

Abstract
Background and Purpose
The aim of this study was to compare the abilities of cannabidiolic acid methyl ester (HU‐580) and cannabidiolic acid (CBDA) to enhance 5‐HT1A receptor activation in vitro and produce 5‐HT1A‐mediated reductions in nausea and anxiety in vivo.
Experimental Approach
We investigated the effects of HU‐580 and CBDA on (i) activation by 8‐hydroxy‐2‐(di‐n‐propylamino)tetralin of human 5‐HT1A receptors in CHO cell membranes, using [35S]‐GTPγS binding assays, (ii) gaping by rats in acute and anticipatory nausea models, and (iii) stress‐induced anxiety‐like behaviour, as indicated by exit time from the light compartment of a light–dark box of rats subjected 24 h earlier to six tone‐paired foot shocks.
Key Results
HU‐580 and CBDA increased the Emax of 8‐hydroxy‐2‐(di‐n‐propylamino) tetralin in vitro at 0.01–10 and 0.1–10 nM, respectively, and reduced signs of (i) acute nausea at 0.1 and 1 μg·kg−1 i.p. and at 1 μg·kg−1 i.p., respectively, and (ii) anticipatory nausea at 0.01 and 0.1 μg·kg−1, and at 0.1 μg·kg−1 i.p. respectively. At 0.01 μg·kg−1, HU‐580, but not CBDA, increased the time foot‐shocked rats spent in the light compartment of a light–dark box. The anti‐nausea and anti‐anxiety effects of 0.01 or 0.1 μg·kg−1 HU‐580 were opposed by the 5‐HT1A antagonist, WAY100635 (0.1 mg·kg−1 i.p.).
Conclusions and Implications
HU‐580 is more potent than CBDA at enhancing 5‐HT1A receptor activation, and inhibiting signs of acute and anticipatory nausea, and anxiety. Consequently, HU‐580 is a potential medicine for treating some nausea and anxiety disorders and possibly other disorders ameliorated by enhancement of 5‐HT1A receptor activation.
Abbreviations
- 8‐OH‐DPAT
8‐hydroxy‐2‐(di‐n‐propylamino)tetralin
- CBD
cannabidiol
- CBDA
cannabidiolic acid
- FS
foot shock
- HU‐580
cannabidiolic acid methyl ester
- LiCl
lithium chloride
- No FS
no foot shock
- WAY100635
N‐[2‐[4‐(2‐methoxyphenyl)‐1‐piperazinyl]ethyl]‐N‐2‐pyridinylcyclohexanecarboxamide maleate salt
Introduction
Cannabidiolic acid (CBDA) is a major constituent of Cannabis sativa. It was first isolated in 1955 (Krejčí and Šantavý, 1955), and its structure (Figure 1) was elucidated in 1965 by analysis of the physical properties of its methyl ester (Mechoulam and Gaoni, 1965). Its synthesis from cannabidiol was subsequently reported (Mechoulam and Ben‐Zvi, 1969). CBDA gradually decarboxylates, while still in the plant, to cannabidiol (Figure 1) (Mechoulam, 1973), a process that is speeded up by heat. Whereas cannabidiol has been the topic of a large number of publications and its biological/therapeutic properties have now been reasonably well identified (Mechoulam et al., 2002; Zhornitsky and Potvin, 2012; Cascio and Pertwee, 2014), our knowledge of the pharmacology of CBDA is much more limited. However, even the limited amount of information on this phytocannabinoid that has been published suggests that it may have a wide variety of actions and effects. Thus, it has been shown to inhibit breast cancer cell migration (Takeda et al., 2017) and to cause a down‐regulation of COX‐2 (Takeda et al., 2014).
Figure 1.
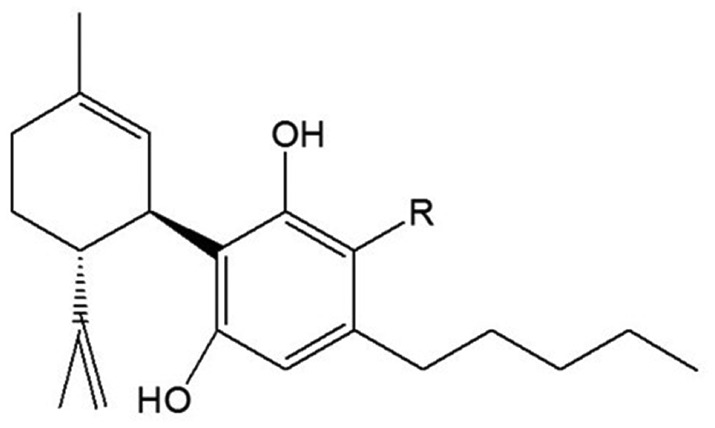
Structure of cannabidiol (R = H), CBDA (R = COOH) and HU‐580 (R = COOCH3).
Considerable recent evidence suggests that CBDA (at a dose as low as 1 μg·kg−1 i.p.) can induce potent 5‐HT1A receptor‐medated anti‐nausea effects as indicated by its apparent ability to prevent both vomiting in Suncus murinus and acute nausea‐induced behaviour of conditioned gaping in rats (Grill and Norgren, 1978) by enhancing 5‐HT1A receptor activation (Bolognini et al., 2013; Rock and Parker, 2013; Rock and Parker, 2015). As well as reducing acute nausea, CBDA has the potential to reduce anticipatory (conditioned) nausea, an effect experienced by chemotherapy patients upon returning to the clinic in which they received their nauseating treatment (Rock et al., 2014; 2015; 2016). There are currently no effective selective treatments for anticipatory nausea once it develops in these patients. It is noteworthy, therefore, that Rock et al. (2014; 2015; 2016) have demonstrated that CBDA reduces contextually elicited conditioned gaping (a model of anticipatory nausea), also by a 5‐HT1A‐dependent mechanism of action. Finally, like cannabidiol, CBDA has also been shown to produce anxiolytic‐like effects under conditions of high stress at doses as low as 0.1 μg·kg−1, i.p (Rock et al., 2017).
The instability of CBDA (Crombie and Crombie, 1977), especially when subjected to heat, weakens the case for developing it as a medicine. Hence, we decided to search for a more stable analogue with a similar biological profile. We eventually decided to compare CBDA with its methyl ester (HU‐580, Figure 1), since decarboxylation of phenolic acids is known to proceed through the carboxyl anion (RCOO−−) of the acid (Norman and Coxon, 1993) which is not readily formed by the methyl ester, and indeed found that when kept at 4°C for 21 days, HU‐580 remained unchanged, contrary to CBDA which partly decomposed. Here, we present evidence that HU‐580 is even more potent than CBDA at producing signs of 5‐HT1A receptor‐mediated suppression of nausea. Our initial experiments explored the possibility that HU‐580 can enhance the ability of the selective 5‐HT1A agonist, 8‐hydroxy‐2‐(di‐n‐propylamino)tetralin (8‐OH‐DPAT), to stimulate [35S]‐GTPγS binding to membranes obtained from human 5‐HT1A receptor‐expressing CHO cells, with a potency equal to or even greater than that shown previously to be displayed by CBDA (Bolognini et al., 2013). Since these experiments yielded positive results, we went on to evaluate the relative potency of CBDA and HU‐580 at inducing apparent 5‐HT1A receptor‐mediated suppression of acute and anticipatory nausea‐induced behaviour in the rat gaping models. Furthermore, we evaluated the relative abilities of extremely low doses of HU‐580 and CBDA to produce anxiolytic‐like behaviour in a model of stress‐induced anxiogenic responding.
Methods
In vitro procedures
CHO cells
CHO cells stably transfected with cDNA encoding human 5‐HT1A receptors (a generous gift from Dr Keith Parker) were maintained at 37°C and 5% CO2 in Gibco™ Ham's F‐12 Nutrient Mix supplied by Fisher Scientific UK Ltd that was supplemented both with 2 mM L‐glutamine, 10% FBS and 0.6% penicillin‐streptomycin, all also supplied by Fisher Scientific UK Ltd, and with the disulphate salt of G418 [(2R,3S,4R,5R,6S)‐5‐amino‐6‐{[(1R,2S,3S,4R,6S)‐4,6‐diamino‐3‐{[(2R,3R,4R,5R)‐3,5‐dihydroxy‐5‐methyl‐4‐(methylamino)oxan‐2‐yl]oxy}‐2‐hydroxycyclohexyl]oxy}‐2‐[(1R)‐1‐hydroxyethyl]oxane‐3,4‐diol; 600 mg·mL−1] supplied by Sigma‐Aldrich UK.
[35S]‐GTPγS binding assay
Each assay was carried out with human 5‐HT1A CHO cell membranes (50 μg protein per well), GTPγS‐binding buffer (50 mM Tris–HCl; 50 mM Tris‐Base; 5 mM MgCl2; 1 mM EDTA; 100 mM NaCl; 1 mM DTT and 0.1% BSA), 0.1 nM [35S]‐GTPγS and 30 μM GDP, in a final volume of 500 μL (Cascio et al., 2010). Binding was initiated by the addition of [35S]‐GTPγS to the wells. Non‐specific binding was measured in the presence of 30 μM GTPγS. Assays were performed at 30°C for 60 min (Cascio et al., 2010). The reaction was terminated by a rapid vacuum filtration method using Tris‐binding buffer as described previously by Cascio et al. (2010), and the radioactivity was quantified by liquid scintillation spectrometry. In all the [35S]‐GTPγS‐binding assays, we used 0.1 nM [35S]‐GTPγS, 30 mM GDP and a protein concentration of 5 μg per well. CBDA, HU‐580, 8‐OH‐DPAT and WAY100635 were stored at −20°C as 10 mM stock solutions dissolved in DMSO.
In vivo procedures
Animals
Animal procedures complied with the Canadian Council on Animal Care, and the protocols were approved by the Institutional Animal Care Committee at University of Guelph. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). A total of 200 näive male Sprague–Dawley rats, obtained from Charles River Laboratories (St Constant, Quebec), were used for all in vivo studies. Rats were individually housed (for acute nausea studies) or pair‐housed [for anticipatory nausea and light–dark emergence studies] in home cages made of opaque white plastic (48 × 26 × 20 cm), containing bed‐o‐cob bedding from Harlan Laboratories, Inc. (Mississauga, Ontario), a brown paper towel, and Crink‐l'Nest™ from The Andersons, Inc. (Maumee, Ohio). Additionally, in the home cage, rats were provided with a soft white paper container that was 14 cm long and 12 cm in diameter. All rats were subjected to an ambient temperature of 21°C and a 12/12 h light‐dark schedule (lights off at 07:00 h) and maintained on food (Highland Rat Chow [8640]) and water ad libitum. For the acute and anticipatory nausea studies, their body weights ranged from 263 to 329 g on the day of conditioning. For the light‐dark emergence studies, their body weights ranged from 320 to 387 g on the day of test.
Apparatus
For the studies of acute nausea (in vivo experiment 1), rats were placed in taste reactivity (Grill and Norgren, 1978) chambers with their cannula attached to an infusion pump (Model KDS100, KD Scientific, Holliston, MA, USA) for fluid delivery. The taste reactivity chambers were made of clear Plexiglas (22.5 × 26 × 20 cm) that sat on a table with a clear glass top. A mirror beneath the chamber at a 45° angle facilitated viewing of the ventral surface of the rat to observe orofacial responses. The conditioning chamber was in a dark room next to a 25 W light source. A video camera (Sony DCR‐HC48, Henry's Cameras, Waterloo, ON, Canada) fire‐wired into a computer was focused on the mirror and used to record each rat's orofacial reactions during the 2 min taste reactivity test. The video tapes were later scored using ‘The Observer’ (Noldus Information Technology Inc., Leesburg, VA, USA) software.
For in vivo experiment 2, contextually elicited conditioned gaping (a model of anticipatory nausea) was measured using a distinctive conditioning chamber made of opaque black Plexiglass (22.5 × 26 × 20 cm) with an opaque lid that sat on a table with a clear glass top. A mirror beneath the chamber at a 45° angle facilitated viewing of the ventral surface of the rat to observe orofacial responses. The conditioning chamber was in a dark room next to a 25 W light source. A video camera that was fire‐wired into a computer was focused on the mirror to record each rat's orofacial reactions during the 5 min test trial. The video tapes were later scored using ‘The Observer’ software. To assess activity, an activity chamber made of white Plexiglas (60 × 25 × 25 cm) was used, illuminated by a red light found in a different room that the contextual chamber was used to create a different context from the AN chamber. The activity of each rat was captured by video camera and sent to the Ethovision software programme (Noldus, Inc., NL) to measure distance (cm) travelled.
For the in vivo experiment 3, anxiolytic‐like responding was evaluated using the light‐dark emergence apparatus, which consisted of an opaque white plastic rectangular box that was divided into two compartments: a small (25 cm wide × 20.5 cm long × 20.5 cm high) enclosed dark box built of opaque black plastic with a door (8 cm wide × 10 cm high) leading to a larger (39.5 cm long × 25 cm wide) open lit box. The open lit box was illuminated by one lamp (with a 60 W bulb, 180 lux in the light chamber) positioned 115 cm above the centre of the lit box. A video camera was mounted over the top of the light‐dark box, and the video tapes were analysed by the Ethovision software (Noldus Information Technology, Leesburg, VA, USA) for the duration of time spent in the light box for the 5 min test. For the foot shock (FS) session, the rats were placed in sound attenuating MED Associates fear conditioning chambers (St. Albans, VT, USA). The 6 min FS session consisted of six 0.8 mA foot shocks delivered 1 min apart. Each 0.5 s shock was preceded by a 30 s auditory tone (90 Db, 5000 Hz) as described by Bluett et al. (2014).
In vivo procedures
In vivo experiment 1: dose‐related effects of CBDA and HU‐580 on acute nausea and 5‐HT1A receptor mediation of HU‐580 effects
All rats were surgically implanted with an intraoral cannula according the procedures described by Limebeer et al. (2010). On the day of surgery, the rats were injected with an antibiotic (Derapin: 100 mg·kg−1 s.c.; Pfizer Animal Health, Pfizer Canada Inc, Kirkland, Quebec, Canada) 30 min prior to being anaesthetized with isoflurane (4 −5% induction, 1.5% maintenance in O2). Surgical plane anaesthesia, as indicated by absence of the hind limb withdrawal reflex and defined by the Canadian Council of Animal Care, was induced before any surgery began, and was adjusted as necessary. Once sufficient anaesthesia had been induced, a 2 cm2 section of skin was shaved at the back of the neck at the level of the scapula. The skin was prepared by cleaning with soap (Bactistat; Ecolab, St. Paul, MN, USA) and wiping with 70% isopropyl alcohol followed by 7% Betadine solution (Purdue Products L.P., Stamford, CT, USA). Each rat was then administered a 5 mg·kg−1 injection (i.p.) of the anti‐inflammatory/analgesic drug carprofen (Rimadyl; Pfizer Canada Inc., Kirkland, Quebec, Canada). A thin‐walled 15‐gauge stainless steel needle was inserted into the shaved area on the neck, directed subcutaneously around the ear and brought out behind the first molar inside the mouth. A 10 cm length of Intra Medic PE90 tubing (Clay Adams Brand; Becton Dickinson and Co., Sparks, MD, USA) with an inner diameter of 0.86 mm and an outer diameter of 1.27 mm was then inserted through the needle after which the needle was removed. Betadine (10%) was applied to the puncture site and three elastic discs (2 cm2) were placed over the exposed end of the tubing and drawn to the skin at the back of the neck for the purpose of stabilizing the cannula. The cannula was held secure in the oral cavity by a 6 mm disc of polypropylene mesh (297 micron; Small Parts Inc., Miramar, FL, USA) secured behind the heat flanged intraoral opening. The rats were then returned to their home cage and monitored daily for 3 days. For 3 days following surgery the rats were weighed and their cannulae were flushed with an antiseptic mouth wash. During this time, the rats were also monitored for activity, vocalization, dehydration, rigidity, and presence of porphyrin staining around the eyes. On the first post‐surgical day, the rats were also given an analgesic/anti‐inflammatory injection of Rimadyl (5 mg·kg−1 i.p.).
Following post‐surgical monitoring, the rats received an adaptation trial in which they were placed in the taste reactivity chamber with each rat's cannula attached to the infusion pump. During adaptation, water was infused into their intraoral cannulae for 2 min at a rate of 1 mL·min−1. On the day following the adaptation trial, the rats received a conditioning trial in which they were administered a pretreatment injection of vehicle (VEH) (n = 8), CBDA (0.01, 0.1, 1 μg·kg−1; n = 8 per group) or HU‐580 (0.01, 0.1, 1 μg·kg−1; n = 8 per group). Forty‐five minutes after the pretreatment injection, the rats were individually placed in the chamber and infused, p.o., with 0.1% saccharin solution for 2 min at the rate of 1 mL·min−1. Immediately after the saccharin infusion, all rats were injected with 20 mL·kg−1 of 0.15 M LiCl and returned to their home cage. Seventy‐two hours later, rats were tested drug‐free. Rats were again infused p.o. with 0.1% saccharin solution for 2 min at the rate of 1 mL·min−1 while the orofacial responses were video recorded from a mirror at a 45° angle beneath the chambers. Rats were then returned to their home cages. Two additional groups were added to determine the mechanism of action. These rats were injected with WAY100635 (0.1 mg·kg−1) 15 min prior to an injection of either vehicle (n = 8) or 0.1 μg·kg−1 HU‐580 (n = 6). The video tapes were later scored by an observer blind to the experimental conditions using ‘The Observer’ for the behaviours of gaping (large openings of the mouth and jaw, with lower incisors exposed).
In vivo experiment 2: effect of CBDA and HU‐580 on anticipatory nausea and 5‐HT1A receptor mediation of HU‐580 effects
To compare the potential of HU‐580 and CBDA to reduce anticipatory nausea, the contextually elicited conditioned gaping paradigm was used (e.g. Limebeer et al., 2010; Rock et al., 2014; see also Figure 2B). Rats underwent four conditioning trials during which the distinctive context was paired with 127 mg·kg−1 LiCl. On each trial, rats were injected with LiCl and then immediately placed in the conditioning chamber for 30 min. This procedure was repeated four times with a 48 h interval between conditioning trials. For the test trial, rats were randomly assigned to one of five treatment groups (n = 6 per group): VEH, 0.1 μg·kg−1 CBDA, 0.1 μg·kg−1 HU‐580, 0.01 μg·kg−1 CBDA, 0.01 μg·kg−1 HU‐580. Pretreatments were injected 45 min before the rats were given an saline injection (20 mL·kg−1 i.p.) and individually placed in the conditioning (contextual) chamber for 5 min, and orofacial responses were video recorded. To investigate the mechanism of action of HU‐580, two additional groups of rats were administered 0.1 mg·kg−1 WAY‐VEH (n = 8), 0.1 mg·kg−1 WAY‐0.1 μg·kg−1 HU‐580 (n = 8). VEH or WAY100635 were administered 15 min before HU‐308 or VEH. The video tapes from the test trial were scored by an observer blind to the experimental conditions using ‘The Observer’ for the behaviours of gaping (large openings of the mouth and jaw, with lower incisors exposed).
Figure 2.
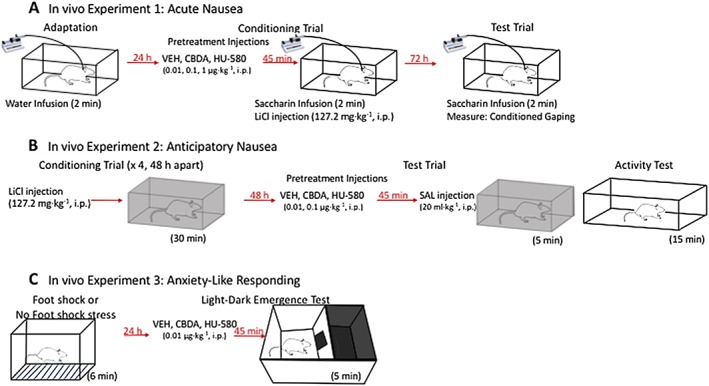
An illustration of the in vivo experimental procedures. (A) For the acute nausea model, rats undergo intra‐oral cannulation surgery and after recovery receive an adaptation trial consisting of a 2 min water infusion in the taste reactivity chamber. Twenty‐four hours later, they receive a single conditioning trial. Rats are pretreated with vehicle (VEH), CBDA or HU‐580 and, 45 min later, are placed in the taste reactivity chamber where they are infused with 0.1% saccharin for 2 min. To investigate the mechanism of action of HU‐580, additional groups of rats received WAY100635 15 min prior to HU‐580 or VEH. Immediately after the saccharin infusion, rats are injected with LiCl and returned to the homecage. Seventy‐two hours later, rats were subjected to a test trial during which they are returned to the taste reactivity chamber where they are infused with 0.1% saccharin for 2 min, and the number of gapes expressed are quantified. (B) For the anticipatory nausea model, rats receive four conditioning trials, 48 h apart, during which they are injected with LiCl and placed in the black conditioning chamber for 30 min, and then returned to the homecage. Forty‐eight hours after the final conditioning trial, rats receive a test trial where they are pretreated with VEH, CBDA or HU‐580 and, 45 min later, are injected with saline (SAL) and returned to the black conditioning chamber for 5 min while the number of gapes expressed are quantified. To investigate the mechanism of action of HU‐580, additional groups of rats received WAY100635 15 min prior to HU‐580 or VEH. Immediately after the anticipatory nausea test, rats are placed in a white activity chamber, and their distance travelled is tracked for 15 min. (C) For the anxiety‐like responding model, rats receive either footshock (FS) or No FS (remain in home cage) stress. Those that receive footshock stress are placed in sound‐attenuating MED Associates fear conditioning chambers. The 6 min FS session consists of six 0.8 mA foot shocks delivered 1 min apart. Each 0.5 s shock is preceded by a 30 s auditory tone (90 Db, 5000 Hz). Twenty‐four hours later, rats are pretreated with VEH, CBDA or HU‐580 45 min prior to placement in the dark chamber of the light‐dark box, and time spent in the light box is tracked. To investigate the mechanism of action of HU‐580, additional groups of rats received WAY100635 15 min prior to HU‐580 or VEH.
Immediately following the test trial, rats were put in the activity chamber (white Plexiglas, 60 × 25 × 25 cm, illuminated by a red light) for 15 min, and locomotor activity was captured by a video camera and sent to a computer using EthoVision software (Noldus, Inc, NL) to measure distance (cm) travelled.
In vivo experiment 3: effect of CBDA and HU‐580 on anxiety‐like responding and 5‐HT1A receptor mediation of HU‐580 effects
The effect of CBDA and HU‐580 on anxiety‐like responding was evaluated using the light‐dark box emergence test following either foot shock stress or no foot shock (No FS) stress (Figure 2C). Bluett et al. (2014) have demonstrated that anxiety‐like responding in this test is greatly enhanced 24 h following foot shock stress. Also, Rock et al. (2017) have shown that CBDA (at doses as low as 0.1 μg·kg−1 i.p.) prevents the enhanced anxiety‐like responding following foot shock stress, by a 5‐HT1A‐dependent mechanism of action. Therefore, we compared the relative effectiveness of an even lower dose (0.01 μg·kg−1, i.p) of CBDA and HU‐508 in this paradigm. Since we found that HU‐580 was anxiolytic at this low dose, we subsequently evaluated the ability of the 5HT1A receptor antagonist, WAY100635, to reverse the suppression of anxiety‐like responding by HU‐580.
All rats were acclimatized to the facility for 13 days prior to experimental manipulations, with weighing and handling occurring for eight of these days. After this acclimatization, the rats received a single FS stress session or No FS stress session 24 h before the light‐dark emergence test (Bluett et al., 2014). For the FS group, the rats were placed in sound‐attenuating MED Associates fear conditioning chambers (St. Albans, VT, USA). The 6 min FS session consisted of six 0.8 mA FSs delivered 1 min apart. Each 0.5 s shock was preceded by a 30 s auditory tone (90 Db, 5000 Hz) as described by Bluett et al. (2014). The No FS stress group remained in their home cage during this session.
Twenty‐four hours later, the rats were subjected to the light‐dark emergence test. Rats in the FS group and the No FS group were pretreated with VEH, 0.01 μg·kg−1 CBDA or 0.01 μg·kg−1 HU‐580. Forty‐five minutes later, they were placed in the dark chamber of the light‐dark box, and their movement was tracked for a 5 min test. To investigate the possibility that the effect of HU‐580 was 5‐HT1A receptor‐mediated, additional groups were injected with WAY100635, 15 min prior to VEH or 0.01 μg·kg−1 HU‐580. The number of seconds spent in the light box was measured. Groups were as follows: No FS–VEH (n = 9), FS‐VEH (n = 12), No FS‐0.01 μg·kg−1 CBDA (n = 8), FS‐0.01 μg·kg−1 CBDA (n = 8), No FS‐ 0.01 HU‐580 (n = 8), FS‐0.01 HU‐580 (n = 8), No FS‐0.1 μg·kg−1 WAY‐VEH (n = 8), FS‐0.1 μg·kg−1 WAY‐VEH (n = 7), No FS‐0.1 μg·kg−1 WAY‐0.01 μg·kg−1 HU580 (n = 8), FS‐ 0.1 μg·kg−1 WAY‐0.01 μg·kg−1 HU‐580 (n = 8).
In vitro and in vivo data analysis
Net agonist‐stimulated [35S]‐GTPγS binding values were calculated by subtracting basal binding values (obtained in the absence of agonist) from agonist‐stimulated values (obtained in the presence of agonist) (Cascio et al., 2010). Values are expressed as means and variability as SEM or as 95% confidence limits. Mean EC50 and mean maximal effect (Emax) values, and SEM or 95% confidence limits of these values, have been calculated by nonlinear regression analysis using the equation for a sigmoid concentration–response curve (GraphPad Prism). P values <0.05 were considered significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
For analysis of data from the acute nausea experiment (In vivo experiment 1), a single factor ANOVA was conducted for the mean number of gapes in the 2 min test, and subsequent pairwise comparisons were assessed with least significant difference (LSD) post hoc tests.
For analysis of data from the anticipatory nausea (AN) experiment (In vivo experiment 2), a single factor ANOVA was conducted for the number of gapes in the 5 min AN test and for the total distance travelled in the activity test, and subsequent pairwise comparisons were assessed with LSD post hoc tests.
For analysis of data from the anxiety‐like responding experiment (In vivo experiment 3), the amount of time spent in the light box during the light‐dark emergence test was entered into a 2 × 5 between factors ANOVA with the factors of FS stress/No FS stress and each pretreatment and μg·kg−1 i.p. dose condition (VEH, 0.01 μg·kg−1 CBDA, 0.01 μg·kg−1 HU‐580, WAY‐VEH or WAY‐HU‐580). Subsequent independent t‐tests were conducted to explore the interaction. Significance levels were set at P < 0.05.
Drugs and materials used in vitro
8‐OH‐DPAT and WAY100635 were supplied by Bio‐Techne (Abingdon, UK). [35S]‐GTPγS (1250 Ci mmol−1) was purchased from PerkinElmer Life Sciences, Inc. (Boston, MA, USA), and GTPγS, GDP and DMSO from Sigma‐Aldrich UK. CBDA and its methyl ester (HU‐580) were provided by Raphael Mechoulam.
Drugs used in vivo
Lithium chloride (LiCl; Sigma Aldrich) was prepared in a 0.15 M solution with sterile water and was administered i.p. at a volume of 20 mL·kg−1 (127.2 mg·kg−1 dose). CBDA and its methyl ester (HU‐580), both provided by Raphael Mechoulam, were dissolved in a glass graduated tube in 1 mL ethanol with 1 mL Tween80 (Sigma) added to the solution, and the ethanol was evaporated off with a nitrogen stream, after which 9 mL of saline was added (final Tween80:saline ratio = 1:9). CBDA or HU‐580 were administered to rats i.p. at a dose of 0.01, 0.1 or 1.0 μg·kg−1, in a volume of 1 mL·kg−1, using a stock solution containing one or other of these compounds at a concentration of 0.01, 0.1 or 1.0 μg·mL−1 respectively. WAY100635 (Sigma, St Louis, MO, USA) was dissolved in saline at a concentration of 0.1 mg·mL−1 and administered to rats i.p. at a dose of 0.1 mg·kg−1 (1 mL·kg−1).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
CBDA and HU‐580 enhance the ability of a 5‐HT1A receptor agonist to stimulate [35S]GTPγS binding to human 5‐HT1A receptors in vitro
As found previously in [35S]‐GTPγS binding experiments performed with rat brainstem membranes (Bolognini et al., 2013), CBDA enhanced the stimulation of [35S]‐GTPγS binding induced by the selective 5‐HT1A receptor agonist, 8‐OH‐DPAT, to membranes obtained from CHO cells stably transfected with human 5‐HT1A receptors (Figure 3 and Table 1). Concentrations of CBDA in the sub‐micromolar range, producing significant increases in the mean E max of 8‐OH‐DPAT at 0.1, 1.0 and 10 nM, but not at 0.01 or 100 nM. None of these increases in mean E max was accompanied by any significant change in the mean EC50 of 8‐OH‐DPAT (P > 0.05; Table 1). The methyl ester of CBDA, HU‐580, was even more potent than CBDA at enhancing 8‐OH‐DPAT‐induced stimulation of [35S]‐GTPγS binding to human 5‐HT1A receptor‐expressing CHO cell membranes (Figure 4 and Table 2). Thus, it produced a significant increase in the mean E max of 8‐OH‐DPAT not only at 0.1, 1.0 and 10 nM (like CBDA) but also at 0.01 nM (unlike CBDA). HU‐580 did not increase the mean E max of 8‐OH‐DPAT either at 100 nM (like CBDA) or at 0.001 nM and did not significantly affect the mean EC50 of 8‐OH‐DPAT at any of the concentrations investigated (Table 2). When administered by itself, at concentrations of 0.01, 0.1, 1, 10 or 100 nM, HU‐580 did not behave as a 5‐HT1A receptor agonist or inverse agonist as indicated by the lack of a detectable effect of any of these concentrations on [35S]‐GTPγS binding to membranes obtained from human 5‐HT1A receptor‐transfected CHO cells (n = 6; data not shown).
Figure 3.
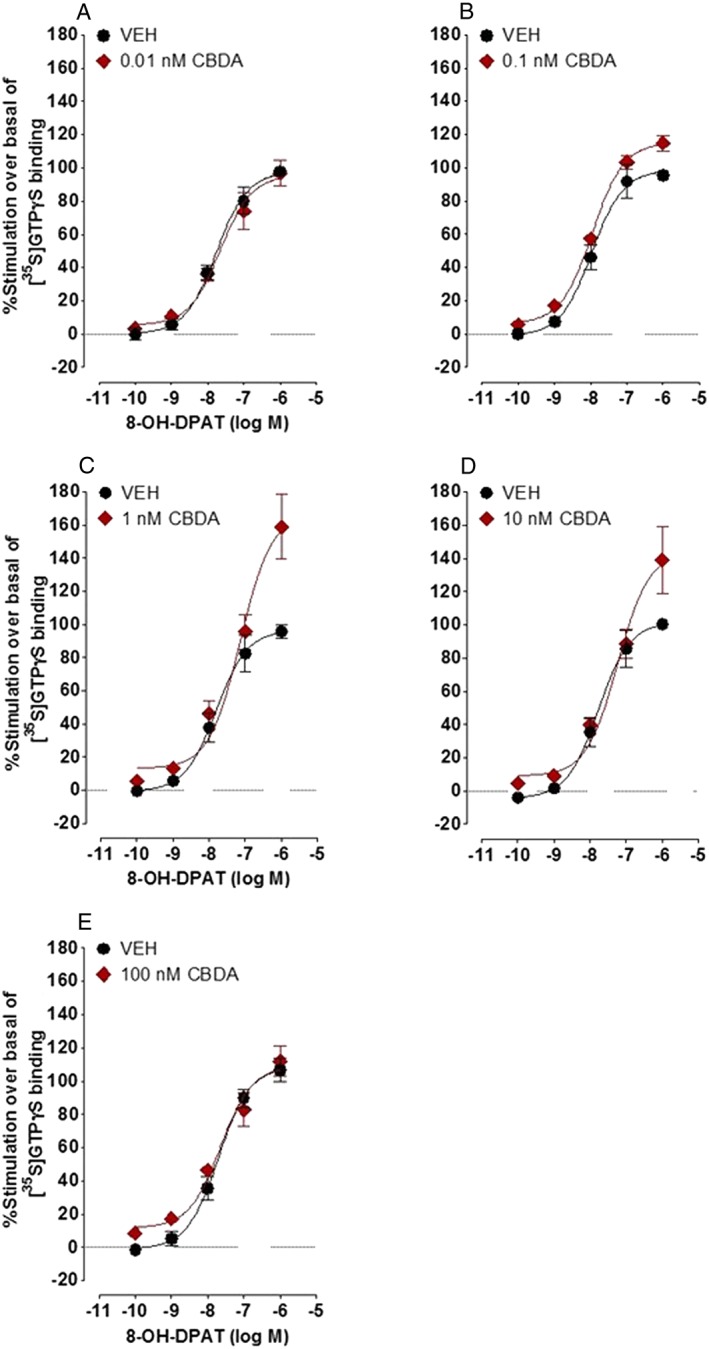
Effect of CBDA (0.01, 0.1, 1.0, 10 or 100 nM) on 8‐OH‐DPAT‐induced stimulation of [35S]‐GTPγS binding to membranes obtained from CHO cells stably transfected with human 5‐HT1A receptors. Symbols represent mean values ± SEM (n = 6). Mean E max and EC50 values for 8‐OH‐DPAT determined in the presence of CBDA or just of its vehicle (VEH), DMSO, together with the 95% confidence limits of these values, are listed in Table 1.
Table 1.
Effects of CBDA on the mean EC50 and E max values of 8‐OH‐DPAT for its stimulation of [35S]‐GTPγS binding to membranes obtained from CHO cells stably transfected with human 5‐HT1A receptors
| Pretreatment | Mean EC50 (nM) | 95% confidence limits (nM) | Mean E max (%) | 95% confidence limits (%) | n |
|---|---|---|---|---|---|
| Vehicle | 18 | 10 and 30 | 98 | 89 and 107 | 6 |
| 0.01 nM CBDA | 23 | 10 and 50 | 96 | 83 and 108 | 6 |
| Vehicle | 11 | 6 and 20 | 99 | 88 and 109 | 6 |
| 0.1 nM CBDA | 11 | 8 and 15 | 115* | 110 and 121 | 6 |
| Vehicle | 16 | 8 and 32 | 97 | 85 and 109 | 6 |
| 1.0 nM CBDA | 73 | 32 and 166 | 167* | 138 and 196 | 6 |
| Vehicle | 17 | 9 and 33 | 102 | 90 and 114 | 6 |
| 10 nM CBDA | 57 | 24 and 140 | 144* | 118 and 169 | 6 |
| Vehicle | 20 | 12 and 35 | 109 | 98 and 119 | 6 |
| 100 nM CBDA | 23 | 11 and 49 | 109 | 96 and 122 | 6 |
See also Figure 3.
Each asterisk indicates a significant difference (*P < 0.05) between a mean E max value of 8‐OH‐DPAT determined in the presence of a particular concentration of CBDA and the mean E max value of 8‐OH‐DPAT displayed in the previous row that was determined in the same experiment in the presence of vehicle (DMSO) instead of CBDA. Significant differences are indicated by non‐overlapping 95% confidence limits.
Figure 4.
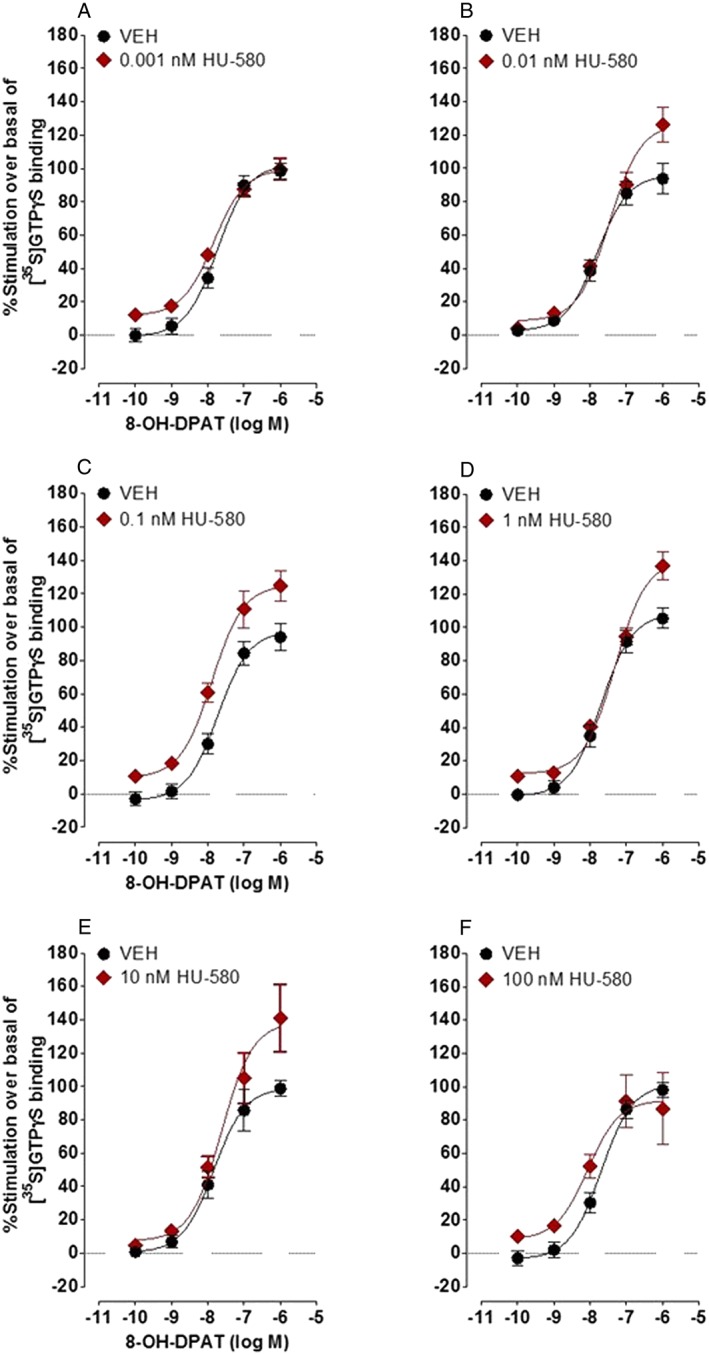
Effect of HU‐580 (0.001, 0.01, 0.1, 1.0, 10 or 100 nM) on 8‐OH‐DPAT‐induced stimulation of [35S]‐GTPγS binding to membranes obtained from CHO cells stably transfected with human 5‐HT1A receptors. Symbols represent mean values ± SEM (n = 6). Mean E max and EC50 values for 8‐OH‐DPAT determined in the presence of HU‐580 or just of its vehicle (VEH), DMSO, together with the 95% confidence limits of these values, are listed in Table 2.
Table 2.
Effects of HU‐580 on the mean EC50 and E max values of 8‐OH‐DPAT for its stimulation of [35S]‐GTPγS binding to membranes obtained from CHO cells stably transfected with human 5‐HT1A receptors
| Pretreatment | Mean EC50 (nM) | 95% confidence limits (nM) | Mean E max (%) | 95% confidence limits (%) | n |
|---|---|---|---|---|---|
| Vehicle | 18 | 11 and 30 | 102 | 93 and 112 | 6 |
| 0.001 nM HU‐580 | 14 | 9 and 22 | 100 | 98 and 106 | 6 |
| Vehicle | 16 | 8 and 31 | 96 | 85 and 107 | 6 |
| 0.01 nM HU‐580 | 34 | 19 and 63 | 127* | 114 and 140 | 6 |
| Vehicle | 19 | 10 and 36 | 98 | 86 and 109 | 6 |
| 0.1 nM HU‐580 | 13 | 7 and 24 | 125* | 113 and 138 | 6 |
| Vehicle | 20 | 11 and 34 | 108 | 98 and 119 | 6 |
| 1.0 nM HU‐580 | 48 | 31 and 74 | 141* | 129 and 152 | 6 |
| Vehicle | 15 | 7 and 31 | 99 | 87 and 112 | 6 |
| 10 nM HU‐580 | 24 | 9 and 65 | 139* | 116 and 162 | 6 |
| Vehicle | 20 | 12 and 34 | 102 | 92 and 111 | 6 |
| 100 nM HU‐580 | 9 | 2 and 39 | 92 | 72 and 112 | 6 |
See also Figure 4.
Each asterisk indicates a significant difference (*P < 0.05) between a mean E max value of 8‐OH‐DPAT determined in the presence of a particular concentration of HU‐580 and the mean E max value of 8‐OH‐DPAT displayed in the previous row that was determined in the same experiment in the presence of vehicle (DMSO) instead of HU‐580. Significant differences are indicated by non‐overlapping 95% confidence limits.
In vivo experiment 1: dose‐related effects of CBDA and HU‐580 on acute nausea and 5‐HT1A receptor mediation of HU‐580 effects
At a dose of 0.1 μg·kg−1, but not at 0.01 or 1 μg·kg−1, HU‐580 was more effective than CBDA in reducing acute nausea as assessed by the rat gaping model. HU‐580's suppressive effect on acute nausea (0.1 μg·kg−1) was blocked by WAY100635. A single factor ANOVA revealed a significant group effect F(8, 61) = 3.9; P <0.05. Figure 5 presents the mean number of gapes displayed by the various pretreatment groups. Subsequent LSD post hoc comparison tests revealed that both compounds reduced LiCl‐induced gaping responses relative to vehicle at a dose of 1 μg·kg−1 (P < 0.05), replicating our previous findings (Limebeer et al., 2010; Rock and Parker, 2013). However, at the even lower dose of 0.1 μg·kg−1, that is, subthreshold for a CBDA‐induced reduction of nausea‐like behaviour, HU‐580 reduced LiCl‐induced conditioned gaping behaviour relative to vehicle (P < 0.05). Rats pretreated with HU‐580 (0.1 μg·kg−1) also gaped significantly less than group WAY‐0.1 μg·kg−1 HU‐580 (P < 0.05), indicating a 5‐HT1A receptor‐mediated effect.
Figure 5.
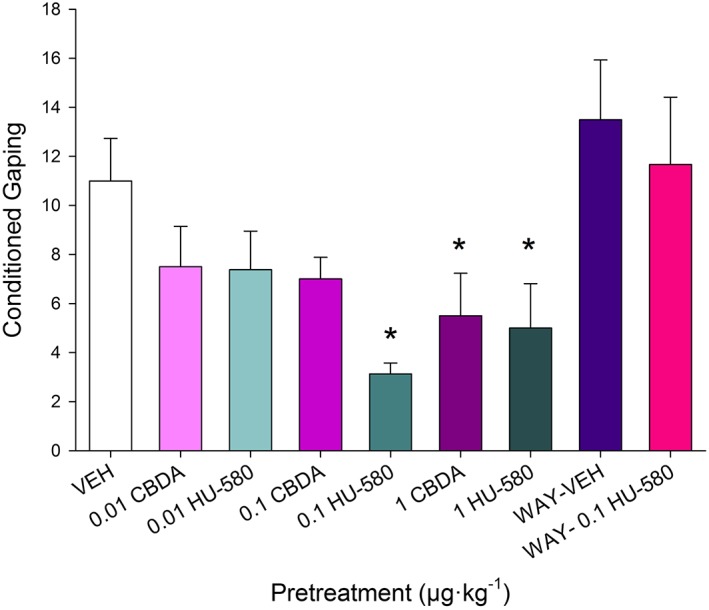
Mean number of conditioned gapes elicited by a LiCl‐paired saccharin solution among the rats pretreated with various doses of CBDA (n = 8 per group) or HU‐580 (n = 8 per group) or just with vehicle (VEH; n = 8). Additional groups were administered a pretreatment of WAY100635 (0.1 mg·kg−1) 15 min prior to 0.1 mg·kg−1 HU‐580 (n = 6) or VEH (n = 8). Results are presented as mean ±SEM and *P < 0.05, depicts mean responses to CBDA or HU‐580, which differed significantly from mean responses to VEH.
In vivo experiment 2: effect of CBDA and HU‐580 on anticipatory nausea and 5‐HT1A receptor mediation of HU‐580 effects
At an extremely low dose of 0.01 μg·kg−1, but not at 0.1 μg·kg−1, HU‐580 was more effective than CBDA in reducing anticipatory nausea as assessed by the contextually elicited conditioned gaping model. The suppressive effect of HU‐580 (0.1 μg·kg−1) was blocked by pretreatment with WAY100635. A single factor ANOVA revealed a significant group effect F(6, 39) = 8.7; P < 0.05. Figure 6A presents the mean number of gapes displayed. Subsequent LSD post hoc comparisons revealed that compared to VEH controls, at a dose of 0.1 μg·kg−1, both CBDA and HU‐580 reduced conditioned gaping (P values < 0.05); however, the groups did differ at a dose of 0.01 μg·kg−1, with group HU‐580 gaping significantly less than VEH controls (P < 0.05) and group 0.01 CBDA (P = 0.05). Rats pretreated with HU‐580 (0.1 μg·kg−1) also gaped significantly less than group WAY‐0.1 μg·kg−1 HU‐580 (P < 0.05), indicating a 5‐HT1A receptor‐mediated effect.
Figure 6.
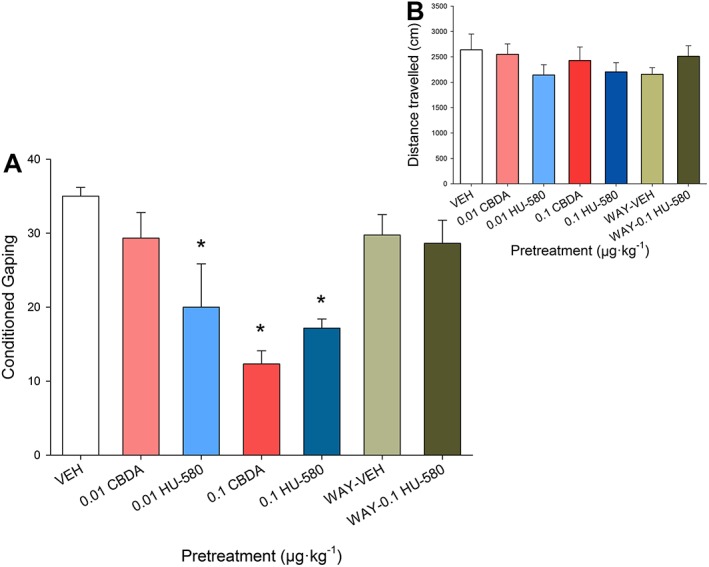
(A) Effect of CBDA or HU‐580 (0.01, 0.1 μg·kg−1) or vehicle (VEH) administered i.p. 45 min prior to the anticipatory nausea test (n = 6 per group). Additional groups were administered a pretreatment of WAY100635 (0.1 mg·kg−1) 15 min prior to 0.1 mg·kg−1 HU‐580 (n = 8) or VEH (n = 8). The mean number of conditioned gaping responses was measured during the anticipatory nausea test trial. Each bar represents the mean ± SEM. *P < 0.05, significant difference from the VEH‐treated control animals. (B) The mean distance (cm) travelled was measured in an activity test performed after the anticipatory nausea test. Each bar represents the mean ± SEM.
A single factor ANOVA for the locomotor activity test (Figure 6B) revealed no significant effect on distance moved, F(6, 39) = 0.9, P > 0.05.
In vivo experiment 3: anxiolytic effects of CBDA and HU‐580
Figure 7 presents the mean number of seconds spent by rats in the light box for each of the various pretreatment groups that received FS or No FS 24 h prior the light‐dark test. As can be seen, FS stress greatly enhanced the anxiety‐like responding of decreased time spent in the light box. At a low dose of 0.01 μg·kg−1, HU‐580, but not CBDA, reversed the effect of FS on the anxiety‐like responding of decreased time spent in the light box. The 2 × 5 ANOVA for the number of seconds spent in the light box revealed a significant main effect of FS stress, F(1, 84) = 25.6; P < 0.05, and a FS stress × pretreatment interaction, F(4, 84) = 3.2; P , 0.05). To analyse the interaction, subsequent independent t‐tests revealed that rats pretreated with VEH (P < 0.05), 0.01 μg·kg−1 CBDA (P < 0.05), WAY‐VEH (P < 0.05) or WAY‐0.01 μg·kg−1 HU‐580 (P = 0.05) spent less time in the light box following FS stress than following No FS stress, but rats pretreated with 0.01 μg·kg−1 HU‐580 did not display this anxiogenic‐like response. Furthermore, subsequent single factor ANOVAs of the time spent in the light box revealed a significant pretreatment effect among the FS groups, F(4, 38) = 4.6; P < 0.05, but not among the No FS groups. Among the FS groups, subsequent Bonferroni tests revealed that only group 0.01 μg·kg−1 HU‐580 spent significantly more time in the light box than group VEH (P < 0.05).
Figure 7.
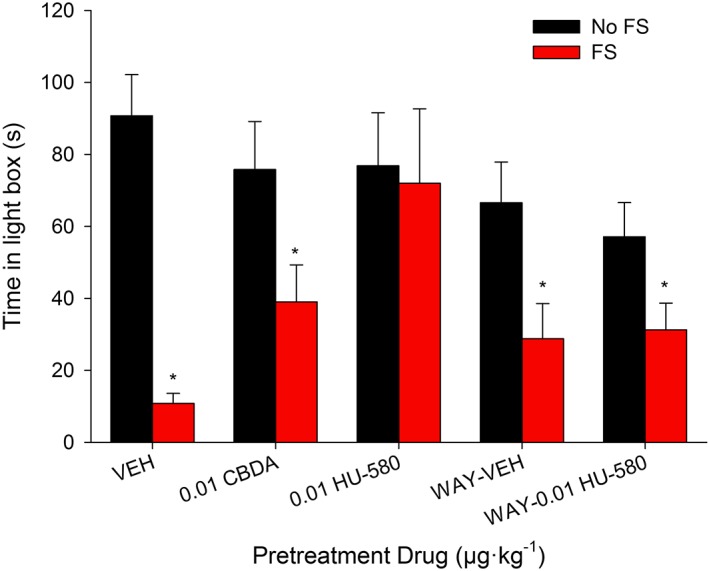
The mean time spent by rats in the light box, 24 h following exposure to no foot shocks (No‐FS) or to FSs. All rats were injected i.p. with vehicle (VEH; n = 9 or 12), 0.01 μg·kg−1, CBDA (n = 8) or 0.01 μg·kg−1 HU‐580 (n = 8), 45 min prior to a 5 min light‐dark box emergence test. Additional groups were injected with 0.1 mg·kg−1 WAY100635 15 min prior to VEH (n = 7 or 8) or 0.01 μg·kg−1 HU‐580 (n = 8). Each bar represents the mean ± SEM. *P < 0.05, indicates a significant difference between FS and No FS stress groups.
Discussion
The results obtained in this investigation confirm our previous findings (Bolognini et al., 2013) that CBDA displays significant potency both at producing an apparent enhancement of the activation of 5‐HT1A receptors, by the direct 5‐HT1A receptor agonist, 8‐OH‐DPAT, in vitro, and at producing a 5‐HT1A receptor‐mediated reduction of both acute and anticipatory nausea in rats, in vivo (Bolognini et al., 2013; Rock et al., 2014, 2015).
The new in vitro data we have now obtained suggest, first, that CBDA can enhance the activation not only of rat brain stem 5‐HT1A receptors, as shown previously (Bolognini et al., 2013), but also of human 5‐HT1A receptors (this paper) and, second, that at both rat brain stem (Bolognini et al., 2013) and human 5‐HT1A receptors (this paper), CBDA induces such enhancement with a bell‐shaped concentration–response curve in the sub‐micromolar range.
The in vitro data described in this paper also reveal an important similarity between the pharmacological effects of CBDA and its methyl ester, HU‐580. More specifically, these data have provided convincing evidence that HU‐580 shares the ability of CBDA to produce an apparent enhancement of the activation of human 5‐HT1A receptors by 8‐OH‐DPAT in the [35S]‐GTPγS binding assay. Importantly, HU‐580 produced such enhancement both with greater potency and with an even broader bell‐shaped concentration–response curve than CBDA. Thus, significant enhancement was induced by HU‐580 at concentrations of 0.01 to 10 nM (Table 2) and by CBDA at concentrations of 0.1 to 10 nM (Table 1). Whereas, at concentrations of 1, 10 and 100 nM, HU‐580 produced slightly less enhancement of 8‐OH‐DPAT‐induced 5‐HT1A receptor activation than CBDA, HU‐580 produced slightly greater enhancement of this activation than CBDA, at concentrations of 0.01 and 0.1 nM (Tables 1 and 2).
It is noteworthy that none of the concentrations of CBDA and HU‐580 that significantly increased Emax values of 8‐OH‐DPAT for its stimulation of [35S]‐GTPγS binding to 5‐HT1A receptors produced any significant change in the EC50 of 8‐OH‐DPAT (Tables 1 and 2). This finding suggests that CBDA and HU‐580 may have been acting as positive allosteric modulators of the activation of these receptors by 8‐OH‐DPAT, there being evidence that some positive allosteric modulators do indeed increase the Emax values but not the potencies of agonists at certain receptors (Christopoulos et al., 2014). The possibility that CBDA and HU‐580 target an allosteric site on the 5‐HT1A receptor, as positive allosteric modulators, merits further investigation. It is also noteworthy that the positive in vitro data for CBDA and HU‐580 we obtained in this investigation all came from experiments performed with CHO cells transfected with human 5‐HT1A receptors. Consequently, it will be of interest to establish, in a follow‐up investigation, whether similar results would be obtained in experiments performed with a human‐derived cell line that expresses human HT1A receptors constitutively.
Moving on to our new in vivo data, these too reveal similarities between the pharmacological effects of HU‐580 and CBDA. Thus, these data show that the ability of CBDA to reduce acute and anticipatory nausea in rats extends to HU‐580. Importantly, as also found in our in vitro experiments, HU‐580 displayed even greater potency than CBDA. More specifically, effective suppression of acute nausea‐induced conditioned gaping was induced by HU‐580 at a dose as low as 0.1 μg·kg−1 i.p., whereas the lowest effective dose of CBDA for the production of such suppression was 1 μg·kg−1 i.p. (Figure 5). Indeed, we found that at a dose as low as 0.01 μg·kg−1 i.p., HU‐580, but not CBDA, suppressed contextually elicited conditioned gaping. We have also shown that, as demonstrated previously in experiments with CBDA (Bolognini et al., 2013; Rock and Parker, 2013), and indeed with cannabidiol (Rock et al., 2012), suppression of LiCl‐induced gaping and contextually elicited conditioned gaping by HU‐580 can be completely prevented by the 5‐HT1A receptor‐selective antagonist, WAY100635. Finally, although CBDA has recently been found to reduce FS enhancement of anxiogenic‐like behaviour in the light‐dark box emergence test at doses of 0.1, 1 and 100 μg·kg−1 i.p. (Rock et al., 2017), in the present investigation, we found that it did not share the ability of HU‐580 to reduce FS enhancement of anxiogenic‐like behaviour in the light‐dark emergence test at the lower dose of 0.01 μg·kg−1 i.p., suggesting that HU‐580 may be even more potent than CBDA in reducing stress‐induced anxiety. Furthermore, we also obtained convincing evidence that the ability of HU‐580 to reduce FS enhancement of anxiogenic‐like behaviour is 5‐HT1A receptor‐mediated, evidence similar to that already obtained from experiments with CBDA (Rock et al., 2017) and cannabidiol (Campos and Guimarães, 2008). Future studies should extend this finding to other measures of anxiogenic‐like behaviour and examine other potential sites of action of CBDA and HU580 in their anti‐nausea and anti‐anxiety‐like effects, such as antagonism of the allosteric site of the CB1 receptor (Laprairie et al., 2015). It will also be important to establish whether, as found in our in vitro experiments, HU‐580 is effective over a broader range of doses than CBDA. Thus, such a finding, together with our discoveries that HU‐580 is both more stable than CBDA, and more potent than CBDA, at least versus signs of acute and anticipatory nausea, would strengthen the hypothesis that HU‐580 has markedly greater potential than CBDA as a new medicine for the management of unwanted symptoms such as nausea.
Ideally, drugs used as medicines should, when stored, display stability over a reasonable period of time. Hence, since stored CBDA undergoes significant decomposition, even at 4°C, a major aim of this project was to develop a compound that produces no less potency than CBDA in the assays described in this paper but displays much greater stability over a reasonable length of time when stored at this temperature. It is noteworthy, therefore, that we found that HU‐580 is, indeed, more stable than CBDA when stored at 4°C for 21 days. In addition, our finding that HU‐580 seems to be more potent than CBDA both in vitro and in vivo supports the hypothesis that the pharmacological effects produced by HU‐580 in our experiments did not depend on its decomposition or metabolism to CBDA. It will be of interest to establish in a follow‐up investigation, both why HU‐580 displayed greater potency than CBDA in our assays and whether it is HU‐580 itself that produces the effects we observed in our experiments with this compound or whether HU‐580 is a promising ‘pro‐drug’ that was converted in our in vitro and/or in vivo assays to one or more compounds that are more active than HU‐580 itself.
In conclusion, this investigation has provided evidence that the methyl ester of CBDA, HU‐580, displays even greater potency than CBDA at suppressing signs both of acute and anticipatory nausea, and of stress‐induced anxiety in rats, and that it produces these effects in a 5‐HT1A receptor‐dependent manner. Further experiments are still needed to determine the extent to which this apparent potency difference occurred (i) because CBDA had undergone at least some conversion to one or more less potent enhancers of 5‐HT1A receptor activation, such as CBD (Bolognini et al., 2013), or to one or more inactive compounds, during our experiments, and/or (ii) because CBDA itself really is less potent than HU‐580 at enhancing 5‐HT1A receptor activation. In addition, since HU‐580 is a relatively stable compound, our findings also prompt a need for human clinical research with this compound, especially since there is already evidence that CBD is anxiolytic in human subjects and that the ability of cannabidiol to decrease anxiety induced in healthy human volunteers by simulated public speaking is shared by the 5‐HT1A receptor agonist, ipsapirone (Zuardi et al., 1993; Patel et al., 2017). Such clinical research, performed with HU‐580, and possibly subsequently with cannabidiol and CBDA as well, so that the therapeutic potential of these three cannabinoids can be compared, should be directed at establishing, firstly, whether HU‐580 really does display therapeutic potential for the treatment of particular kinds of nausea and anxiety and, secondly, whether it could possibly be effective against any other disorders that might be ameliorated by enhancing the activation of 5‐HT1A receptors, for example, cerebral infarction, pain and depression (Bolognini et al., 2013). The effectiveness of HU‐580 versus anticipatory nausea will be particularly important to investigate as no specific therapy for this disorder is currently available. It will also be important both to identify the precise pharmacological action(s) through which HU‐580, as well as CBDA and cannabidiol, appear to enhance agonist‐induced activation of the 5‐HT1A receptor and to seek out any 5‐HT1A receptor‐independent pharmacological actions of HU‐580, particularly any which might affect its benefit‐to‐risk ratio in the clinic, for example, for its possible treatment of nausea or anxiety. Finally, in view of our findings that HU‐580 seems to strengthen 5‐HT1A receptor activation and to produce 5‐HT1A‐mediated amelioration of stress‐induced anxiety in rats, the evidence that the 5‐HT1A receptor direct agonist, ipsapirone, is anxiolytic in human subjects (Zuardi et al., 1993) prompts a need to investigate whether HU‐580, or indeed CBDA or cannabidiol, interact synergistically with an exogenously administered 5‐HT1A receptor direct agonist to reduce signs of anxiety in rats or humans, more effectively, potently and/or selectively than HU‐580, CBDA, cannabidiol or a 5‐HT1A receptor direct agonist administered by itself.
Author contributions
All authors contributed equally: R.G.P., L.A.P., E.M.R. and R.M. planned and designed the research; R.M., R.S. and C.H. designed and synthesized HU‐580; L.A.S. performed the in vitro experiments; E.M.R., K.G. and C.L.L. performed the in vivo experiments; R.G.P., L.A.P. and E.M.R. analysed the data and the literature; R.G.P., L.A.P., E.M.R. and R.M. co‐wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The in vivo research described in this paper was supported by a grant from Natural Sciences and Engineering Council of Canada to L.A.P. The authors would like to thank Dr. Keith Parker for supplying CHO cells stably transfected with human 5‐HT1A receptors.
Pertwee, R. G. , Rock, E. M. , Guenther, K. , Limebeer, C. L. , Stevenson, L. A. , Haj, C. , Smoum, R. , Parker, L. A. , and Mechoulam, R. (2018) Cannabidiolic acid methyl ester, a stable synthetic analogue of cannabidiolic acid, can produce 5‐HT1A receptor‐mediated suppression of nausea and anxiety in rats. British Journal of Pharmacology, 175: 100–112. doi: 10.1111/bph.14073.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017). The Concise Guide To PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluett RJ, Gamble‐George JC, Hermanson DJ, Hartley ND, Marnett LJ, Patel S (2014). Central anandamide deficiency predicts stress-induced anxiety: behavioral reversal through endocannabinoid augmentation. Transl Psychiatry 4: e408. https://doi.org/10.1038/tp.2014.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognini D, Rock EM, Cluny NL, Cascio MG, Limebeer CL, Duncan M et al (2013). Cannabidiolic acid prevents vomiting in Suncus murinus and nausea‐induced behaviour in rats by enhancing 5‐HT1A receptor activation. Br J Pharmacol 168: 1456–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AC, Guimarães FS (2008). Involvement of 5HT1A receptors in the anxiolytic‐like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology (Berl) 199: 223–230. [DOI] [PubMed] [Google Scholar]
- Cascio MG, Pertwee RG (2014). Known pharmacological actions of nine non‐psychotropic phytocannabinoids In: Pertwee RG. (ed). Handbook of Cannabis. Oxford University Press: Oxford, pp. 137–156. [Google Scholar]
- Cascio MG, Gauson LA, Stevenson LA, Ross RA, Pertwee RG (2010). Evidence that the plant cannabinoid cannabigerol is a highly potent α2‐adrenoceptor agonist and moderately potent 5HT1A receptor antagonist. Br J Pharmacol 159: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopoulos A, Changeux J‐P, Catterall WA, Fabbro D, Burris TP, Cidlowski JA et al (2014). International Union of Basic and Clinical Pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66: 918–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crombie L, Crombie WML (1977). Cannabinoid acids and esters: miniaturized synthesis and chromatographic study. Phytochemistry 16: 1413–1420. [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill HJ, Norgren R (1978). Chronically decerebrate rats demonstrate satiation but not bait shyness. Science 201: 267–269. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krejčí Z, Šantavý F (1955). Isolace dalsˇích látek z listí indického konopí Cannabis sativa L. Acta Univ Palacki Olomuc 6: 59–66. [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan‐Wright EM (2015). Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol 172: 4790–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limebeer CL, Vemuri VK, Bedard H, Lang ST, Ossenkopp KP, Makriyannis A et al (2010). Inverse agonism of cannabinoid CB1 receptors potentiates LiCl‐induced nausea in the conditioned gaping model in rats. Br J Pharmacol 161: 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R (1973). Cannabinoid chemistry In: Marijuana Chemistry, Metabolism, Pharmacology and Clinical Effects. Ed Mechoulam R Academic Press: New York, pp. 1–99. [Google Scholar]
- Mechoulam R, Ben‐Zvi Z (1969). Carboxylation of rescorcinols and methyl magnesium carbonate. Synthesis of cannabinoid acids. J Chem Soc D ‐ Chem Commun Issue 7: 343–344. [Google Scholar]
- Mechoulam R, Gaoni Y (1965). Hashish – IV. The isolation and structure of cannabinolic, cannabidiolic and cannabigerolic acids. Tetrahedron 21: 1223–1229. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Parker LA, Gallily R (2002). Cannabidiol: an overview of some pharmacological aspects. J Clin Pharmacol 42: 11S–19S. [DOI] [PubMed] [Google Scholar]
- Norman ROC, Coxon JM (1993). Principles of organic synthesis. Blackie Academic: London, p. 389. [Google Scholar]
- Patel S, Hill MN, Cheer JF, Wotjak CT, Holmes A (2017). The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci Biobehav Rev 76: 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Bolognini D, Limebeer CL, Cascio MG, Anavi‐Goffer S, Fletcher PJ et al (2012). Cannabidiol, a non‐psychotropic component of cannabis, attenuates vomiting and nausea‐like behaviour via indirect agonism of 5‐HT1A somatodendritic autoreceptors in the dorsal raphe nucleus. Br J Pharmacol 165: 2620-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Parker LA (2013). Effect of low doses of cannabidiolic acid and ondansetron on LiCl‐induced conditioned gaping (a model of nausea‐induced behaviour) in rats. Br J Pharmacol 169: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Parker LA (2015). Synergy between cannabidiol, cannabidiolic acid, and Δ9‐tetrahydrocannabinol in the regulation of emesis in the Suncus murinus (house musk shrew). Behav Neurosci 129: 368–370. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Navaratnam R, Sticht MA, Bonner N, Engeland K et al (2014). A comparison of cannabidiolic acid with other treatments for anticipatory nausea using a rat model of contextually elicited conditioned gaping. Psychopharmacology (Berl) 231: 3207–3215. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Parker LA (2015). Effect of combined doses of ∆9‐tetrahydrocannabinol (THC) and cannabidiolic acid (CBDA) on acute and anticipatory nausea using rat (Sprague‐Dawley) models of conditioned gaping. Psychopharmacology (Berl) 232: 4445–4454. [DOI] [PubMed] [Google Scholar]
- Rock EM, Connolly C, Limebeer CL, Parker LA (2016). Effect of combined oral doses of Δ9‐tetrahydrocannabinol (THC) and cannabidiolic acid (CBDA) on acute and anticipatory nausea in rat models. Psychopharmacology (Berl) 233: 3353–3360. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Petrie GN, Williams LA, Mechoulam R, Parker LA (2017). Effect of prior foot shock stress and Δ9‐tetrahydrocannabinol, cannabidiolic acid, and cannabidiol on anxiety‐like responding in the light‐dark emergence test in rats. Psychopharmacology (Berl) 234: 2207–2217. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Okazaki H, Ikeda E, Abe S, Yoshioka Y, Watanabe K et al (2014). Down‐regulation of cyclooxygenase‐2 (COX‐2) by cannabidiolic acid in human breast cancer cells. J Toxicol Sci 39: 711–716. [DOI] [PubMed] [Google Scholar]
- Takeda S, Himeno T, Kakizoe K, Okazaki H, Okada T, Watanabe K et al (2017). Cannadidiolic acid‐mediated selective down‐regulation of c‐fos in highly agressive breast cancer MDA‐MB‐231 cells: possible involvement of its down‐regulation in the abrogation of aggressiveness. J Nat Med 71: 286–291. [DOI] [PubMed] [Google Scholar]
- Zhornitsky S, Potvin S (2012). Cannabidiol in humans – the quest for therapeutic targets. Pharmaceuticals 21: 529–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuardi AW, Cosme RA, Graeff FG, Guimarães FS (1993). Effect of ipsapirone and cannabidiol on human experimental anxiety. J Psychopharmacol 7: 82–88. [DOI] [PubMed] [Google Scholar]