

Abstract
Objective
Delayed cerebral ischemia (DCI) is an independent risk factor for poor outcome after aneurysmal subarachnoid hemorrhage (SAH) and is multifactorial in etiology. While prior studies have suggested a role for matrix metalloproteinase‐9 (MMP‐9) in early brain injury after SAH, its contribution to the pathophysiology of DCI is unclear.
Methods
In the first experiment, wild‐type (WT) and MMP‐9−/− mice were subjected to sham or endovascular perforation SAH surgery. In separate experiments, WT and MMP‐9−/−mice were administered vehicle or minocycline either pre‐ or post‐SAH. All mice underwent assessment of multiple components of DCI including vasospasm, neurobehavioral function, and microvessel thrombosis. In another experiment, rabbits were subjected to sham or cisterna magna injection SAH surgery, and administered vehicle or minocycline followed by vasospasm assessment.
Results
MMP‐9 expression and activity was increased after SAH. Genetic (MMP‐9−/− mice) and pharmacological (pre‐SAH minocycline administration) inhibition of MMP‐9 resulted in decreased vasospasm and neurobehavioral deficits. A therapeutically feasible strategy of post‐SAH administration of minocycline resulted in attenuation of multiple components of DCI. Minocycline administration to MMP‐9−/− mice did not yield additional protection. Consistent with experiments in mice, both pre‐ and post‐SAH administration of minocycline attenuated SAH‐induced vasospasm in rabbits.
Interpretation
MMP‐9 is a key player in the pathogenesis of DCI. The consistent attenuation of multiple components of DCI with both pre‐ and post‐SAH administration of minocycline across different species and experimental models of SAH, combined with the excellent safety profile of minocycline in humans suggest that a clinical trial in SAH patients is warranted.
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) is a form of stroke associated with major morbidity and mortality. Multiple pathophysiological processes occurring after SAH contribute to the poor outcome seen in this patient population. These injurious processes can broadly be categorized into early brain injury (EBI) and delayed cerebral ischemia (DCI). EBI occurs in 12% of patients after SAH1 and is caused by a combination of transient global ischemia associated with abrupt increase in intracranial pressure plus the toxic effects of blood in the subarachnoid space. It is characterized by neuroinflammation, blood–brain barrier (BBB) disruption, cerebral edema, and neuronal cell death.2 DCI occurs in ~30–40% of patients after SAH, is multifactorial in etiology, and is characterized by delayed onset of ischemic neurological deficits and/or radiographic evidence of cerebral infarction. The best characterized contributor to DCI is cerebral vasospasm, wherein narrowing of large cerebral arteries occurs days after SAH leading to reduced cerebral blood flow, cerebral ischemia, and in many cases frank cerebral infarction.3 In recent years, however, several lines of evidence suggest additional contributors to DCI likely exist, including microcirculatory autoregulatory dysfunction, microvessel thrombosis, cortical spreading depression, and delayed neuronal cell death.4, 5 This more thorough understanding of the numerous pathophysiological processes contributing to secondary brain injury after SAH has led many to conclude that development of a truly efficacious SAH therapy will require identification of molecular target(s) that contribute to numerous injurious pathways including ideally both DCI and EBI.4, 5 It is therefore essential that experimental studies explore whether molecules(s) implicated in one injurious process (e.g., EBI) also causally contribute to other injurious processes (e.g., vasospasm‐induced DCI and/or nonvasospasm contributors to DCI).
MMP‐9 is a type IV collagenase that is involved in the cleavage of a variety of substrates on the cell membrane and extracellular matrix. It has been strongly implicated in the pathophysiology of BBB disruption and cerebral edema in several acute CNS injury paradigms including ischemic stroke,6 traumatic brain injury,7, 8 and more recently SAH.9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 However, the role of MMP‐9 in SAH‐induced cerebral vasospasm and DCI is poorly understood. Numerous studies have shown that MMP‐9 levels and/or activity are increased following SAH, both in patients and in animal models.9, 10, 17, 18, 20, 21, 22 Regarding its role in EBI, several investigators have found that pharmacologically inhibiting MMP‐9 via SB3CT and minocycline reduces EBI and improves neurological deficits after SAH,13, 15, 16, 19 although both these pharmacologic agents are known to inhibit other matrix metalloproteinases such as MMP‐2. MMP‐9 has been more directly implicated in EBI after SAH when studies found that mice having genetic deletion of MMP‐9 developed markedly less BBB disruption, cerebral edema, and neurological deficits after SAH.14
Regarding the potential role of MMP‐9 in DCI after SAH, several clinical studies have examined the correlation between MMP‐9 levels and radiographic vasospasm and/or clinical DCI, with conflicting results. Two laboratory studies have also examined the potential role of MMP‐9 in cerebral vasospasm. The first reported a decrease in hemolysate‐induced contractility of cultured smooth muscle cells (SMCs) with the MMP‐2/MMP‐9 inhibitor, SB‐3CT.12 The second reported an increase in MMP‐9 expression in the basilar artery of rats subjected to cisterna magna injection SAH and a decrease in basilar artery vasospasm with pre‐SAH intracisternal injection of MMP‐2/MMP‐9 inhibitor, SB‐3CT.23 Neither study utilized genetic approaches to directly interrogate the role of MMP‐9 (a critical point given the lack of an available specific MMP‐9 pharmacological inhibitor), and neither examined anti‐MMP‐9 agents when delivered at clinically relevant time points after SAH (an important point given the overall hope that these observations might ultimately lead to a novel therapy for SAH patients). Overall, the aforementioned preclinical and clinical investigations into the role of MMP‐9 in SAH‐induced cerebral vasospasm and DCI certainly suggest that MMP‐9 may play a key pathophysiological role; however, a definitive causal contribution for MMP‐9 in these injurious processes is yet to be firmly established.
Therefore, in this study, we critically examined the mechanistic role of MMP‐9 in the pathogenesis of cerebral vasospasm and DCI after SAH using transgenic mice having targeted genetic deletion of MMP‐9. We then evaluated the translational potential of MMP‐9‐directed therapy in two complementary animal models of SAH by administering the broad MMP inhibitor, minocycline (an FDA‐approved drug with a proven safety profile in humans including those with ischemic stroke) at clinically relevant time points (up to 2 h after SAH) in wild‐type mice and rabbits.24, 25, 26
Methods
Experimental animals
Twelve‐ to fourteen‐week‐old male wild‐type C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). MMP‐9−/− mouse breeders were obtained from Jackson Laboratories (Strain #007084) and a colony was established at our institution. Twelve‐ to fourteen‐week‐old male MMP‐9−/− mice on C57BL/6 background were used for experiments, with age‐ and gender‐matched wild‐type C57BL/6 mice serving as controls. All studies were approved by the Animal Studies Committee at Washington University School of Medicine.
Twelve‐week‐old male New Zealand white rabbits were purchased from the Animal Center of the Chinese Academy of Sciences (Shanghai, China). All studies involving rabbits were approved by the Animal Care and Use Committee at Nanjing University and performed at that institution.
Experimental SAH
Experimental SAH was induced in mice by the endovascular perforation technique as previously described.27, 28, 29 Briefly, mice were anesthetized with isoflurane (2% induction, 1.5% maintenance). A 5‐0 nylon suture was introduced into the external carotid artery (ECA) and advanced through the internal carotid artery (ICA) until the ICA bifurcation was encountered. The suture was then advanced 5 mm further to cause SAH. The suture was then removed and the ECA was ligated. Mice were then allowed to recover from anesthesia in an incubator and then returned to their cages. Mice that underwent sham surgery had all the steps performed except for suture advancement beyond the ICA bifurcation.
Experimental SAH was induced in rabbits according to the cisterna magna double‐hemorrhage method as previously described.30, 31 The rabbits were anesthetized with intramuscular injection of ketamine (25 mg/kg) and droperidol (1.0 mg/kg). A 23‐gauge butterfly needle was then inserted percutaneously into the cisterna magna. After withdrawal of 1.5 mL of cerebrospinal fluid, the same amount of nonheparinized fresh autologous auricular arterial blood was slowly injected into the cisterna magna over 1 minute. The animals were then kept in a 30° head‐down position for 30 minutes. After recovery from anesthesia, they were returned to their housing. Forty‐eight hours after the first SAH, a second injection was performed in the same manner as the first. In rabbits that underwent sham surgery, the same technique was applied with injection of sterile saline instead of blood.
Drug administration
Mice (45 mg/kg in PBS) and rabbits (3 mg/kg in PBS) were administered minocycline every 12 h, starting 2 h prior to SAH for the pre‐SAH treatment groups. For post‐SAH treatment groups, the same dose of minocycline was administered every 12 h starting 1 h after SAH. Vehicle‐treated animals were administered equal volumes of PBS. Drugs were administered intraperitoneally in mice and subcutaneously in rabbits. In pilot studies, minocycline administration to Sham‐operated mice and rabbits did not induce any changes in body weight, vessel diameter, and neurological function, and this group was therefore not included in subsequent experiments.
Neurobehavioral tests
Neurobehavioral outcome in mice was examined daily using Neuroscore and Rotarod tests, as previously described.27, 28, 29 Briefly, neurological function was graded based on a motor score (0–12) that evaluated spontaneous activity, symmetry of limb movements, climbing, balance and coordination, and a sensory score (4–12) that evaluated body proprioception and vibrissae, visual, and tactile responses. Balance and coordination were assessed by performance on rotarod (Rotamex‐5, Columbus Instruments, Columbus, OH). Mice were pretrained on the rotarod 1 day prior to surgery. Latency on three trials of 180 sec was averaged daily.
Vasospasm assessment
Vasospasm assessment in mice was performed on postsurgery day 3 via cerebrovascular casting, as previously described.27, 28, 29 Briefly, mice were anesthetized with isoflurane, and transcardially perfused with PBS, 4% formalin, and 3% gelatin‐India ink solution. Brains were removed, SAH was graded as previously described, and blood vessels imaged under a microscope using a CCD camera. The narrowest diameter within the first 1000 μm of the middle cerebral artery (MCA) outside the lateral sulcus was measured to assess vasospasm.
Vasospasm assessment in rabbits was performed by measurement of basilar artery lumen area as previously described.30, 31 Briefly, rabbits were anesthetized with an intramuscular injection of ketamine (40 mg/kg) and droperidol (2.5 mg/kg), endotracheally intubated, and mechanically ventilated. A thoracotomy was performed, and transcardiac perfusion was initiated with 500 ml of physiologic PBS (pH 7.4) at 37°C, followed by 500 mL of 10% buffered formaldehyde. A constant perfusion pressure of 120 cm H2O was maintained. After perfusion‐fixation, the whole brain with intact circle of Willis was removed and fixed further by immersion in 10% buffered formaldehyde for 24 h. The formalin‐fixed and paraffin‐embedded basilar artery sections (4 μm in thickness) were stained with hematoxylin and eosin. Cross‐sectional areas were then obtained from photomicrographs of three regions of the basilar artery (proximal, middle, and distal) by a blinded investigator using the High Definition Medical Image Analysis Program (HMIAP‐2000, developed by Tongji Medical University, Wuhan, China).
MMP‐9 zymography
MMP‐9 activity was determined by the gelatin zymography method as previously described.32 Briefly, frozen brain tissue samples were homogenized and centrifuged (10,000 g), and the resulting supernatant was used for analysis. The supernatant was incubated with Gelatin‐Sepharose 4B (Pharmacia Biotech, Uppsala, Sweden) for 1 h with constant shaking. After centrifugation, the Gelatin‐Sepharose pellet was rinsed with 500 μL of buffer containing 50 mm Tris‐HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl, 0.05% Brij‐35, and 0.02% NaN3. The solution was centrifuged again and the pellet was incubated for 30 min with 50 μL of elution buffer consisting of 50 mm Tris‐HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl, 0.05% Brij‐35, 0.02% NaN3, and 10% dimethylsulfoxide (DMSO). Each 25 μL of the eluted sample was mixed with 25 μL of 2X nonreducing sample buffer (0.125 mol/L Tris‐HCl, 20% glycerol, 4% SDS, 0.003% bromophenol blue, pH 6.8), loaded onto 7.5% SDS‐PAGE containing 0.1% gelatin, and electrophoresed at 100 V. Recombinant human MMP‐2 and MMP‐9 (0.1–0.5 ng; EMD Bioscience) was used as positive controls to identify the pro‐ and active forms of MMP‐2 and MMP‐9. The gels were then washed twice in 2.5% Triton X‐100 for 20 min each followed by incubation for 20 h at 37°C in a buffer containing 20 mmol/L Tris‐HCl pH 7.6, 10 mmol/L CaCl2, and 0.04% NaN3. After incubation, the gels were stained for 1 h with 0.1% Coomassie Blue (diluted with 40% methanol and 10% acetic acid) and destained until clear proteolytic bands appeared on a contrasting blue background.
Quantitative polymerase chain reaction (qPCR)
qPCR was performed as previously described.29 Briefly, messenger RNA (mRNA) was extracted from the ipsilateral cerebral cortex using TRIzol (ThermoFisher Scientific, Waltham, MA), followed by cDNA synthesis using High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems, Foster City, CA). The following oligonucleotide primer pairs were used for qPCR: MMP‐2 (Forward: 5′‐AACACCGAGGACTATGACCG‐3′, Reverse: 5′‐CCACAC CTTGCCATCGTT‐3′), MMP‐9 (Forward: 5′‐GATCCCCAGAGCGTCATTC‐3′, Reverse: 5′‐CCACCTTGTTCACCTCATTTTG‐3′), and GAPDH (Forward: 5′‐ CTTTGTCAAGCTCATTTCCTGG‐3′, Reverse: 5′‐TCTTGCTCAGTGTCCTTGC‐3′). The qPCR reactions were performed on a 7500 real‐time PCR System (Applied Biosystems, Foster City, CA) using SYBR Green PCR Master Mix reagents (Applied Biosystems, Foster City, CA). The expression of GAPDH served as an internal control. The delta‐delta calculation method was used to calculate fold change relative to sham controls.
Immunohistochemistry
Immunohistochemistry for assessment of microvessel thrombi was performed as previously described.27, 28, 29 Briefly, mice were transcardially perfused with heparinized PBS, whole brains were extracted and fixed by immersion in 4% paraformaldehyde for 48 h, followed by coronal slicing with 50 μmol/L thickness. Nine equally spaced coronal sections from the frontal lobe to posterior parietal lobe were then blocked in PBS containing 1% bovine serum albumin, 0.2% dry milk, and 0.1% Triton‐X100 for 1 h, followed by overnight incubation at 4°C with rabbit anti‐fibrinogen antibody (1:1000 dilution; Abcam, Cambridge, MA). The free‐floating sections were then incubated with biotinylated goat‐anti Rabbit antibody (Biorad, Hercules, CA) at room temperature for 2 h, followed by incubation with VECTASTAIN Elite ABC Kit solution (Vector Laboratories, Inc., Burlingame, CA), and 3,3′‐Diaminobenzidine tetrachloride (DAB) solution. Images of the brain sections were obtained using Hamamatsu NanoZoomer 2.0 HT system (Hamamatsu Photonics, Shizuoka, Japan).
Statistical analysis
Data represent individual animals and are expressed as mean ± SEM. After testing for normality, data from qPCR, vessel diameter and area measurements, MMP zymography, and microvessel thrombosis quantification were analyzed by Student's t‐test for experiments involving two groups and by one‐way ANOVA followed by Tukey's multiple comparisons post hoc test for experiments involving three or more groups. Neuroscore data were analyzed using repeated measures ANOVA and Newman–Keuls post hoc test for multiple comparisons. Nonparametric data with two groups were analyzed using Mann–Whitney U test. A P < 0.05 was considered as statistically significant.
Results
SAH increases MMP‐9 expression and activity
WT mice subjected to SAH demonstrated 2.9 ± 0.2‐fold increase in MMP‐9 mRNA at 24 h after SAH, and 3.2 ± 0.3‐fold increase in MMP‐9 mRNA at 72 h after SAH (P < 0.05 vs. Sham, Fig. 1A). On zymography, there was a 4.0 ± 1.4‐fold increase in MMP‐9 activity in the ipsilateral hemisphere of mice subjected to SAH compared to sham‐operated mice (P < 0.05, Fig. 1B).
Figure 1.
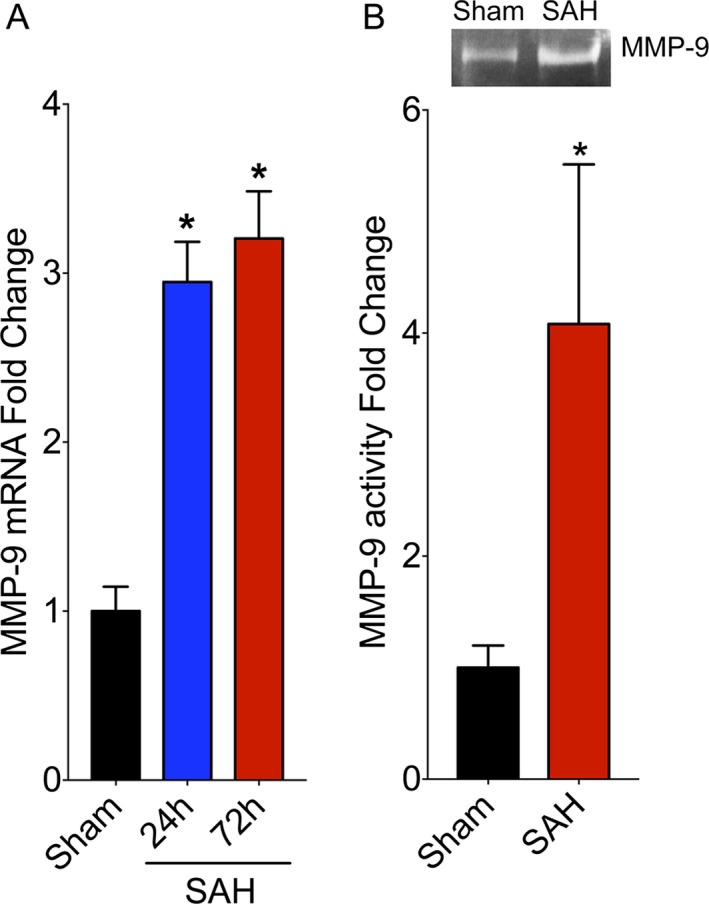
SAH induces an increase in MMP‐9 expression and activity. (A) Wild‐type mice were subjected to sham or endovascular perforation SAH surgery. MMP‐9 RNA levels were assessed by qPCR in the ipsilateral cortex at 24 and 72 h after SAH. Data represent mean ± SEM. *P < 0.05 vs. Sham, by one‐way ANOVA with Tukey's multiple comparisons test. N = 5 per group. (B) WT mice were subjected to sham or endovascular perforation SAH. MMP‐9 activity was assessed in the ipsilateral cortex at 72 h after SAH by gelatin zymography. Representative zymogram images are at the top. Data represent mean ± SEM. *P < 0.05 vs. Sham, by Mann–Whitney U test. N = 6 and 5 per group, respectively. WT, wild type.
Genetic inhibition of MMP‐9 attenuates SAH‐induced vasospasm and neurobehavioral deficits
WT mice subjected to SAH exhibited significant cerebral vasospasm on Day 3 after endovascular perforation SAH (23 ± 11%, P = 0.04) (Fig. 2A and B). In contrast, MMP‐9−/− mice did not develop significant vasospasm (2 ± 11%, P = 0.8 vs. Sham; P = 0.02 vs. MMP‐9+/+ SAH) (Fig. 2A and B). Similarly, WT mice subjected to SAH exhibited significant neurobehavioral deficits on post‐SAH days 1, 2, and 3 (P < 0.05) (Fig. 2C). In contrast, neurobehavioral deficits after SAH were milder on post‐SAH days 2 and 3 in MMP‐9−/− mice when compared to WT mice that underwent SAH (P < 0.05 vs. MMP‐9+/+:SAH). The degree of neurobehavioral deficits in these mice was not statistically significant on post‐SAH days 2 and 3 when compared to mice that underwent sham surgery (P > 0.05 vs. MMP‐9−/− Sham) (Fig. 2C).
Figure 2.
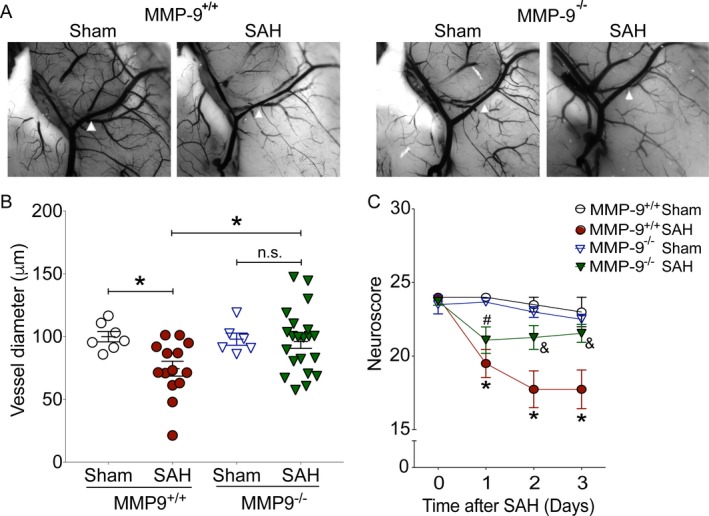
Genetic inhibition of MMP‐9 attenuates SAH‐induced neurovascular dysfunction. Wild‐type (MMP‐9+/+) and MMP‐9 knockout (MMP‐9−/−) mice were subjected to sham or endovascular perforation SAH surgery. Large artery vasospasm was assessed on post‐SAH Day 3 via measurement of left middle cerebral artery (MCA) diameter. (A) Representative images of left MCA ipsilateral to endovascular perforation (white arrowhead) (B) Left MCA vessel diameter measurements. Data represent mean ± SEM. *P < 0.05 and n.s. P> 0.05 by one‐way ANOVA with Tukey's multiple comparisons test. (C) Neurobehavioral testing was performed at baseline prior to SAH (Day 0) and daily for 3 days via Neuroscore test. Data represent mean ± SEM. *P < 0.05 vs. MMP‐9+/+ Sham, #P < 0.05 vs. MMP‐9−/− Sham, &P < 0.05 vs. MMP‐9+/+ SAH by repeated measures ANOVA with Newman–Keuls multiple comparisons test. N = 7, 14, 6, and 21 for MMP‐9+/+ Sham, MMP‐9+/+ SAH, MMP‐9−/− Sham, and MMP‐9−/− SAH, respectively.
Minocycline attenuates SAH‐induced increase in MMP‐9 activity
Vehicle‐treated WT mice that were subjected to SAH exhibited a 4.8 ± 1.0‐fold increase in MMP‐9 activity compared to mice that underwent sham surgery (P < 0.05) (Fig. 3A and B). The increase in MMP‐9 activity after SAH was attenuated (2.7 ± 0.6‐fold increase, P < 0.05 vs. SAH‐vehicle, P > 0.05 vs. Sham) in minocycline‐treated mice (Fig. 3A and B). MMP‐2 activity did not differ between the three groups (P > 0.05) (Fig. 3A). Taken together, these results suggest that the effect of minocycline in experimental SAH primarily involves MMP‐9 inhibition.
Figure 3.
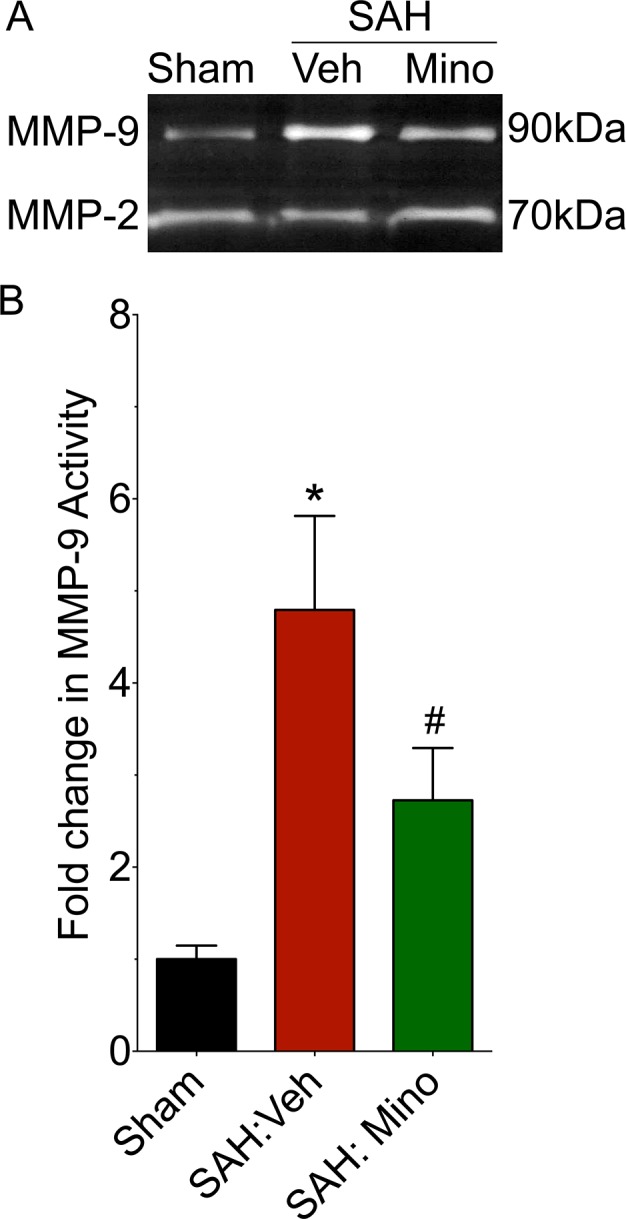
Minocycline attenuates SAH‐induced exacerbation in MMP‐9 activity. WT mice were subjected to sham surgery or endovascular perforation SAH. MMP‐9 activity was assessed in the ipsilateral cortex at 72 h after SAH by gelatin zymography. (A) Representative zymogram (B) Data represent mean ± SEM. *P < 0.05 vs. Sham #P < 0.05 vs. SAH:Veh,, by Mann–Whitney U test. N = 6 and 5 per group, respectively.
Pre‐SAH minocycline treatment attenuates SAH‐induced vasospasm and neurobehavioral deficits
Vehicle‐treated WT mice that were subjected to SAH exhibited significant cerebral vasospasm on post‐SAH Day 3 (P < 0.05 vs. Sham, Fig. 4A and B). In contrast, treatment with minocycline, starting 1 h prior to SAH, significantly attenuated SAH‐induced vasospasm (P > 0.05 vs. Sham, P < 0.05 vs. SAH, Fig. 4A and B) in WT mice. Similarly, vehicle‐treated WT mice that were subjected to SAH experienced marked neurobehavioral deficits (P < 0.05 vs. Sham on post‐SAH days 1, 2, and 3), whereas minocycline‐treated mice experienced significantly less neurobehavioral deficits (P < 0.05 vs. SAH:Vehicle on post‐SAH days 1, 2, 3; Fig. 4C).
Figure 4.
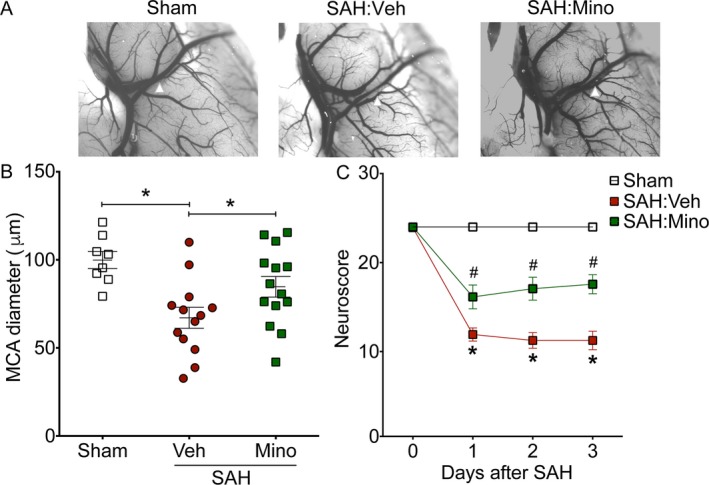
Pre‐SAH administration of minocycline attenuates SAH‐induced neurovascular dysfunction. WT mice treated with vehicle (Veh) or minocycline (Mino) were subjected to endovascular perforation SAH. Large artery vasospasm was assessed on post‐SAH Day 3 via measurement of left middle cerebral artery (MCA) diameter. (A) Representative images of left MCA ipsilateral to endovascular perforation (white arrowhead). (B) Left MCA vessel diameter measurements. Data represent mean±SEM. *P < 0.05 by one‐way ANOVA with Tukey's multiple comparisons test. (C) Neurobehavioral testing was performed at baseline prior to SAH (Day 0) and daily for 3 days via Neuroscore test. Data represent mean ± SEM. *P < 0.05 vs. Sham, and #P < 0.05 vs. SAH:Veh by repeated measures ANOVA with Newman–Keuls multiple comparisons test. N = 8, 13, and 14 for sham, SAH:Veh, and SAH:Mino, respectively.
MMP‐9 mediates the protective effect of minocycline against SAH‐induced vasospasm and neurobehavioral deficits
Consistent with our prior results, WT mice subjected to SAH exhibited significant cerebral vasospasm and neurobehavioral deficits after endovascular perforation SAH, while vehicle‐treated MMP‐9−/− mice did not develop significant vasospasm and had milder neurobehavioral deficits after SAH (Fig. 5A and B). Importantly, the degree of protection against vasospasm and neurobehavioral deficits in minocycline‐treated MMP‐9−/− mice was similar to vehicle‐treated MMP‐9−/− mice (MMP‐9−/−:SAH:Veh vs. MMP‐9−/−:SAH:Mino, P < 0.05 for vasospasm and neurobehavioral deficits, Fig. 5A and B). This lack of additional protection with minocycline treatment in MMP‐9−/− mice suggests that majority of the protective effect of minocycline is exerted through MMP‐9.
Figure 5.
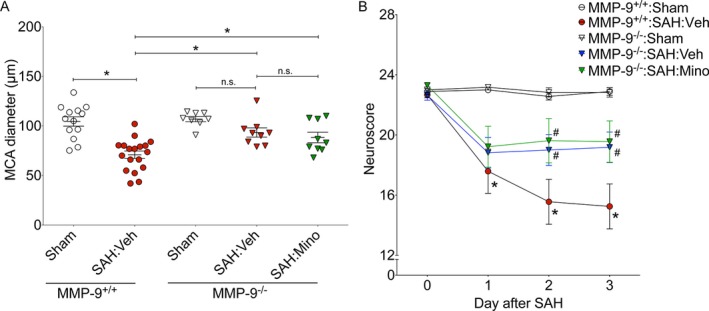
MMP‐9 mediates minocycline‐induced neurovascular protection in SAH. Wild‐type (MMP‐9+/+) and MMP‐9 knockout (MMP‐9−/−) mice were subjected to sham or endovascular perforation SAH surgery. Mice undergoing SAH were treated with vehicle (Veh) or minocycline (Mino) starting 2 h prior to SAH. Large artery vasospasm was assessed on post‐SAH Day 3 via measurement of left middle cerebral artery (MCA) diameter. (A) Left MCA vessel diameter measurements. Data represent mean±SEM. *P < 0.05 and n.s. P > 0.05 by one‐way ANOVA with Tukey's multiple comparisons test. (B) Neurobehavioral testing was performed at baseline prior to SAH (Day 0) and daily for 3 days via Neuroscore test. Data represent mean ± SEM. *P < 0.05 vs. MMP‐9+/+ Sham, and #P < 0.05 vs. MMP‐9+/+:SAH:Veh by repeated measures ANOVA with Newman–Keuls multiple comparisons test. N = 13, 18, 8, 9, and 9 for MMP‐9+/+:Sham, MMP‐9+/+:SAH:Veh, MMP‐9−/−:Sham, and MMP‐9−/−:SAH:Veh and MMP‐9−/−:SAH:Mino, respectively.
Post‐SAH minocycline treatment attenuates SAH‐induced vasospasm and neurobehavioral deficits in mice
To assess the utility of minocycline as a therapeutic agent, WT mice were administered vehicle or minocycline 2 h after sham or SAH surgery. Minocycline treatment afforded significant protection against SAH‐induced vasospasm (P < 0.05 vs. SAH:Vehicle, Fig. 6A) and neurobehavioral deficits (P < 0.05 vs. SAH:Vehicle on post‐SAH days 2 and 3, Fig. 6B). Taken together, these results suggest that treatment with minocycline is a promising therapeutic strategy against neurovascular dysfunction following SAH.
Figure 6.
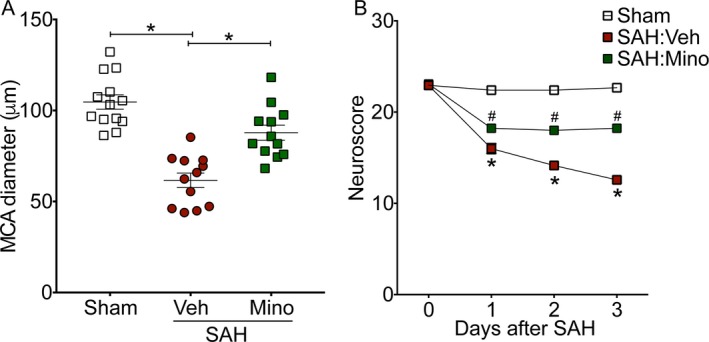
Post‐SAH administration of minocycline attenuates SAH‐induced neurovascular dysfunction. Wild‐type mice were subjected to sham or endovascular perforation SAH surgery. Mice undergoing SAH were treated with vehicle (Veh) or minocycline (Mino) starting 1 h after SAH. Large artery vasospasm was assessed on post‐SAH Day 3 via measurement of left middle cerebral artery (MCA) diameter. (A) Left MCA vessel diameter measurements. Data represent mean ± SEM. *P < 0.05 by one‐way ANOVA with Tukey's multiple comparisons test. (B) Neurobehavioral testing was performed at baseline prior to SAH (Day 0) and daily for 3 days via Neuroscore test. Data represent mean±SEM. *P < 0.05 vs. Sham, and #P < 0.05 vs. SAH:Veh by repeated measures ANOVA with Newman–Keuls multiple comparisons test. N = 13, 12, and 12 for Sham, SAH:Veh, and SAH:Mino, respectively.
Post‐SAH minocycline treatment attenuates SAH‐induced microvessel thrombosis
To assess the effect of minocycline on SAH‐induced microvessel thrombosis, WT mice that underwent SAH were administered vehicle or minocycline every 12 h, beginning 2 h after surgery. Minocycline‐treated mice developed significantly lesser microvessel thrombosis after SAH (P < 0.05 vs. SAH:Vehicle, Fig. 7). This result suggests that the protective effect of minocycline against SAH‐induced DCI is through its effect on multiple components.
Figure 7.
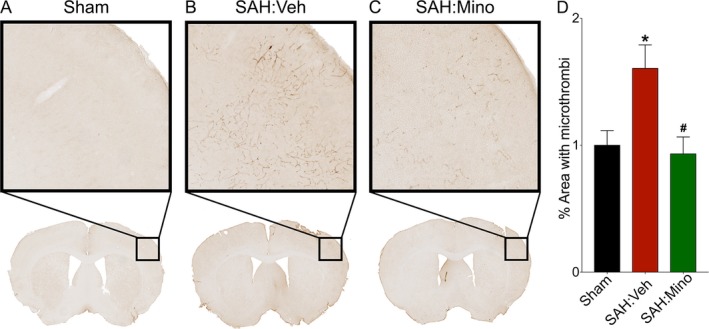
Post‐SAH administration of minocycline attenuates SAH‐induced microvessel thrombosis. Wild‐type mice were subjected to sham or endovascular perforation SAH surgery. Mice undergoing SAH were treated with vehicle (Veh) or minocycline (Mino) starting 1 h after SAH. Microvessel thrombosis was assessed by immunohistochemical staining for fibrinogen in brain sections on post‐SAH Day 3. (A) Representative images of brain sections. (B) Microvessel thrombosis was determined as percent coverage of ipsilateral cortex with thrombi. N = 6, 11, and 10 for Sham, SAH:Veh, and SAH:Mino, respectively. Data represent mean ± SEM. *P < 0.05 vs. Sham, #P < 0.05 vs. SAH:Veh, by one‐way ANOVA with Tukey's multiple comparisons test.
Minocycline treatment attenuates vasospasm in a rabbit cisterna magna injection model of SAH
To further assess the translational potential of minocycline, we examined its effect in a larger animal model – the rabbit cisterna magna injection model of SAH. Vehicle‐treated rabbits experienced significant SAH‐induced vasospasm as assessed by basilar artery (BA) area (P < 0.05 vs. Sham, Fig. 8B) and BA wall thickness (P < 0.05 vs. Sham, Fig. 8C). In contrast, rabbits that underwent pre‐ or post‐SAH treatment with minocycline demonstrated significantly lesser vasospasm (P < 0.05 vs. SAH:Vehicle, Fig. 8 B and C). These results reaffirm the therapeutic potential of minocycline.
Figure 8.
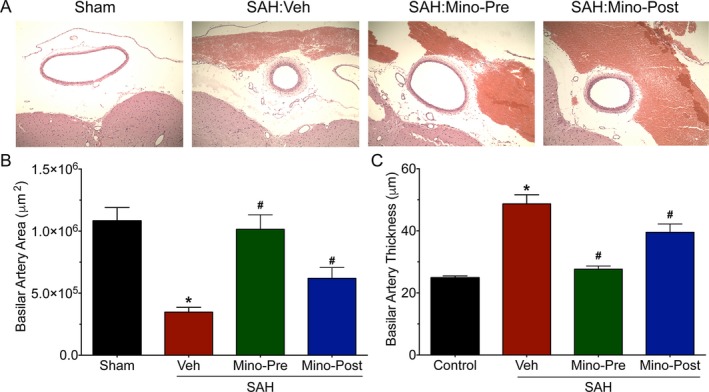
Pre‐ and Post‐SAH minocycline treatment attenuates vasospasm in rabbits. Rabbits were subjected to sham surgery or cisterna magna double‐hemorrhage SAH. The rabbits undergoing SAH were administered vehicle (Veh), minocycline 2 h prior to SAH (Mino‐Pre), and 1 h after SAH (Mino‐Post). Vasospasm was assessed by measurement of basilar artery cross‐sectional area and wall thickness 5 days after the second hemorrhage. (A) Representative cross‐sectional images of basilar artery. (B) Basilar artery area measurements. Data represent mean±SEM. *P < 0.05 vs. Sham, and # P < 0.05 vs. SAH:Veh by one‐way ANOVA with Tukey's multiple comparisons test. (B) Basilar artery wall thickness measurements. Data represent mean ± SEM. *P < 0.05 vs. Sham, and #P < 0.05 vs. SAH:Veh by one‐way ANOVA with Tukey's multiple comparisons test.
Discussion
Our study has several new and important findings. First, genetic deletion of MMP‐9 results in significantly less vasospasm and neurobehavioral deficits in a mouse model of SAH. Second, pre‐SAH administration of the MMP inhibitor, minocycline, attenuates cerebral vasospasm and neurobehavioral deficits in murine SAH – a protective effect that is lost in MMP‐9 knockout mice. Third, post‐SAH administration of minocycline attenuates SAH‐induced vasospasm and neurological dysfunction in murine SAH. Fourth, post‐SAH administration of minocycline affords protection against microvessel thrombosis in murine SAH and therefore protects against multiple components of DCI. Fifth, pre‐SAH and post‐SAH administration of minocycline affords protection against cerebral vasospasm in a larger, gyrencephalic animal model of SAH. Taken together, our results provide direct mechanistic evidence that MMP‐9 is a critical player in the development of cerebral vasospasm and neurobehavioral deficits after SAH, and that minocycline – an already FDA‐approved drug for the treatment of other diseases – carries significant therapeutic potential to prevent or reduce the incidence of DCI after SAH.
Two prior laboratory studies have evaluated the role of MMP‐9 in the pathophysiology of cerebral vasospasm after experimental SAH. Dang et al. examined cultured rat cerebrovascular smooth muscle cells (SMC) and observed increased MMP‐9 expression and SMC contraction after exposure to hemolysate.12 The hemolysate‐induced increase in MMP‐9 expression and SMC contraction was reduced after pretreatment with SB‐3CT, a non‐selective MMP‐2 and MMP‐9 inhibitor. In a separate publication, their group also reported increased MMP‐9 expression in the basilar artery of rats subjected to cisterna magna injection model of SAH, and showed that pre‐SAH administration of SB‐3CT into the cisterna magna, resulted in decreased vasospasm on Day 3 after SAH.23 In addition to these two preliminary laboratory studies, several groups have evaluated the potential role of MMP‐9 in DCI and/or radiographic vasospasm in SAH patients. Two studies reported higher MMP‐9 activity in the blood,22 CSF,22 and interstitial fluid18 of SAH patients who developed clinical DCI compared to those who did not; while another study did not find an association between blood MMP‐9 levels and clinical DCI.21 Four other studies examined the relationship between MMP‐9 levels and radiographic vasospasm in SAH patients. Of these, three reported an association between higher levels of blood MMP‐9 and cerebral vasospasm,9, 17, 33 while the other reported a nonsignificant trend toward higher CSF MMP‐9 levels and cerebral vasospasm. Taken together, these preclinical and clinical studies suggest, but do not definitively establish, a role for MMP‐9 in the pathophysiology of vasospasm and DCI after SAH. In contrast, our finding that SAH‐induced cerebral vasospasm and neurological deficits are attenuated in MMP‐9−/− mice provides the most direct evidence to date that MMP‐9 plays a causal role in the pathogenesis of SAH‐induced vasospasm and DCI.
While our study examined the role of MMP‐9 in DCI after SAH, two prior experimental studies have directly examined the contribution of MMP‐9 to EBI after SAH via MMP‐9 knockout mice. Feiler et al. reported decreased intracranial pressure and cerebral edema in MMP‐9−/− mice after endovascular perforation SAH.14 They also observed a decrease in neurobehavioral deficits on day 2, and a trend toward decreased neurobehavioral deficits on days 1 and 3 after endovascular perforation SAH in MMP‐9−/− mice. Egashira et al. reported decreased T2 hyperintensity and myelin breakdown in the white matter of MMP‐9−/− mice after SAH.13 Their study did not observe any difference in neurobehavioral deficits between WT and MMP‐9−/− on days 1 and 8 after endovascular perforation SAH. Our observation that MMP‐9−/− mice develop significantly less neurobehavioral deficits after SAH on post‐SAH days 2 and 3 (but not on post‐SAH day 1) is generally consistent with these prior studies. While Feiler et al. suggest that the attenuation of SAH‐induced neurobehavioral deficits in MMP‐9−/− mice is a manifestation of decreased cerebral edema and intracranial pressure seen in their study, our results suggest that the protection against delayed onset vasospasm noted in MMP‐9−/− mice is also a major contributor to the decreased neurobehavioral deficits seen in these mice after SAH.
Taken together, our finding that MMP‐9 directly contributes to the pathogenesis of vasospasm and neurobehavioral deficits after SAH, and prior studies demonstrating a key role for MMP‐9 in pathogenesis of EBI after SAH, suggest that MMP‐9 inhibition represents an attractive multifaceted therapeutic strategy to combat SAH‐induced neurovascular dysfunction. To examine this hypothesis, we utilized minocycline, an FDA‐approved semi‐synthetic tetracycline antibiotic that exhibits strong MMP‐9 inhibition. In a proof‐of‐concept experiment, we found that pre‐SAH treatment with minocycline markedly attenuates cerebral vasospasm and neurobehavioral deficits after SAH. To determine its translational potential, mice were administered minocycline 2 h after endovascular perforation SAH. Consistent with the initial proof‐of‐concept experiment, post‐SAH administration of minocycline significantly attenuated cerebral vasospasm and neurological deficits. Importantly, it also reduced SAH‐induced microvessel thrombosis, thus suggesting a protective effect on nonvasospasm components of DCI that are likely critical for successful translation of a therapeutic strategy for SAH. To further examine its translational potential, we utilized post‐SAH administration of minocycline in a rabbit cisterna magna injection model of SAH. Consistent with our findings in murine SAH, pre‐SAH as well as post‐SAH administration of minocycline significantly attenuated basilar artery vasospasm. These results, in a larger gyrencephalic animal utilizing a fundamentally different experimental technique for induction of SAH, provide important cross‐validation of the strong therapeutic potential of minocycline in combating vasospasm and DCI after SAH. When combined with prior studies demonstrating the attenuation of EBI16, 19, 34 and cognitive deficits19 after SAH with MMP‐9 inhibition, the results of our study suggest that a clinical trial to assess the therapeutic effect of minocycline in patients with SAH is warranted.
Conclusion
In this study, we provide definitive mechanistic evidence that MMP‐9 is a key player in the development of cerebral vasospasm and DCI after SAH. We also demonstrate consistent attenuation of SAH‐induced neurovascular deficits with both pre‐ and post‐SAH administration of minocycline across different species and experimental models of SAH, and against multiple components of DCI. Although additional preclinical testing would be useful, the excellent safety profile of minocycline in humans and promising results against both EBI and DCI in experimental SAH studies suggest that a clinical trial in patients with aneurysmal SAH is the next logical step.
Author Contributions
Conception and design of experiments: A.K.V., M‐L.Z., I.S., B.H.H., G.J.Z.; Acquisition of data: A.K.V., M‐L.Z., I.S., D.J.A., J.W.N., B.H.H.; Analysis and interpretation of data: A.K.V., M‐L.Z., I.S., D.J.A., J.W.N., U.A., B.H.H., G.J.Z.; Critical revisions of manuscript: A.K.V., G.J.Z.; Copyediting and approval of manuscript: all authors.
Conflict of Interest
None declared.
Acknowledgments
The authors thank Ernesto Gonzales for performing subarachnoid hemorrhage surgery.
This work was supported by the National Institutes of Health grants R01 NS091603 awarded to G.J.Z., R25 NS090978 to G.J.Z. and A.K.V., P30 NS057105 to Alafi Neuroimaging Laboratory at Washington University School of Medicine, American Heart Association Postdoctoral Fellowship Grant (11POST7650038) awarded to A.K.V., Neurosurgery Research and Education Foundation Research Fellowship Grant awarded to A.K.V., and Brain Aneurysm Foundation Research Grant to I.S. and B.H.H.
Funding Statement
This work was funded by National Institutes of Health grants R01 NS091603, R25 NS090978, and P30 NS057105; Alafi Neuroimaging Laboratory at Washington University School of Medicine, American Heart Association Postdoctoral Fellowship grant 11POST7650038; Neurosurgery Research and Education Foundation Research Fellowship grant ; Brain Aneurysm Foundation Research grant .
References
- 1. Claassen J, Carhuapoma JR, Kreiter KT, et al. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke 2002;33:1225–1232. [DOI] [PubMed] [Google Scholar]
- 2. Turan N, Miller BA, Heider RA, et al. Neurobehavioral testing in subarachnoid hemorrhage: A review of methods and current findings in rodents. J Cereb Blood Flow Metab 2016;1:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Francoeur CL, Mayer SA. Management of delayed cerebral ischemia after subarachnoid hemorrhage. Crit Care 2016;20:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet 2017;389:655–666. [DOI] [PubMed] [Google Scholar]
- 5. Terpolilli NA, Brem C, Buhler D, Plesnila N. Are we barking up the wrong vessels? cerebral microcirculation after subarachnoid hemorrhage Stroke 2015;46:3014–3019. [DOI] [PubMed] [Google Scholar]
- 6. Chaturvedi M, Kaczmarek L. Mmp‐9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol 2014;49:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang H, Adwanikar H, Werb Z, Noble‐Haeusslein LJ. Matrix metalloproteinases and neurotrauma: evolving roles in injury and reparative processes. Neuroscientist 2010;16:156–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hadass O, Tomlinson BN, Gooyit M, et al. Selective inhibition of matrix metalloproteinase‐9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS ONE 2013;8:e76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Akpinar A, Ucler N, Erdogan U, et al. Measuring serum matrix metalloproteinase‐9 levels in peripheral blood after subarachnoid hemorrhage to predict cerebral vasospasm. Springerplus 2016;5:1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chou SH, Feske SK, Simmons SL, et al. Elevated peripheral neutrophils and matrix metalloproteinase 9 as biomarkers of functional outcome following subarachnoid hemorrhage. Transl Stroke Res 2011;2:600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chou SH, Lee PS, Konigsberg RG, et al. Plasma‐type gelsolin is decreased in human blood and cerebrospinal fluid after subarachnoid hemorrhage. Stroke 2011;42:3624–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dang B, Shen H, Li H, et al. Matrix metalloproteinase 9 may be involved in contraction of vascular smooth muscle cells in an in vitro rat model of subarachnoid hemorrhage. Mol Med Rep 2016;14:4279–4284. [DOI] [PubMed] [Google Scholar]
- 13. Egashira Y, Zhao H, Hua Y, et al. White matter injury after subarachnoid hemorrhage: role of blood‐brain barrier disruption and matrix metalloproteinase‐9. Stroke 2015;46:2909–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feiler S, Plesnila N, Thal SC, et al. Contribution of matrix metalloproteinase‐9 to cerebral edema and functional outcome following experimental subarachnoid hemorrhage. Cerebrovasc Dis 2011;32(3):289–295. [DOI] [PubMed] [Google Scholar]
- 15. Guo Z, Sun X, He Z, et al. Role of matrix metalloproteinase‐9 in apoptosis of hippocampal neurons in rats during early brain injury after subarachnoid hemorrhage. Neuro Sci 2010;31:143–149. [DOI] [PubMed] [Google Scholar]
- 16. Guo ZD, Zhang XD, Wu HT, et al. Matrix metalloproteinase 9 inhibition reduces early brain injury in cortex after subarachnoid hemorrhage. Acta Neurochir Suppl 2011;110(Pt 1):81–84. [DOI] [PubMed] [Google Scholar]
- 17. McGirt MJ, Lynch JR, Blessing R, et al. Serum von Willebrand factor, matrix metalloproteinase‐9, and vascular endothelial growth factor levels predict the onset of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery 2002;51:1128–1134; discussion 34‐5. [DOI] [PubMed] [Google Scholar]
- 18. Sarrafzadeh A, Copin JC, Bengualid DJ, et al. Matrix metalloproteinase‐9 concentration in the cerebral extracellular fluid of patients during the acute phase of aneurysmal subarachnoid hemorrhage. Neurol Res 2012;34:455–461. [DOI] [PubMed] [Google Scholar]
- 19. Sherchan P, Lekic T, Suzuki H, et al. Minocycline improves functional outcomes, memory deficits, and histopathology after endovascular perforation‐induced subarachnoid hemorrhage in rats. J Neurotrauma 2011;28:2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horstmann S, Su Y, Koziol J, et al. MMP‐2 and MMP‐9 levels in peripheral blood after subarachnoid hemorrhage. J Neurol Sci 2006;251(1–2):82–86. [DOI] [PubMed] [Google Scholar]
- 21. Lago A, Tembl JI, Lopez‐Cuevas R, et al. Characterisation of DWI‐MRI confirmed cerebral infarcts in patients with subarachnoid haemorrhage and their association with MMP‐9 levels. Neurol Res 2015;37:688–692. [DOI] [PubMed] [Google Scholar]
- 22. Triglia T, Mezzapesa A, Martin JC, et al. Early matrix metalloproteinase‐9 concentration in the first 48 h after aneurysmal subarachnoid haemorrhage predicts delayed cerebral ischaemia: An observational study. Eur J Anaesthesiol 2016;33:662–669. [DOI] [PubMed] [Google Scholar]
- 23. Wang Z, Fang Q, Dang BQ, et al. Potential contribution of matrix metalloproteinase‐9 (mmp‐9) to cerebral vasospasm after experimental subarachnoid hemorrhage in rats. Ann Clin Lab Sci 2012;Winter 42:14–20. [PubMed] [Google Scholar]
- 24. Fagan SC, Waller JL, Nichols FT, et al. Minocycline to improve neurologic outcome in stroke (MINOS): a dose‐finding study. Stroke 2010;41:2283–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kohler E, Prentice DA, Bates TR, et al. Intravenous minocycline in acute stroke: a randomized, controlled pilot study and meta‐analysis. Stroke 2013;44:2493–2499. [DOI] [PubMed] [Google Scholar]
- 26. Switzer JA, Hess DC, Ergul A, et al. Matrix metalloproteinase‐9 in an exploratory trial of intravenous minocycline for acute ischemic stroke. Stroke 2011;42:2633–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Han BH, Vellimana AK, Zhou ML, et al. Phosphodiesterase 5 inhibition attenuates cerebral vasospasm and improves functional recovery after experimental subarachnoid hemorrhage. Neurosurgery 2012;70:178–186; discussion 86‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vellimana AK, Milner E, Azad TD, et al. Endothelial nitric oxide synthase mediates endogenous protection against subarachnoid hemorrhage‐induced cerebral vasospasm. Stroke 2011;42:776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Milner E, Johnson AW, Nelson JW, et al. HIF‐1alpha mediates isoflurane‐induced vascular protection in subarachnoid hemorrhage. Ann Clin Transl Neurol 2015;2:325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou ML, Shi JX, Hang CH, et al. Potential contribution of nuclear factor‐kappaB to cerebral vasospasm after experimental subarachnoid hemorrhage in rabbits. J Cereb Blood Flow Metab 2007;27:1583–1592. [DOI] [PubMed] [Google Scholar]
- 31. Zhou ML, Shi JX, Zhu JQ, et al. Comparison between one‐ and two‐hemorrhage models of cerebral vasospasm in rabbits. J Neurosci Methods 2007;159:318–324. [DOI] [PubMed] [Google Scholar]
- 32. Yin KJ, Cirrito JR, Yan P, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid‐beta peptide catabolism. J Neurosci 2006;26:10939–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer M, Dietmann A, Beer R, et al. Differential regulation of matrix‐metalloproteinases and their tissue inhibitors in patients with aneurysmal subarachnoid hemorrhage. PLoS ONE 2013;8:e59952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo Z, Sun X, He Z, et al. Matrix metalloproteinase‐9 potentiates early brain injury after subarachnoid hemorrhage. Neurol Res 2010;32:715–720. [DOI] [PubMed] [Google Scholar]