

Abstract
The clinical phenotypes of human CoQ10‐deficiency caused by COQ2 mutations range from fatal neonatal disease to adult‐onset multisystem atrophy. So far, treatment options for these diseases are unsatisfactory. Here, we demonstrate that supplementation of 4‐hydroxybenzoic acid (4‐HBA) fully restores endogenous CoQ10‐biosynthesis in COQ2‐deficient cell lines. This was accompanied by increased protein expression of CoQ10‐biosynthesis‐enzymes as well as a rescue of cell viability during stress conditions. In silico analysis suggested a ligand transportation path for 4‐HBA through the COQ2 protein towards the mitochondrial matrix side. This process is apparently hindered by disease‐causing mutations, which can be overcome by increasing 4‐HBA concentrations.
Introduction
The mitochondrial respiratory chain requires several cofactors for normal functioning.1 Among these cofactors, coenzyme Q10 (CoQ10) features prominently. CoQ10 acts as a lipid‐soluble electron carrier from mitochondrial complex I/II to complex III.2 In addition, CoQ10 participates in a number of aspects of cellular metabolism including redox homeostasis and membrane stability.2, 3
CoQ10 is mainly derived via endogenous biosynthesis, depending on the interplay of at least 12 different enzymes.4 One of these enzymes is COQ2 (4‐HBA‐polyprenyltransferase), which catalyzes the prenylation of 4‐hydroxybenzoic acid (4‐HBA) with an all‐trans polyprenyl chain.5 Human COQ2‐deficiency was first identified in 2006 in two siblings with infantile‐onset nephropathy and psychomotor regression.6 In the following, several other patients were reported. The disease spectrum ranges from neonatal‐onset multisystem diseases to adult‐onset ataxia and cerebellar atrophy (Table S1).
Of note, clinical response to supplementation of CoQ10 was unsatisfactory in most COQ2‐deficient patients reported so far, which might be related to its poor oral bioavailability (Table S1).7, 8 Additional factors are that CoQ10 is incorporated in all cell membranes (e.g., not specifically mitochondrial) and its subcellular distribution requires the action of the chaperone‐like proteins COQ10A and COQ10B.4 Therefore, it remains unclear if orally administered CoQ10 effectively reaches the respiratory chain.
Methods
Cell culture
Fibroblasts cell lines were cultured in Dulbecco's modified Eagle's medium (Life technologies) supplemented with 10% fetal bovine serum (life technologies) and 1% penicillin/streptomycin (life technologies) at 37°C in a humidified atmosphere of 5% CO2. The use of patient‐derived cell lines was approved by the ethical committee of the Medical Faculty, Heinrich‐Heine‐University Düsseldorf (#5238). Biochemical details regarding the COQ2 and the COQ9 cell lines were published previously (COQ2‐def.1 and COQ9‐def. see Danhauser et al., 2015; COQ2‐def.2 and COQ2‐def.3 see Jakobs et al., 2013; see also Table S1).9, 10
UPLC‐ESI‐MS/MS analysis
UPLC‐ESI‐MS/MS analysis was performed using an Acquity UPLC‐I Class (Waters, UK) coupled to a Waters Xevo TQ‐S tandem mass spectrometer (Waters, UK), which was equipped with an ESI source operating in the positive ion mode. Methodological details were described previously.11
Compound supplementation studies
For compound testing, 400,000 cells/T75 flask were cultured and medium was changed every third day containing one of the following substances: 4‐hydroxybenzoic acid (4‐HBA), 4‐hydroxyphenylpyruvic acid (4‐HPPA), 4‐hydroxybenzaldehyde (4‐HBAL), L‐tyrosin and mevalonic acid. Chemicals were purchased from Sigma‐Aldrich. L‐tyrosin was dissolved in 1N NaOH. All other compounds were dissolved in 0.03% DMSO (Sigma). After 14 days of culturing, cells were harvested for UPLC‐ESI‐MS/MS analysis.
Immunoblot analyses
Methodological details of immunoblot analyses were described previously.11 The following primary antibodies were used: COQ2 (anti‐chicken; 1:1000; AS132713; Agrisera), COQ4 (anti‐rabbit; 1:500; 16654‐1‐AP; Proteintech), COQ7 (anti‐rabbit; 1:1000; 15083‐1‐AP; Proteintech) or SDHA (anti‐mouse; 1:1000; ab14715; Abcam).
Cell proliferation
Cell proliferation was determined using the crystal violet assay as described previously.12
Live/dead assay
Cells viability was measured using Life/Dead assay® (Invitrogen) according to the manufactures protocol.
In silico analysis
The apo structure of the archaeal homolog of COQ2 from Aeropyrum pernix was used as a template to build a model of human COQ2 using the modeling server Phyre 2.13, 14 The resulting model was manually inspected using the program COOT.15 4‐HBA binding sites were determined using the program AUTODOCK.16 After inspection of protein ligand interactions, putative binding sites were visualized using the program Pymol (www.pymol.org).
Results
Supplementation of 4‐hydroxybenzoic acid restores CoQ10 biosynthesis
4‐hydroxybenzoic acid (4‐HBA) is a small molecule, which is derived from L‐tyrosine (Fig. 1A). As depicted in Figure 1B, 4‐HBA as well as its precursor compounds 4‐HPPA, 4‐HBAL and, to a lesser extent, L‐tyrosine rescued the biochemical defect in three COQ2‐deficient fibroblast lines. This phenomenon was dose‐dependent with significant effects of 4‐HBA, 4‐HPPA, and 4‐HBAL down to concentrations of 1 μmol/L (Fig. 1C). Of note, treatment of fibroblasts with mevalonic acid (1000 μmol/L for 2 weeks) had no effect on CoQ10 biosynthesis (not shown).
Figure 1.
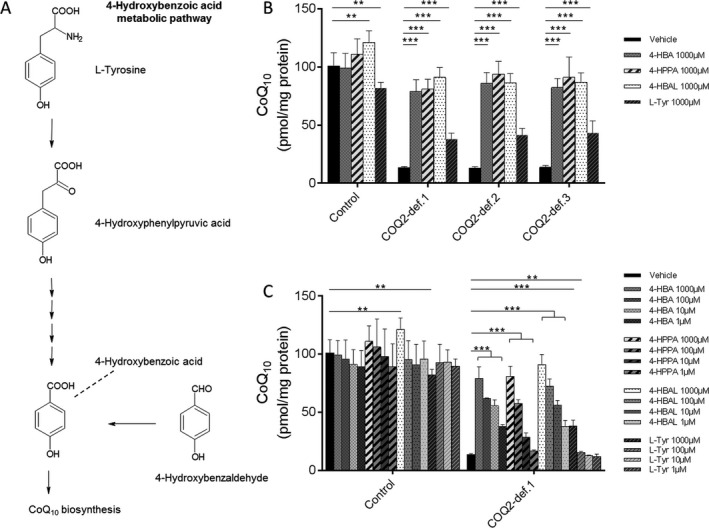
4‐hydroxybenzoic acid (4‐HBA) metabolic pathway and effects of 4‐HBA on CoQ10 biosynthesis in human COQ2 deficiency. (A) Schematic illustration of the 4‐hydroxybenzoic acid (4‐HBA) metabolic pathway in human cells. (B) Quantitative analysis of CoQ10 levels in control and COQ2 deficient fibroblast cell lines measured by UPLC‐MS/MS. For details regarding the cell lines COQ2‐def.1, COQ2‐def.2 and COQ2‐def.3 see Table S1. The different cell lines were treated with 4‐hydroxybenzoic acid (4‐HBA), 4‐hydroxyphenylpyruvic acid (4‐HPPA), 4‐hydroxybenzaldehyde (4‐HBAL), and L‐tyrosin (L‐Tyr), respectively. The effect of each concentration was expressed relative to values obtained with vehicle‐treated control cells, which was set at 100%, measured on the same day. (C) Quantitative analysis of CoQ10 levels in control and COQ2‐def.1 fibroblast using different concentrations of 4‐HBA, 4‐HPPA, 4‐HBAL and L‐Tyr. Statistics: ***P < 0.001) and **P < 0.01) relative to the indicated condition. Statistical significance was assessed using Student's t‐test. All experimental data was obtained in at least three independent experiments.
4‐hydroxybenzoic acid stimulates the expression of CoQ10 biosynthesis enzymes
As depicted in Figure 2A and B, COQ2 protein levels were normal in COQ2 patient cells compared to controls. This phenomenon has been described before and indicates that COQ2 patients express normal levels of a dysfunctional COQ2 protein.17 4‐HBA did not alter COQ2 levels in control or patient fibroblasts. Regarding COQ4 and COQ7, which are both crucial CoQ10 biosynthesis enzymes, we observed reduced protein levels in the COQ2‐deficient patient fibroblasts. These abnormalities significantly improved upon 4‐HBA supplementation.
Figure 2.
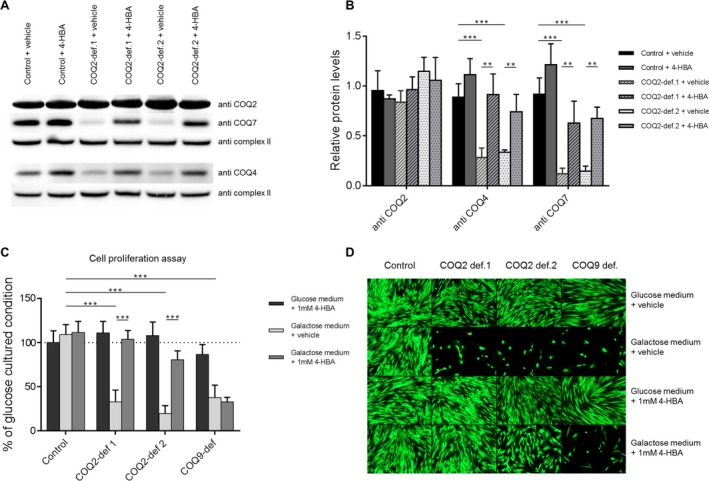
4‐HBA stimulates the expression of CoQ10 biosynthesis enzymes and rescues cell viability in COQ2‐deficient fibroblasts. (A) Representative immunoblot analysis in control as well as two COQ2‐deficient fibroblast cell lines. Protein levels of COQ4 and COQ7 are reduced in cell lines with COQ2 deficiency. Supplementation of 4‐hydroxybenzoic acid (4‐HBA) rescued this abnormality. Complex II (SDHA) was used as loading control. Results shown are representative for three independent experiments. (B) Quantitative data of immunoblot analyses confirming the results depicted in A). (C) Quantitative analysis of cell proliferation using the crystal violet assay. Results were expressed relative to values obtained with the vehicle‐treated condition cultured in glucose medium, which was set at 100% for each individual condition (dotted line). A COQ9‐deficient fibroblast cell line was used as an additional control. Results indicate a severe growth defect of all CoQ10‐deficient cell lines in galactose medium, which was specifically rescued by 4‐HBA treatment in COQ2‐deficient cell lines. (D) Live/Dead assay with similar conditions as depicted in C). Also in this assay, 4‐HBA specifically rescued cell viability of galactose‐cultured COQ2 cells. Statistics: ***P < 0.001 and **P < 0.01 relative to the indicated condition. Statistical significance was assessed using Student's t‐test. All experimental data was obtained in at least three independent experiments.
4‐hydroxybenzoic acid rescues cell viability of COQ2‐deficient fibroblasts
We previously demonstrated that culturing of CoQ10‐deficent fibroblasts in galactose medium impairs cell viability.11 In accordance, determination of cell proliferation using the crystal violet assay showed severely impaired cell growth of COQ2‐deficient cells during galactose culture stress conditions (Fig. 2C). Comparable findings were observed in fibroblasts with COQ9 deficiency. 4‐HBA treatment fully normalized cell proliferation in COQ2‐deficient cells but had no effects on COQ9‐deficient fibroblasts. In keeping with this observation, 4‐HBA rescued cell viability in COQ2‐deficient cells during galactose culture (Fig. 2D).
In silico analysis of human COQ2
By applying homology modeling using the structure of ApUbiA in its apo‐state13 and subsequent ligand docking, we obtained a model of human COQ2 in complex with the 4‐HBA ligand (Fig. 3A; sequence alignments see Figure S2). We verified our docking results with the structure, which was reported in the presence of the 4‐HBA ligand.13 The program AUTODOCK identified several putative 4‐HBA binding sites. One of these binding sites corresponded well with the binding site in the ApUbiA crystal structure.13 The crystalized ligands are shown in cyan, whereas the docked ligand is highlighted in white (Fig. 3A; left panel, magnified region). The amino acids interacting with the ligand as observed in the crystal structure are completely conserved in human COQ2 (highlighted in cyan, Fig. 3B, left panel and Fig. S1, right panel). Of note, Cheng & Li (2014) demonstrated for the ApUbiA protein that mutations of these interacting residues result in an almost complete loss of activity, indicating that this is the main catalytic site.13 Interestingly, we further obtained several additional putative 4‐HBA binding sites (Fig. 3A). It appears that these binding sites are located along a tunnel‐like structure (Fig. 3A, right panel). We then mapped the clinically‐described mutants of COQ2 onto our model. As shown in Figure 3B, the majority of these mutants are located near the putative tunnel where 4‐HBA passes through the protein.
Figure 3.
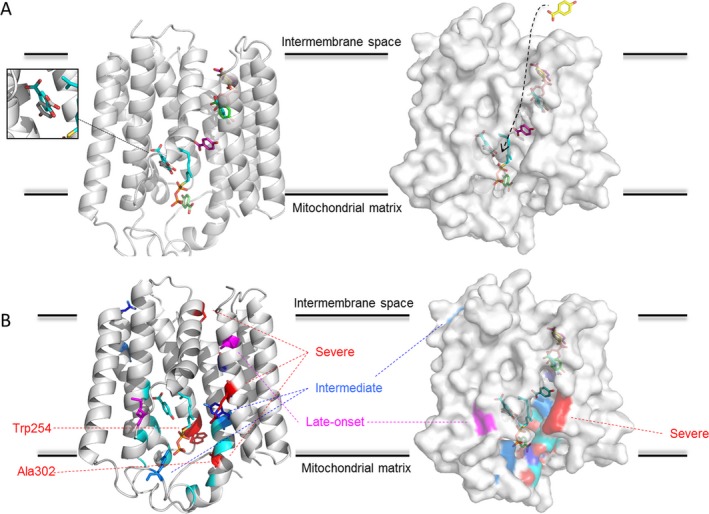
In silico analysis with homology modeling and ligand docking of the human COQ2 protein. (A) Structural model of human COQ2 based on the ApUbiA crystal structure (PDB entry: 4OD5). Left panel: cartoon representation with the 4‐HBA ligand bound. The binding site of 4‐HBA as identified in the ApUbiA crystal structure is highlighted in cyan. The magnified region demonstrates the co‐localization of the binding site in the crystal structure with one of the binding sites identified in this study. In addition, ligand docking revealed several additional putative binding sites, which are colored differently. Right panel: surface representation of the human COQ2 model. The identified ligand binding sites are shown in sticks. Ligand docking appears to follow a tunnel‐like structure, which directs 4‐HBA to the final activity site for conversion (the ligand transportation path is highlighted by an arrow). (B) Known mutations are docked onto the human COQ2 model. Left panel: the mutants published by Cheng & Li (2014) in ApUbiA, which almost completely abolish the enzymatic activity are shown in cyan.13 The patient mutations as described in Table S1 are shown as stick representation. Severity of the mutations in the clinical context is highlighted as follows: severe (Trp254Arg Ala302Val Ser146Asn and Arg387Xp) in red, intermediate (Gly390Ala Tyr297Cys and Arg197His) in blue and late‐onset (Met128Val and Val393Ala) in magenta. The mutations that were investigated in this study are indicated by dotted lines. Right panel: surface representation of the COQ2 model with the possible 4‐HBA binding sites. The mutations are color‐coded as described above. Please note that the mutations described so far are mainly clustering around the proposed tunnel‐like structure.
Discussion
Clinical response to CoQ10 substitution in CoQ10 deficiency disorders varies greatly among the different underlying genetic defects. As discussed above, especially in the severe forms, results of CoQ10 treatment are unsatisfactory. Therefore, the idea of metabolic bypass treatment using CoQ precursor compounds emerged.11, 18
Here, we identified 4‐HBA supplementation as an effective treatment for COQ2‐deficient fibroblasts. 4‐HBA is a water‐soluble small molecule, which is derived from L‐tyrosine (Fig. 1A). The finding that 4‐HBA rescues COQ2 deficiency initially appeared to be surprising since the concept of metabolic bypass strategies mainly implies that a substrate is provided, which enters the metabolic pathway distal of the enzymatic block.11 In contrast, 4‐HBA is a direct substrate of COQ2, which mediates its condensation with the polyisoprenoid side chain. This suggests that clinically‐relevant COQ2 mutations are associated with a residual enzyme activity that can be stimulated by increasing substrate availability. Interestingly, as shown in Figure 2A, COQ2 protein levels in several patient‐derived fibroblast lines were not reduced. This indicates that mutant COQ2 is still active but operates at a slow rate.
Another surprising finding of our study was the full restoration of CoQ10 levels in COQ2 patient fibroblasts with 4‐HBA treatment. Metabolic bypass strategies that we performed previously in the context of COQ7 and COQ9 deficiency yielded only moderate effects on CoQ10 biosynthesis.11 This treatment response reminded us of defects involving cofactor transport processes (e.g., thiamin or riboflavin metabolism disorders), which can be compensated by high‐dose supplementation of the substrate. Interestingly, COQ2 is a transmembrane protein of the inner mitochondrial membrane that is in contact with the mitochondrial intermembrane space as well as the mitochondrial matrix.19 However, no transport functions have been described for COQ2 so far. Importantly, in our experiments 4‐HBA seemed to be the rate‐limiting compound since stimulation of the isoprenoid pathway using mevalonic acid was ineffective.
Of note, although COQ2 was already described by Winrow et al. in 1969, its exact function is still not fully understood.20 In order to gain more insights into 4‐HBA metabolism via COQ2 we performed in silico analysis with homology modeling and ligand docking. As shown in Figure 3, our analysis revealed several interesting aspects: First of all, the putative catalytic site of COQ2, which binds 4‐HBA and mediates the condensation with the polyprenyl side chain is highly conserved. No patient‐related mutations directly affecting these residues were described so far suggesting that such defects may be incompatible with life. Moreover, we identified several additional putative 4‐HBA binding sites appearing to be located along a tunnel‐like structure (Fig. 3A). It is tempting to speculate that this structure constitutes a ligand transportation path for 4‐HBA through the COQ2 protein. This idea is further supported by the observation that clinically‐relevant COQ2 mutations cluster around this tunnel‐like structure, indicating that they hinder 4‐HBA transport. We suggest that increasing 4‐HBA concentrations might accelerate substrate flow through the COQ2 protein thereby reactivating CoQ10 biosynthesis. This reactivation is actually highlighted by our immunoblot analyses that revealed a drastic increase in CoQ10 biosynthesis protein expression levels in COQ2‐deficient cell lines upon 4‐HBA treatment.
In view of the above findings, further in vivo studies will be of major importance to establish a drug profile analysis for 4‐HBA and to evaluate its therapeutic potential under clinical conditions. Moreover, testing 4‐HBA in the context of different clinically relevant mutations will be helpful to investigate differences in drug response based on the underlying genetic defect.
Author Contributions
D. Herebian and A. Seibt contributed to the study design, performed experiments and analyzed data. S. Smits performed in silico analysis. R. Rodenburg provided the fibroblasts lines COQ2‐def.2 and COQ2‐def.3. E. Mayatepek provided intellectual input and laboratory resources. F. Distelmaier contributed to the study design, analyzed data and wrote the manuscript. All authors commented on the manuscript at all stages.
Conflict of Interest
The authors declare that they have no conflict of interest with the contents of this article.
Supporting information
Figure S1. It depicts a cartoon representation of the crystal structure of ApUbiA in the substrate bound state. The essential residues are shown, which most likely constitute the main catalytic site of the enzyme.
Figure S2. It depicts a sequence alignment of the ApUbiA protein with the human COQ2 sequence.
Table S1. It provides an overview of individuals with COQ2 defects published in the literature. The table illustrates the different clinical phenotypes associated with COQ2 deficiency and indicates the poor response of this disorder to oral CoQ10 supplementation.
Acknowledgments
This study was supported by a grant of the German Research Foundation/Deutsche Forschungsgemeinschaft (DI 1731/2‐1 to FD). The authors thank G. Bünning, Department of General Pediatrics, Neonatology and Pediatric Cardiology, University Children's Hospital Düsseldorf, for technical support. FD dedicates this work to Professor Ernst Otto Fischer, 1973 Nobel Prize laureate for chemistry.
Funding Statement
This work was funded by German Research Foundation/Deutsche Forschungsgemeinschaft grant DI 1731/2‐1.
References
- 1. Distelmaier F, Haack TB, Wortmann SB, et al. Treatable mitochondrial diseases: cofactor metabolism and beyond. Brain 2017;140(Pt 2):e11. [DOI] [PubMed] [Google Scholar]
- 2. Bentinger M, Tekle M, Dallner G. Coenzyme Q–biosynthesis and functions. Biochem Biophys Res Commun 2010;396:74–79. [DOI] [PubMed] [Google Scholar]
- 3. Luna‐Sánchez M, Hidalgo‐Gutiérrez A, Hildebrandt TM, et al. CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol Med 2017;9:78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Acosta MJ, Vazquez Fonseca L, Desbats MA, et al. Coenzyme Q biosynthesis in health and disease. Biochim Biophys Acta 2016;1857:1079–1085. [DOI] [PubMed] [Google Scholar]
- 5. Ashby MN, Kutsunai SY, Ackerman S, et al. COQ2 is a candidate for the structural gene encoding para hydroxybenzoate:polyprenyltransferase. J Biol Chem 1992;267:4128–4136. [PubMed] [Google Scholar]
- 6. Quinzii C, Naini A, Salviati L, et al. A mutation in para‐hydroxybenzoate‐polyprenyl transferase (COQ2) causes primary coenzyme Q10 deficiency. Am J Hum Genet 2006;78:345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kommuru TR, Gurley B, Khan MA, Reddy IK. Self‐emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm 2001;212:233–246. [DOI] [PubMed] [Google Scholar]
- 8. Beg S, Javed S, Kohli K. Bioavailability enhancement of coenzyme Q10: an extensive review of patents. Recent Pat Drug Deliv Formul 2010;4:245–255. [DOI] [PubMed] [Google Scholar]
- 9. Danhauser K, Herebian D, Haack TB, et al. Fatal neonatal encephalopathy and lactic acidosis caused by a homozygous loss‐of‐function variant in COQ9. Eur J Hum Genet 2016;24:450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jakobs BS, van den Heuvel LP, Smeets RJ, et al. A novel mutation in COQ2 leading to fatal infantile multisystem disease. J Neurol Sci 2013;326:24–28. [DOI] [PubMed] [Google Scholar]
- 11. Herebian D, Seibt A, Smits SHJ, et al. Detection of 6‐demethoxyubiquinone in CoQ10 deficiency disorders: insights into enzyme interactions and identification of potential therapeutics. Mol Genet Metab 2017;121:216–223. [DOI] [PubMed] [Google Scholar]
- 12. Sarenac T, Trapecar M, Gradisnik L, et al. Single‐cell analysis reveals IGF‐1 potentiation of inhibition of the TGF‐β/Smad pathway of fibrosis in human keratocytes in vitro. Sci Rep 2016;6:34373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng W, Li W. Structural insights into ubiquinone biosynthesis in membranes. Science 2014;343:878–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morris GM, Huey R, Lindstrom W, et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J Comput Chem 2009;16:2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. López‐Martín JM, Salviati L, Trevisson E, et al. Missense mutation of the COQ2 gene causes defects of bioenergetics and de novo pyrimidine synthesis. Hum Mol Genet 2007;16:1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Freyer C, Stranneheim H, Naess K, et al. Rescue of primary ubiquinone deficiency due to a novel COQ7 defect using 2,4‐dihydroxybensoic acid. J Med Genet 2015;52:779–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Desbats MA, Morbidoni V, Silic‐Benussi M, et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum Mol Genet 2016;25:4256–4265. [DOI] [PubMed] [Google Scholar]
- 20. Winrow MJ, Rudney H. The incorporation of p‐hydroxybenzoic acid and isopentenyl pyrophphate into ubiquinone precursors by cell‐free preparations of rat tissues. Biochem Biophys Res Commun 1969;37:833–840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. It depicts a cartoon representation of the crystal structure of ApUbiA in the substrate bound state. The essential residues are shown, which most likely constitute the main catalytic site of the enzyme.
Figure S2. It depicts a sequence alignment of the ApUbiA protein with the human COQ2 sequence.
Table S1. It provides an overview of individuals with COQ2 defects published in the literature. The table illustrates the different clinical phenotypes associated with COQ2 deficiency and indicates the poor response of this disorder to oral CoQ10 supplementation.