7‐deficient CD4+ cells (Figure 4B). Moreover, LU‐001i was found to have no effects on Th17 differentiation (Figure 4C).
Figure 4.
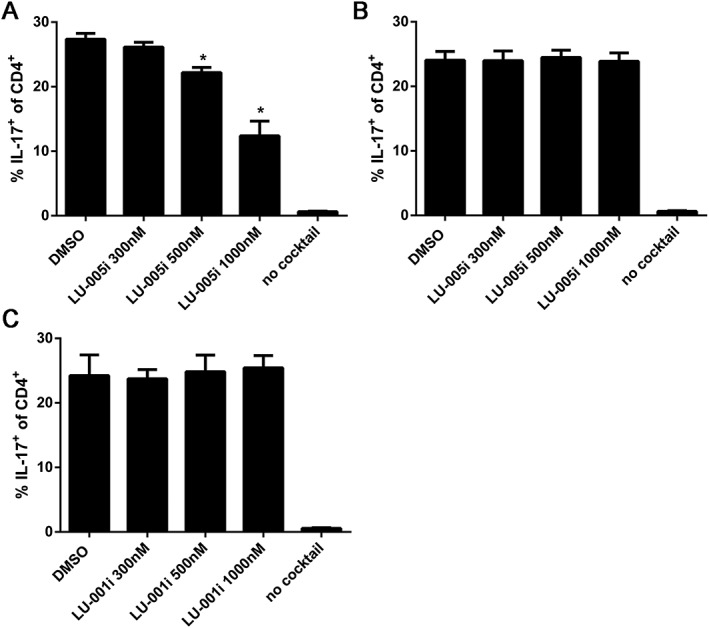
LU‐005i blocks Th17 differentiation. Differentiation of CD4+ T cells from C57BL/6 (A, C) or LMP7−/− (B) mice exposed to the indicated concentrations of LU‐005i (A, B) or LU‐001i (C) and stimulated with antibodies to CD3/CD28 in the presence of TGF‐β and IL‐6 and antibodies to IL‐4 and IFN‐γ was measured in 3 day cultures. IL‐17 expression was detected by intracellular cytokine staining after a short restimulation with PMA/ionomycin. IL‐17 expression in activated T cells cultured in the absence of Th17‐polarizing conditions is shown as a comparison (no cocktail). Pooled data from five independent experiments done in triplicates are shown as means ± SEM. Values reflect the percentage of CD4+ cells that are also IL‐17A+. All data were statistically compared to the DMSO‐treated group. *P < 0.05. The ‘no cocktail’ group was excluded from statistical analysis due to variance inhomogeneity.
LU‐005i reduces the symptoms of DSS‐induced colitis
Immunoproteasome inhibition prevented the production of cytokines driving Th17 generation (Figure 3; Muchamuel et al., 2009; Basler et al., 2014) and IL‐17 production in T cells cultured in the presence of polarizing cytokines (Figure 4; Muchamuel et al., 2009; Kalim et al., 2012). Therefore, we investigated the impact of immunoproteasome inhibition in DSS‐induced colitis in C57BL/6 mice. To analyse whether LU‐005i and LU‐001i were active in mice, C57BL/6 mice were treated with 15 mg·kg−1 LU‐005i or 15 mg·kg−1 LU‐001i, or vehicle. Two and 24 h post treatment, proteasomes were immunoprecipitated from spleen lysates and assessed with a fluorogenic substrate specific for the chymotrypsin‐like activity (Suc‐LLVY‐AMC) and a substrate specific for LMP2‐activity (Ac‐PAL‐AMC; Supporting Information Figure S4). Both inhibitors showed activity in mice and, thus, were used to study the effect of immunoproteasome inhibition on colitis. DSS‐induced colitis was induced by oral administration of 3% DSS in the drinking water for 5 days. Mice were treated daily with LU‐005i (15 mg·kg−1), LU‐001i (15 mg·kg−1) or vehicle. Body weight and disease activity index (DAI) of the mice were recorded for 9 days (Figure 5A, B). Compared to vehicle treatment, the administration of LU‐005i led to a significantly reduced weight loss and DAI. Interestingly, although LMP2‐deficient mice are protected from DSS‐induced colitis (Fitzpatrick et al., 2006; Basler et al., 2010), treatment of mice with the LMP2‐selective inhibitor LU‐001i had no effect on weight loss. The induction of colitis is associated with a reduced colon length in diseased mice. To investigate whether the decreased body weight loss in LU‐005i‐treated mice is accompanied by an altered colon length, this parameter was measured in DSS‐treated mice on day 9 post induction (Figure 5C). Colon length was reduced in DSS‐treated wild‐type mice by approximately 2 cm compared with untreated naive mice. The colon of LU‐005i‐treated mice showed a slight reduction compared to that of naïve mice, whereas length of the colon in the LU‐001i‐treated mice was comparable to that of vehicle‐treated mice. Nevertheless, the colon length of LU‐005i‐treated mice was significantly increased compared with vehicle‐treated mice. Colitis is accompanied by the accumulation of a variety of inflammatory cells and the release of several soluble mediators of inflammation, such as cytokines. To determine whether the inflammatory response is altered in LU‐001i‐ and LU‐005i‐treated mice with colitis, TNF‐α, IFN‐γ, IL‐1β, IL‐6 and IL‐17 expression was determined by real‐time RT‐PCR in colon on day 9 after colitis induction (Figure 5D). Compared to naïve mice, cytokines were up‐regulated in vehicle‐treated mice. Cytokine levels in LU‐001i‐treated mice were comparable to vehicle‐treated mice, whereas the expressions of TNF‐α, IL‐1β and IL‐17 were significantly reduced in LU‐005i‐treated mice. Inflammation severity (0–3), extent of inflammation (0–3) and crypt damage (0–4) were graded semiquantitatively on microscopic cross sections of the colon on day 9 post colitis induction (Figure 5E, F). A massive infiltration of immune cells and a high histological score could be observed in vehicle‐ or LU‐001i‐treated mice. In contrast, LU‐005i‐treated mice had only mild signs of inflammation and crypt damage. Taken together, these finding indicate that LU‐005i strongly attenuates the development of DSS‐induced colitis in mice.
Figure 5.
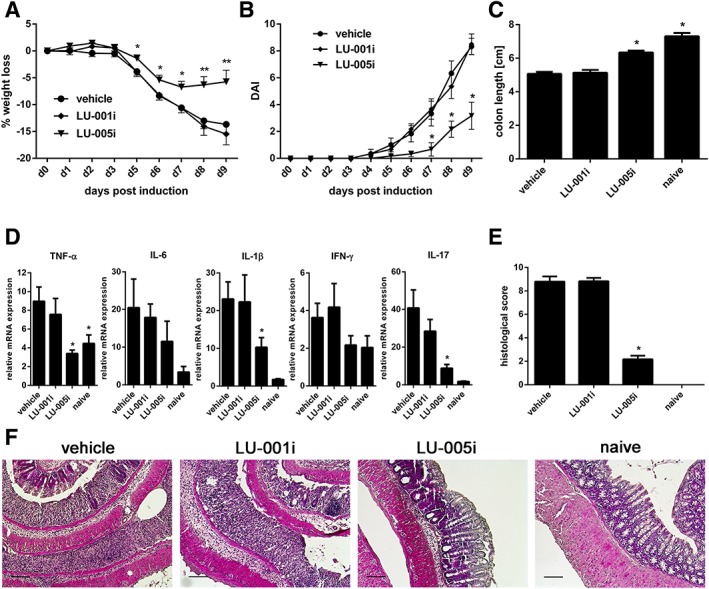
LU‐005i ameliorates DSS‐induced colitis. Colitis was induced by oral administration of 3% DSS for 5 days. Mice were treated daily (s.c.) with LU‐005i (15 mg·kg−1), LU‐001i (15 mg·kg−1), or vehicle as indicated. All data were statistically compared to the vehicle‐treated group. *P < 0.05. (A) Body weight of individual mice was monitored daily: % weight loss (y‐axis) is plotted versus time (x‐axis). Data points represent mean ± SEM of 13 mice. (B) DAI was determined daily. Data points represent mean ± SEM of six mice. (C) On day 9, colon length was analysed. Naïve mice (n = 5) were used as healthy controls. Data points represent mean ± SEM of 13 mice. (D) TNF‐α, IL‐1β, IL‐6, IFN‐γ and IL‐17 mRNA content was analysed by real‐time RT‐PCR in colon on day 9. The values were normalized to the expression of hypoxanthineguanine phosphoribosyl transferase in the same organs, and the relative cytokine expression was calculated, with the value for lowest expression being arbitrarily set to unity. Data points represent mean relative mRNA expression levels ± SEM of six mice or five mice (naïve group). The ‘naive’ group of IL‐1β and IL‐17 was excluded from statistical analysis due to variance inhomogeneity. (E) Semiquantitative histopathological assessment of colitis on day 9. Data points represent mean histopathological score ± SEM of 12 mice or 4 mice (naïve group). The ‘naïve’ group was not statistically analysed due to the low number of mice used. (F) Representative histological colon sections of the indicated groups of mice (H&E, original magnification ×10). Scale bar corresponds to 100 μm.
Discussion
Different studies using preclinical mouse models have demonstrated that the immunoproteasome is an attractive target for the treatment of autoimmune diseases (summarized in Basler et al., 2015). Nevertheless, most of these studies were based on a single compound, ONX 0914, an LMP7‐selective peptide‐ketoepoxide proteasome inhibitor (Muchamuel et al., 2009). Although different selective immunoproteasome inhibitors have been developed, they have not been thoroughly tested in preclinical models. The expectation that the immunoproteasome crystal structure reported in 2012 (Huber et al., 2012; Basler et al., 2015) will promote the structure‐guided design of new immunoproteasome inhibitors was recently realized (de Bruin et al., 2014; Singh et al., 2016; Sula Karreci et al., 2016). De Bruin et al. report the development of new β1i‐ and β5i‐selective inhibitors. Using known inhibitors as starting points and introducing structural features according to the X‐ray structures of the murine constitutive and immunoproteasome 20S core particles, these new inhibitors are the result of a rational design strategy. Some of these inhibitors were further characterized with human and mouse proteasomes in fluorogenic assays in this study (Table 1). These assays indicate that inhibitors selective for human LMP7 are not naturally selective for mouse proteasomes. Indeed, X‐ray structures of humanized yeast proteasome identify unique binding modes of inhibitors for human β5i (Huber et al., 2016). Although the substrate binding pockets and the active site residues are highly conserved among mice and humans, the residues outside the natural substrate binding channel, such as the S3* pocket identified, are subjected to variances, thereby creating species‐specific differences such as Val/Met31 (Huber et al., 2016). Based on the activity in the mouse proteasome, we decided to further investigate two compounds, LU‐005i and LU‐001i, in more detail. In vitro fluorogenic assays with different substrates demonstrated that LU‐005i is over 10 times more selective for MECL‐1, LMP2 and LMP7 than β2c, β1c and β5c (Tables 1, 2, 3). LU‐001i was shown to be an LMP2‐selective inhibitor (Tables 1, 2, 3), confirming previous results (de Bruin et al., 2014). Hence, LU‐005i is the first proteasome inhibitor described that simultaneously inhibits all three immunoproteasome subunits. Immunoproteasomes have been shown to be involved in the generation of MHC‐I ligands. This is evident in cells derived from LMP7‐deficient mice, which have an approximately 50% reduction in MHC‐I surface expression compared to wild‐type cells (Fehling et al., 1994). Since LMP7‐deficient mice also display a reduced LMP2 and MECL‐1 incorporation into immunoproteasomes, the altered MHC‐I surface expression observed in LMP7‐deficient mice cannot be attributed to a single immunoproteasome subunit (De et al., 2003). Treatment of splenocytes with LU‐005i reduced MHC‐I surface expression only in wild‐type cells but not in LMP7‐deficient cells (Figure 1B), demonstrating that LU‐005i is immunoproteasome‐selective. The LMP2‐selective inhibitor LU‐001i did not alter class‐I surface expression, indicating that LMP2 alone is not sufficient to reduce MHC‐I surface expression.
Cytokines play crucial roles in the regulation of the immune response. LU‐005i treatment of endotoxin‐ or TCR‐ stimulated mouse splenocytes strongly reduced IL‐6 or IFN‐γ secretion in an immunoproteasome‐dependent manner (Figure 3). Whereas LU‐005i was less potent at inhibiting IL‐6 production from LPS‐stimulated human PBMCs, it completely inhibited IL‐23 secretion (Figure 3E). In line with previous results (Muchamuel et al., 2009; Basler et al., 2014), immunoproteasome inhibition in IL‐23‐producing cells, probably monocytes, is extremely efficient at preventing IL‐23 secretion. The reason why IL‐23 secretion is strongly affected by immunoproteasome inhibition remains to be investigated. Whereas IL‐6 and IL‐1β are necessary for the induction of Th17 cells, IL‐23 is responsible for the maintenance of Th17 and for the production of IL‐17 (Aggarwal et al., 2003; Bettelli et al., 2006). Hence, immunoproteasome inhibition is a promising strategy for reducing IL‐23 secretion and, therefore, suppressing Th17 development. Indeed, in vitro Th17 differentiation was reduced with LU‐005i‐treated T helper cells (Figure 4), confirming previous observations, obtained both in vitro and in vivo (Muchamuel et al., 2009; Schmidt et al., 2010; Kalim et al., 2012; Basler et al., 2014; Vachharajani et al., 2017). Th17 cells are known to express high levels the multi‐drug transporter MDR1 (also known as P‐glycoprotein and ABCB1), an ATP‐dependent membrane efflux pump with broad substrate specificity (Ramesh et al., 2014). It has been shown that MDR1 can extrude epoxyketone‐peptide proteasome inhibitors such as carfilzomib (CFZ/PR‐171, trade name Kyprolis), ONX 0912 (PR‐047) and ONX 0914 (Verbrugge et al., 2012). Whether the epoxyketone‐peptide proteasome inhibitors, LU‐001i or LU‐005i, are affected by MDR1 remains to be determined. It has been found that inhibiting the immunoproteasome prevents the development of autoimmune diseases and of colitis‐associated cancer (Koerner et al., 2017; Vachharajani et al., 2017) in several preclinical animal models (summarized in Basler et al., 2015). To elucidate the potency of LU‐005i in vivo in an inflammatory disease, we chose to use the DSS‐induced colitis model as a proof of principle. Treatment of mice with LU‐005i in this colitis model resulted in reduced body weight loss, DAI, colon length shortening, cytokine expression in the colon and histological scores compared to vehicle‐treated mice (Figure 5). Hence, as previously reported (Basler et al., 2010), targeting the immunoproteasome in DSS‐induced colitis ameliorates the symptoms of this disease. In contrast, the inhibition of LMP2 with LU‐001i was not effective in this colitis model (Figure 5). This finding conflicts with that obtained in LMP2‐deficient mice, which were found to be partially protected from DSS‐induced colitis (Fitzpatrick et al., 2006; Basler et al., 2010). Hence, it seems that the effects seen in mice lacking LMP2 is based on a mechanism not relying on the peptidolytic activity of LMP2. An opposite effect has been shown in experimental autoimmune encephalomyelitis (EAE). Although LMP7‐deficient mice were not protected from EAE, LMP7 inhibition strongly reduced the clinical symptoms of this disease (Nathan et al., 2013; Basler et al., 2014). Interestingly, in LMP2‐ but not LMP7‐deficient mice the development of thyroid oncocytes and primary hypothyroidism was ameliorated (Kimura et al., 2009). Furthermore, pharmacological blockade of the proteasome with ML‐273 prevented the development of the thyroid oncocytes in mice expressing murine IFN‐γ specifically in thyroid follicular cells. Since LMP7 inhibition is effective in an animal model of Hashimoto's thyroiditis (Nagayama et al., 2012) and the development of thyroid oncocytes and primary hypothyroidism is ameliorated in LMP2‐deficient mice (Kimura et al., 2009), it would be interesting to determine whether LMP2 inhibition alone is also effective in these models. In our study, we used LU‐005i at 15 mg·kg−1 in mice. Carfilzomib, which is also a peptide‐epoxyketone, when given to mice at a bolus dose of 5 mg·kg−1, was found to induce 80% inhibition of chymotrypsin‐like activity 1 h after application in adrenal, heart, liver and whole blood (Demo et al., 2007). In humans, carfilzomib is approved for treatment of multiple myeloma at a dose of 20 mg·m−2. Since carfilzomib achieves, in humans, an 80% inhibition of ß5c and LMP7 activity in whole blood and PBMCs at this dose (Lee et al., 2016), therapeutically effective doses of LU‐005i should also be feasible in humans to treat autoimmunity. Apart from autoimmune diseases, the immunoproteasome has been shown to be involved in other processes. LMP7 deficiency improves obesity and metabolic disorders (Kimura et al., 2015), as LMP7 is involved in regulating adipogenesis (Kitamura et al., 2011; Arimochi et al., 2016), and the immunoproteasome is a regulator of skeletal muscle differentiation (Cui et al., 2014). The dysregulation of such processes leads to disorders which might potentially be treated with immunoproteasome inhibitors. Whether LU‐005i or LU‐001i is effective in the treatment of metabolic disorders remains to be investigated.
The mechanism through which immunoproteasome inhibition can prevent autoimmune diseases still remains elusive. It has been suggested that the immunoproteasome might selectively process a factor that is required for regulating cytokine production and T helper cell differentiation (Basler et al., 2013), but, so far, such a factor has not been identified. The involvement of the immunoproteasome in NF‐kB activation has remained controversial (discussed in Bitzer et al., 2017). Using a reporter cell line, Muchamuel et al. (2009) showed no difference in NF‐kB activation in immunoproteasome‐inhibited cells. However, it has recently been reported that the mTORC1‐immunoproteasome pathway is important for cell survival against stress (Yun et al., 2016). Whether this pathway is affected in immunoproteasome‐targeted mice in autoimmune diseases remains to be determined.
Bortezomib, a proteasome inhibitor that reduces the activities of CP and immunoproteasome, prevents chronic active antibody‐mediated rejection in experimental renal transplantation in the rat (Vogelbacher et al., 2010). However, the toxicological profiles of bortezomib suggest that proteasome inhibitors with reduced toxicity need to be developed for long‐term treatment of transplant patients. Indeed, in a recent study, Sula Karreci et al. (2016) describe a novel highly selective LMP7 inhibitor named DPLG3. This non‐covalent inhibitor promoted long‐term cardiac allograft acceptance in mice across a major histocompatibility barrier. DPLG3 suppressed the cytokine release from PBMCs and the activation of T cells and dendritic cells. The numbers of effector T cells in the allograft and draining lymph nodes were reduced, while exhaustion markers on T cells were increased. Hence, the inhibition of the immunoproteasome is a potential target for the management of transplant rejection.
Taken together, our findings show that a new inhibitor targeting all three catalytically active immunoproteasome subunits is efficient at preventing cytokine release, Th17 differentiation and experimental colitis. Targeting the catalytically active immunoproteasome subunit LMP2 alone was shown not to be effective. These results will be useful to guide the design of new generations of immunoproteasome inhibitors with optimal therapeutic efficacy for the suppression of autoimmunity and transplant rejection. This study confirms that the immunoproteasome is a clinically attractive target in inflammatory diseases and that immunoproteasome inhibitors targeting all immunoproteasome subunits hold potential clinical interest for the treatment of these diseases.
Author contributions
M.B. designed and performed experiments, wrote the manuscript and supervised the project. J.K. performed animal experiments and histology. E.M., G.dB. and H.S.O. synthesized and provided immunoproteasome inhibitors. M.G. supervised the project and refined the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Proteasome composition of LCL721.174 cells and LCL721.145 cells. NEPHGE/SDS‐PAGE analysis of 70 μg 20S proteasome purified from LCL721.174 cells (CP) (top) LCL721.145 cells (IP) (bottom). The proteins were visualized by coomassie stain. The positions of proteasome subunits LMP2, LMP7, β1c, and β5c are indicated.
Figure S2 Cell permeability of LU‐005i and LU‐001i. (A) LCL721.174 cells (CP) or LCL721.145 cells (IP) were treated with the indicated concentrations of LU‐005i. The chymotrypsin‐like activity in the cells was determined by the hydrolysis of the cell permeable substrate Meo‐Suc‐GLF‐AMC. Depicted is the mean ± SD % of maximal activity of triplicate cultures. The highest fluorescence value was set to 100%. (B) LCL721.145 (contain IP) cells were incubated with 300 nM LU‐001i or DMSO overnight. Proteasomes of crude lysates of these cells were immunoprecipitated and assayed for the hydrolysis of the fluorogenic substrate Ac‐PAL‐AMC (LMP2‐activity). Depicted is the mean ± SD % of maximal activity of triplicate cultures. The highest fluorescence value was set to 100%. Experiments were performed twice with a similar outcome. These experiments were only performed to demonstrate the cell permeability of LU‐005i and LU‐001i. Therefore, the data were not statistically analysed.
Figure S3 Effect of LU‐005i on cell death. Splenocytes derived from C57BL/6 mice were treated with DMSO, 300 nM LU‐005i, 500 nM LU‐005i, or 1000 nM LU‐005i for 24 h (A), 48 h (B), or 72 h (C). Cell death (PI+) of CD11c+, CD11b+, F4/80+, CD19+, NK1.1+, CD4+, and CD8+ was analysed by flow cytometry. Shown are the means of PI+ populations ± SEM from splenocytes derived from 5 different mice (n = 5). All data were statistically compared to the DMSO treated group. *P < 0.05.
Figure S4 Activity of LU‐005i and LU‐001i in mice. C57BL/6 mice were treated with LU‐005i (A, B) (15 mg kg−1), LU‐001i (C) (15 mg kg−1), or vehicle for 2 h or 24 h. Proteasomes of crude lysates of splenocytes derived from these mice were immunoprecipitated and assayed for the hydrolysis of the fluorogenic substrate Ac‐PAL‐AMC (LMP2‐acitivity) (A, C) or Suc‐LLVY‐AMC (chymotrypsin‐like activity) (B). Depicted is the mean ± SD of the fluorescence of two mice per group measured in duplicates. Experiments were performed twice with a similar outcome. These experiments were only performed to demonstrate the activity of LU‐005i and LU‐001i in mice. Therefore, the data were not statistically analysed.
Acknowledgements
Ulrike Beck, Heike Goebel and Gerardo Alvarez are acknowledged for excellent technical assistance. We thank Christopher J. Kirk for providing ONX 0914 and Sonja Erath for experimental support. Some of the flow cytometry experiments were done at the FlowKon facility of the University of Konstanz. This work was funded by the Deutsche Forschungsgemeinschaft grant nos BA 4199/2‐1 to M.B. and GR 1517/2.4 and GR 1517/10‐2 to M.G.; the Jubiläumsstiftung der Schweizerischen Lebensversicherungs‐ und Rentenanstalt für Volksgesundheit und medizinische Forschung SwissLife Jubiläumsstiftung to M.B.; and Krebsliga Schweiz grant KFS‐3687‐08‐2015 to M.G. Funding from the Netherlands Organization for Scientific Research (TOP‐PUNT grant) is acknowledged by H.S.O. and E.M.
Basler, M. , Maurits, E. , de Bruin, G. , Koerner, J. , Overkleeft, H. S. , and Groettrup, M. (2018) Amelioration of autoimmunity with an inhibitor selectively targeting all active centres of the immunoproteasome. British Journal of Pharmacology, 175: 38–52. doi: 10.1111/bph.14069.
Contributor Information
Michael Basler, Email: michael.basler@uni-konstanz.de.
Marcus Groettrup, Email: marcus.groettrup@uni-konstanz.de.
References
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL (2003). Interleukin‐23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin‐17. J Biol Chem 278: 1910–1914. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimochi H, Sasaki Y, Kitamura A, Yasutomo K (2016). Differentiation of preadipocytes and mature adipocytes requires PSMB8. Sci Rep 6: 26791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M, Groettrup M (2007). No essential role for tripeptidyl peptidase II for the processing of LCMV‐derived T cell epitopes. Eur J Immunol 37: 896–904. [DOI] [PubMed] [Google Scholar]
- Basler M, Groettrup M (2012). Immunoproteasome‐specific inhibitors and their application. Methods Mol Biol 832: 391–401. [DOI] [PubMed] [Google Scholar]
- Basler M, Groettrup M (2013). Using protease inhibitors in antigen presentation assays. Methods Mol Biol 960: 31–39. [DOI] [PubMed] [Google Scholar]
- Basler M, Youhnovski N, Van Den Broek M, Przybylski M, Groettrup M (2004). Immunoproteasomes down‐regulate presentation of a subdominant T cell epitope from lymphocytic choriomeningitis virus. J Immunol 173: 3925–3934. [DOI] [PubMed] [Google Scholar]
- Basler M, Moebius J, Elenich L, Groettrup M, Monaco JJ (2006). An altered T cell repertoire in MECL‐1‐deficient mice. J Immunol 176: 6665–6672. [DOI] [PubMed] [Google Scholar]
- Basler M, Lauer C, Beck U, Groettrup M (2009). The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. J Immunol 183: 6145–6150. [DOI] [PubMed] [Google Scholar]
- Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ (2010). Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J Immunol 185: 634–641. [DOI] [PubMed] [Google Scholar]
- Basler M, Beck U, Kirk CJ, Groettrup M (2011). The antiviral immune response in mice devoid of immunoproteasome activity. J Immunol 187: 5548–5557. [DOI] [PubMed] [Google Scholar]
- Basler M, Lauer C, Moebius J, Weber R, Przybylski M, Kisselev AF et al (2012). Why the structure but not the activity of the immunoproteasome subunit low molecular mass polypeptide 2 rescues antigen presentation. J Immunol 189: 1868–1877. [DOI] [PubMed] [Google Scholar]
- Basler M, Kirk CJ, Groettrup M (2013). The immunoproteasome in antigen processing and other immunological functions. Curr Opin Immunol 25: 74–80. [DOI] [PubMed] [Google Scholar]
- Basler M, Mundt S, Muchamuel T, Moll C, Jiang J, Groettrup M et al (2014). Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol Med 6: 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M (2015). The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol 33: 74–79. [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M et al (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238. [DOI] [PubMed] [Google Scholar]
- Bitzer A, Basler M, Krappmann D, Groettrup M (2017). Immunoproteasome subunit deficiency has no influence on the canonical pathway of NF‐kappaB activation. Mol Immunol 83: 147–153. [DOI] [PubMed] [Google Scholar]
- Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ et al (2010). Characterization of a new series of non‐covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5‐subunit. Biochem J 430: 461–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin G, Huber EM, Xin BT, van Rooden EJ, Al‐Ayed K, Kim KB et al (2014). Structure‐based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J Med Chem 57: 6197–6209. [DOI] [PubMed] [Google Scholar]
- Cui Z, Hwang SM, Gomes AV (2014). Identification of the immunoproteasome as a novel regulator of skeletal muscle differentiation. Mol Cell Biol 34: 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De M, Jayarapu K, Elenich L, Monaco JJ, Colbert RA, Griffin TA (2003). Beta 2 subunit propeptides influence cooperative proteasome assembly. J Biol Chem 278: 6153–6159. [DOI] [PubMed] [Google Scholar]
- Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN et al (2007). Antitumor activity of PR‐171, a novel irreversible inhibitor of the proteasome. Cancer Res 67: 6383–6391. [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39: 175–191. [DOI] [PubMed] [Google Scholar]
- Fehling HJ, Swat W, Laplace C, Kuehn R, Rajewsky K, Mueller U et al (1994). MHC class I expression in mice lacking proteasome subunit LMP‐7. Science 265: 1234–1237. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick LR, Khare V, Small JS, Koltun WA (2006). Dextran sulfate sodium‐induced colitis is associated with enhanced low molecular mass polypeptide 2 (LMP2) expression and is attenuated in LMP2 knockout mice. Dig Dis Sci 51: 1269–1276. [DOI] [PubMed] [Google Scholar]
- Groettrup M, Soza A, Kuckelkorn U, Kloetzel PM (1996). Peptide antigen production by the proteasome: complexity provides efficiency. Immunol Today 17: 429–435. [DOI] [PubMed] [Google Scholar]
- Harding CV, France J, Song R, Farah JM, Chatterjee S, Iqbal M et al (1995). Novel dipeptide aldehydes are proteasome inhibitors and block the MHC‐I antigen‐processing pathway. J Immunol 155: 1767–1775. [PubMed] [Google Scholar]
- Horino J, Fujimoto M, Terabe F, Serada S, Takahashi T, Soma Y et al (2008). Suppressor of cytokine signaling‐1 ameliorates dextran sulfate sodium‐induced colitis in mice. Int Immunol 20: 753–762. [DOI] [PubMed] [Google Scholar]
- Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M et al (2012). Immuno‐ and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148: 727–738. [DOI] [PubMed] [Google Scholar]
- Huber EM, Heinemeyer W, de Bruin G, Overkleeft HS, Groll M (2016). A humanized yeast proteasome identifies unique binding modes of inhibitors for the immunosubunit beta5i. EMBO J 35: 2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T et al (2012). Novel proteasome inhibitors have a beneficial effect in murine lupus via the dual inhibition of type I interferon and autoantibody secreting cells. Arthritis Rheum 64: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalim KW, Basler M, Kirk CJ, Groettrup M (2012). Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J Immunol 189: 4182–4193. [DOI] [PubMed] [Google Scholar]
- Khan S, van den Broek M, Schwarz K, de Giuli R, Diener PA, Groettrup M (2001). Immunoproteasomes largely replace constitutive proteasomes during an antiviral and antibacterial immune response in the liver. J Immunol 167: 6859–6868. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura HJ, Chen CY, Tzou SC, Rocchi R, Landek‐Salgado MA, Suzuki K et al (2009). Immunoproteasome overexpression underlies the pathogenesis of thyroid oncocytes and primary hypothyroidism: studies in humans and mice. PLoS One 4: e7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H, Usui F, Karasawa T, Kawashima A, Shirasuna K, Inoue Y et al (2015). Immunoproteasome subunit LMP7 deficiency improves obesity and metabolic disorders. Sci Rep 5: 15883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid EZ, Che JW, York I, Escobar H, Reyes‐Vargas E, Delgado JC et al (2012). Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol 13: 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M et al (2011). A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest 121: 4150–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner J, Brunner T, Groettrup M (2017). Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 8: 50873–50888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer M, Henn A, Kolb C, Basler M, Moebius J, Guillaume B et al (2010). Reduced immunoproteasome formation and accumulation of immunoproteasomal precursors in the brains of lymphocytic choriomeningitis virus‐infected mice. J Immunol 185: 5549–5560. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Levitsky K, Parlati F, Bennett MK, Arastu‐Kapur S, Kellerman L et al (2016). Clinical activity of carfilzomib correlates with inhibition of multiple proteasome subunits: application of a novel pharmacodynamic assay. Br J Haematol 173: 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wan C, Ding Y, Han R, He Y, Xiao J et al (2017). PR‐957, a selective inhibitor of immunoproteasome subunit low‐MW polypeptide 7, attenuates experimental autoimmune neuritis by suppressing Th17 cell differentiation and regulating cytokine production. FASEB J 31: 1756–1766. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moebius J, van den Broek M, Groettrup M, Basler M (2010). Immunoproteasomes are essential for survival and expansion of T cells in virus‐infected mice. Eur J Immunol 40: 3439–3449. [DOI] [PubMed] [Google Scholar]
- Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C et al (2009). A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 15: 781–787. [DOI] [PubMed] [Google Scholar]
- Mundt S, Engelhardt B, Kirk CJ, Groettrup M, Basler M (2016). Inhibition and deficiency of the immunoproteasome subunit LMP7 attenuates LCMV‐induced meningitis. Eur J Immunol 46: 104–113. [DOI] [PubMed] [Google Scholar]
- Nagayama Y, Nakahara M, Shimamura M, Horie I, Arima K, Abiru N (2012). Prophylactic and therapeutic efficacies of a selective inhibitor of the immunoproteasome for Hashimoto's thyroiditis, but not for Graves' hyperthyroidism, in mice. Clin Exp Immunol 168: 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan JA, Spinnenhirn V, Schmidtke G, Basler M, Groettrup M, Goldberg AL (2013). Immuno‐ and constitutive proteasomes do not differ in their abilities to degrade ubiquitinated proteins. Cell 152: 1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmowski MJ, Gileadi U, Salio M, Gallimore A, Millrain M, James E et al (2006). Role of immunoproteasomes in cross‐presentation. J Immunol 177: 983–990. [DOI] [PubMed] [Google Scholar]
- Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA et al (2014). Pro‐inflammatory human Th17 cells selectively express P‐glycoprotein and are refractory to glucocorticoids. J Exp Med 211: 89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter RD, Cresswell P (1986). Impaired assembly and transport of HLA‐A and ‐B antigens in a mutant TxB cell hybrid. EMBO J 5: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N, Gonzalez E, Visekruna A, Kuhl AA, Loddenkemper C, Mollenkopf H et al (2010). Targeting the proteasome: partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut 59: 896–906. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008). Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schwarz K, Eggers M, Soza A, Koszinowski UH, Kloetzel PM, Groettrup M (2000). The proteasome regulator PA28α/β can enhance antigen presentation without affecting 20S proteasome subunit composition. Eur J Immunol 30: 3672–3679. [DOI] [PubMed] [Google Scholar]
- Singh PK, Fan H, Jiang X, Shi L, Nathan CF, Lin G (2016). Immunoproteasome beta5i‐selective dipeptidomimetic inhibitors. ChemMedChem 11: 2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sula Karreci E, Fan H, Uehara M, Mihali AB, Singh PK, Kurdi AT et al (2016). Brief treatment with a highly selective immunoproteasome inhibitor promotes long‐term cardiac allograft acceptance in mice. Proc Natl Acad Sci U S A 113: E8425–E8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachharajani N, Joeris T, Luu M, Hartmann S, Pautz S, Jenike E et al (2017). Prevention of colitis‐associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 8: 50447–50459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbrugge SE, Assaraf YG, Dijkmans BA, Scheffer GL, Al M, den Uyl D et al (2012). Inactivating PSMB5 mutations and P‐glycoprotein (multidrug resistance‐associated protein/ATP‐binding cassette B1) mediate resistance to proteasome inhibitors: ex vivo efficacy of (immuno)proteasome inhibitors in mononuclear blood cells from patients with rheumatoid arthritis. J Pharmacol Exp Ther 341: 174–182. [DOI] [PubMed] [Google Scholar]
- Vogelbacher R, Meister S, Guckel E, Starke C, Wittmann S, Stief A et al (2010). Bortezomib and sirolimus inhibit the chronic active antibody‐mediated rejection in experimental renal transplantation in the rat. Nephrol Dial Transplant 25: 3764–3773. [DOI] [PubMed] [Google Scholar]
- Yun YS, Kim KH, Tschida B, Sachs Z, Noble‐Orcutt KE, Moriarity BS et al (2016). mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol Cell 61: 625–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DM, Bekker CP, Grone A, Lie BA, Sijts AJ (2011). Proteasome immunosubunits protect against the development of CD8 T cell‐mediated autoimmune diseases. J Immunol 187: 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Proteasome composition of LCL721.174 cells and LCL721.145 cells. NEPHGE/SDS‐PAGE analysis of 70 μg 20S proteasome purified from LCL721.174 cells (CP) (top) LCL721.145 cells (IP) (bottom). The proteins were visualized by coomassie stain. The positions of proteasome subunits LMP2, LMP7, β1c, and β5c are indicated.
Figure S2 Cell permeability of LU‐005i and LU‐001i. (A) LCL721.174 cells (CP) or LCL721.145 cells (IP) were treated with the indicated concentrations of LU‐005i. The chymotrypsin‐like activity in the cells was determined by the hydrolysis of the cell permeable substrate Meo‐Suc‐GLF‐AMC. Depicted is the mean ± SD % of maximal activity of triplicate cultures. The highest fluorescence value was set to 100%. (B) LCL721.145 (contain IP) cells were incubated with 300 nM LU‐001i or DMSO overnight. Proteasomes of crude lysates of these cells were immunoprecipitated and assayed for the hydrolysis of the fluorogenic substrate Ac‐PAL‐AMC (LMP2‐activity). Depicted is the mean ± SD % of maximal activity of triplicate cultures. The highest fluorescence value was set to 100%. Experiments were performed twice with a similar outcome. These experiments were only performed to demonstrate the cell permeability of LU‐005i and LU‐001i. Therefore, the data were not statistically analysed.
Figure S3 Effect of LU‐005i on cell death. Splenocytes derived from C57BL/6 mice were treated with DMSO, 300 nM LU‐005i, 500 nM LU‐005i, or 1000 nM LU‐005i for 24 h (A), 48 h (B), or 72 h (C). Cell death (PI+) of CD11c+, CD11b+, F4/80+, CD19+, NK1.1+, CD4+, and CD8+ was analysed by flow cytometry. Shown are the means of PI+ populations ± SEM from splenocytes derived from 5 different mice (n = 5). All data were statistically compared to the DMSO treated group. *P < 0.05.
Figure S4 Activity of LU‐005i and LU‐001i in mice. C57BL/6 mice were treated with LU‐005i (A, B) (15 mg kg−1), LU‐001i (C) (15 mg kg−1), or vehicle for 2 h or 24 h. Proteasomes of crude lysates of splenocytes derived from these mice were immunoprecipitated and assayed for the hydrolysis of the fluorogenic substrate Ac‐PAL‐AMC (LMP2‐acitivity) (A, C) or Suc‐LLVY‐AMC (chymotrypsin‐like activity) (B). Depicted is the mean ± SD of the fluorescence of two mice per group measured in duplicates. Experiments were performed twice with a similar outcome. These experiments were only performed to demonstrate the activity of LU‐005i and LU‐001i in mice. Therefore, the data were not statistically analysed.