

Abstract
Background and Purpose
The advanced glycation end products (AGEs) participate in the pathogenesis of diabetic nephropathy (DN) by promoting renal inflammation and injury. L‐carnosine acts as a quencher of the AGE precursors reactive carbonyl species (RCS), but is rapidly inactivated by carnosinase. In this study, we evaluated the effect of FL‐926‐16, a carnosinase‐resistant and bioavailable carnosine derivative, on the onset and progression of DN in db/db mice.
Experimental Approach
Adult male db/db mice and coeval db/m controls were left untreated or treated with FL‐926‐16 (30 mg·kg−1 body weight) from weeks 6 to 20 (prevention protocol) or from weeks 20 to 34 (regression protocol).
Key Results
In the prevention protocol, FL‐926‐16 significantly attenuated increases in creatinine (−80%), albuminuria (−77%), proteinuria (−75%), mean glomerular area (−34%), fractional (−40%) and mean (−42%) mesangial area in db/db mice. This protective effect was associated with a reduction in glomerular matrix protein expression and cell apoptosis, circulating and tissue oxidative and carbonyl stress, and renal inflammatory markers, including the NLRP3 inflammasome. In the regression protocol, the progression of DN was completely blocked, although not reversed, by FL‐926‐16. In cultured mesangial cells, FL‐926‐16 prevented NLRP3 expression induced by RCS but not by the AGE Nε‐carboxymethyllysine.
Conclusion and Implications
FL‐926‐16 is effective at preventing the onset of DN and halting its progression in db/db mice by quenching RCS, thereby reducing the accumulation of their protein adducts and the consequent inflammatory response. In a future perspective, this novel compound may represent a promising AGE‐reducing approach for DN therapy.
Abbreviations
- A/C
albumin/creatinine ratio
- AGER
advanced glycosylation end‐product‐specific receptor
- AGEs
advanced glycation end products
- CML
Nε‐carboxymethyllysine
- CNDP
carnosine dipeptidase
- Ctrl
control
- Diab
diabetic
- fMA
fractional mesangial area
- HNE
4‐hydroxynonenal
- IHC
immunohistochemistry
- MCP‐1
monocyte chemoattractant protein‐1
- mGA
mean glomerular area
- mMA
mean mesangial area
- NLRP3
NOD‐like receptor family, pyrin domain containing 3
- Nox4
renal isoform of NAD(P)H oxidase
- P/C
protein/creatinine ratio
- PAS
periodic acid Schiff
- PCOs
total carbonylated proteins
- RCS
reactive carbonyl species
Introduction
Diabetic nephropathy is one of the most common complications of diabetes. As its prevalence has been increasing steeply along with an epidemic in type 2 diabetes (Tuttle et al., 2014), diabetic nephropathy has become the leading cause of end‐stage renal disease in western countries, where it accounts for approximately 50% of cases (Harjutsalo and Groop, 2014). The Diabetes Control and Complications Trial and the UK Prospective Diabetes Study have definitively shown that strict glycaemic control is effective at reducing but not eliminating the risk of microalbuminuria and overt diabetic nephropathy (Molitch et al., 2004). In addition, in patients with type 2 diabetes, other coexisting risk factors, such as obesity, dyslipidaemia and hypertension, can favour progression toward end‐stage renal disease (Molitch et al., 2004).
Several lines of evidence indicate that advanced glycation end products (AGEs), by exerting both direct (physico‐chemical) and indirect (receptor‐mediated) effects, have a major role in the development and progression of diabetic and non‐diabetic renal disease. Receptor‐independent effects include the modification of enzymatic activity, ligand binding, half‐life and immunogenicity, whereas AGE binding to the advanced glycosylation end‐product‐specific receptor (AGER) triggers redox‐sensitive signalling pathways leading to tissue injury via induction of apoptosis, inflammation and fibrosis (Bohlender et al., 2005). AGEs accumulate in sera and tissues of subjects suffering from diabetes and also of those with dyslipidaemia, arterial hypertension and other disease conditions (Ahmed, 2005). Accumulation of these by‐products is mainly due to increased formation of reactive carbonyl species (RCS) derived from oxidation and also from non‐oxidative metabolism of carbohydrates and lipids, of which there is an overabundance in patients with type 2 diabetes (Negre‐Salvayre et al., 2008). In turn, RCS, which include the α‐oxoaldehyde glyoxal and the aldehyde 4‐hydroxynonenal (HNE), react with aminoacid residues on proteins to generate stable adducts and cross links collectively termed as AGEs (Ahmed, 2005; Ellis, 2007; Negre‐Salvayre et al., 2008).
Carnosine (β‐alanyl‐L‐histidine) is an endogenous histidine‐containing dipeptide that acts as a quencher of RCS (Aldini et al., 2005), thus inhibiting AGE formation. L‐Carnosine has been shown to be effective in several disease models in which carbonyl stress is thought to play a central pathogenic role, such as ischaemic and/or toxic injury of the kidney (Kurata et al., 2006; Soliman et al., 2007), lung (Cuzzocrea et al., 2007) and brain (Rajanikant et al., 2007; Tang et al., 2007). In addition, L‐carnosine was shown to protect kidney cells from the toxic effect of high glucose (Köppel et al., 2011). Unfortunately, in humans, L‐carnosine has a short half‐life, due to its rapid inactivation by serum and tissue carnosinases [carnosine dipeptidases (CNDP)]. Recently, Ahluwalia et al. (2011) have shown that common variants in two CNDP genes (CNDP1 and CNDP2) play a role in susceptibility to kidney disease in patients with type 2 diabetes. What is more, an experimental study conducted in db/db mice revealed that renal CNDP1 activity is up‐regulated by post‐translational modifications induced by RCS and ROS. In turn, increased CNDP1 activity reduces the availability of histidine‐containing dipeptides in the diabetic kidney, thus establishing a positive feedback loop resulting in increasing renal carbonyl stress (Peters et al., 2015). Therefore, the search for carnosinase‐resistant carnosine derivatives represents a suitable strategy against carbonyl stress‐dependent disorders. In particular, the vascular and renal complications associated with diabetes may benefit from treatment with these compounds to prevent both long‐term glucose toxicity, resulting from insufficient glucose‐lowering therapy, and lipotoxicity. Accordingly, the carnosinase‐resistant D‐carnosine pro‐drug, D‐carnosine‐octylester (Vistoli et al., 2009), was shown to be effective in attenuating experimental atherosclerosis and renal disease induced by a western diet or streptozotocin‐induced diabetes in the ApoE‐null mice (Menini et al., 2012; 2015).
The aim of the present study was to investigate the effect of carnosinol, that is, (2S)‐2‐(3‐amino propanoylamino)‐3‐(1H‐imidazol‐5‐yl)propanol (FL‐926‐16), a newly synthesized L‐carnosine derivative, on the onset and progression of diabetic nephropathy. To this end, we used the db/db mouse, a well‐characterized model of type 2 diabetes and nephropathy, in which renal CNDP1 over‐activity has been recently recognized (Peters et al., 2015). An additional objective was to investigate the mechanism by which FL‐926‐16 exerts its effect by the use of primary cultures of mouse mesangial cells. The carnosine peptidomimetic FL‐926‐16 is selective and reactive as an RCS sequestering agent. In addition, it is characterized by a lack of toxicity and a favourable pharmacokinetic profile, related to its recognition by human peptide transporter 1 as well as resistance to carnosinase (Negrisoli et al., 2011) and, hence, is not affected by changes in CNDP1 activity (Peters et al., 2015).
Methods
Design
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
In vivo studies
Randomization and treatment
In this study, C57BLKS/JLepr male 6‐week‐old diabetic db/db and control db/m mice were used (see Results for body weights and blood glucose levels at baseline). The db/db mouse is one of the best characterized and most investigated model of diabetic nephropathy (Sharma et al., 2003). Mice were assigned to blocks based on genotype and, within each block, randomized to receive no treatment (Ctrl and Diab, respectively) or FL‐926‐16 at a dose of 30 mg·kg−1 ·day−1 in the drinking water (Ctrl‐FL and Diab‐FL, respectively). The dose of 30 mg·kg−1 ·day−1was chosen on the basis of results from preliminary in vivo and in vitro studies (Negrisoli et al., 2011; Anderson EJ et al, unpublished observations) indicating a good safety profile of the compound and a good oral bioavailability, giving a significant exposure in plasma after oral administration at the dose ranging from 10 to 45 mg·kg−1 ·day−1. In the prevention protocol, FL‐926‐16 therapy was started at week 6, that is, prior to the onset of diabetic nephropathy, and ended at week 20, whereas in the regression protocol, treatment was started at week 20, that is, after the onset of diabetic nephropathy, and ended at week 34.
Group sizes and ethical approval
The study was designed and started in 2014, that is, before power analysis for establishing group size in experimental research was a requirement. The group sizes were established on the basis of previous studies (Pugliese et al., 2000; Iacobini et al., 2004; 2009a; Menini et al., 2007; Solini et al., 2013) indicating that five animals per group provide sufficient power for detecting significant differences in functional (total proteinuria) and structural [fractional mesangial area (fMA)] parameters in experimental settings of diabetic nephropathy. However, in the prevention protocol, since other outcomes were evaluated in addition to renal function and structure in order to get mechanistic insights, the number of mice for each group was doubled (10 animals). The study protocol was approved by the ethical committee of the Catholic University of Rome, Italy (no. 355/2008). The animals were housed in single cages with wood‐derived bedding material in a specific pathogen‐free facility with a 12 h light/dark cycle and in controlled temperatures (20–22°C). Mice were cared for in accordance with the Principles of Laboratory Animal Care (National Institutes of Health publ. no. 85‐23, revised 1985) and with national laws and received water and food ad libitum. At the end of the study periods, mice were placed into metabolic cages overnight to collect urine. The next day, body weights were recorded, then mice were anaesthetised with ketamine (60 mg·kg−1 Imalgene i.p.) and xylazine (7.5 mg·kg−1 Rompum i.p.), as established by the veterinarian of the facility, and a blood sample was obtained by cardiac puncture. Finally, the animals were killed by cervical dislocation, a longitudinal incision of the abdominal wall was performed, and both kidneys were quickly removed and weighed. All the measurements indicated below were performed in the prevention protocol, whereas only metabolic parameters, renal function and structure, and markers of systemic oxidative and carbonyl stress were analysed in the regression protocol. No animal losses occurred, and all animals were used for the analysis of metabolic parameters, kidney weight and renal function and structure, whereas five animals per group, randomly selected, were used for immunohistochemistry (IHC) and quantitative RT‐PCR (qRT‐PCR). All the analyses were performed by investigators blinded to group assignment. To this end, biological samples were recoded by a technician (C.C., see Acknowledgements) at the time of collection.
In vitro studies
Mesangial cells were isolated from male C57BL6 mice as previously described (Pugliese et al., 2000). Cells between the 3rd and the 10th passage were cultured in DMEM supplemented with 10% FBS, 2 mmol·L−1 L‐glutamine, and antibiotics at 37°C in a 95% air/5% CO2‐humidified atmosphere. Cells were incubated for 24 h with saline, Nε‐carboxymethyllysine (CML, 100 μg·mL−1), its dialdehyde precursor, the RCS glyoxal (100 μM) or the lipoxidation‐derived RCS HNE (5 μM), in the presence or absence of FL‐926‐16 (20 mM) (Menini et al., 2012).
Monolayers were then collected, and total RNA was extracted to assess the gene expression of the gene for NOD‐like receptor family, pyrin domain containing 3 (NLRP3) inflammasome (Nlrp3), which was previously shown to participate in renal inflammation and injury in an animal model of metabolic syndrome (Solini et al., 2013).
All experiments were conducted five times in triplicate; technical replicates were used to ensure the reliability of single values.
Measurements
Metabolic parameters
Blood glucose levels were measured with the aid of the automated colorimetric instrument Glucocard™, whereas serum insulin levels were assessed by an elisa kit.
The Homeostasis Model of Assessment – Insulin Resistance index was then calculated from glucose and insulin levels.
Total cholesterol and triglycerides were assessed by enzymatic colorimetric methods.
Renal function
Serum and urine creatinine levels were measured by HPLC, whereas total proteinuria was assessed by the Bradford method and albuminuria by elisa.
Values of proteinuria and albuminuria were normalized by the urine creatinine concentration and expressed as protein/creatinine (P/C) and albumin/creatinine (A/C) ratios respectively (Iacobini et al., 2004).
Renal morphology/morphometry
For the analysis of renal structure (Iacobini et al., 2004; Menini et al., 2015), serial 4 μm kidney sections were stained with periodic acid Schiff (PAS). Then, the areas of at least 100 glomerular tuft profiles per sample were measured by means of the computer‐assisted image analysis system Optimas 6.5 and expressed as mean glomerular area (mGA), and PAS‐positive material in each glomerulus was quantified and expressed as percentage of the glomerular tuft area (fMA). Finally, the mean mesangial area (mMA) was calculated from the following formula: (fMA × mGA)/100.
Glomerular extracellular matrix and cell apoptosis
The mRNA expression of the genes for the extracellular matrix components fibronectin (Fn1) and collagen IV α1 chain (Col4a1) and the pro‐fibrotic cytokine TGF‐β (Tgfβ) were assessed by qRT‐PCR using a StepOne™ RT‐PCR instrument and TaqMan Gene Expression Assays, as previously reported (Menini et al., 2007).
Briefly, total RNA was extracted from mice renal cortex and mesangial cells with the RNAeasy mini kit. Then, 1 μg of RNA was reverse‐transcribed in a 20 μL reaction tube using a High Capacity cDNA Reverse Transcription Kit, and RT‐PCR was performed in triplicate on a StepOne™ RT‐PCR following the standard protocol. Transcripts for Fn1, Col4a1 and Tgfβ were quantified by TaqMan Gene Expression Assays using the assays reported in Supporting Information Table S1.
Amplifications were normalized to the β‐actin gene (Actb), and gene expression was quantified using the ΔΔCT calculation, where CT is the threshold cycle. The amount of the target gene was expressed as fold of control mean (db/m controls or untreated cells) to control for unwanted sources of variation. A standard curve for each gene was constructed using multiple calibrator dilutions. Standard curves were accepted only if the slope was approximately −3, with an r value greater than 0.98, and were used to estimate PCR efficiency. Results were analysed using SDS 2.1 software.
Protein content and distribution of extracellular matrix were assessed by IHC using rabbit polyclonal antibodies to human fibronectin and mouse collagen IV α1 chain (Iacobini et al., 2004; Menini et al., 2007). As for PAS positivity, the percentage of the glomerular area with positive staining was calculated by means of the image analysis system Optimas™6.5. A region of interest was drawn around the glomerulus, then the percentage of positive area for the specific stain (purple‐magenta for PAS and brown colour for IHC) was calculated at a fixed colour threshold, which was set by identifying three to five separate pixels in areas of positive staining. For each kidney specimen, the average of at least 60 glomeruli was used.
Protein levels of collagen IV α1 chain in homogenates of renal cortex were also assessed by Western blot using the same antibody utilized for IHC. As for mRNA expression levels, results are expressed as fold of control mean.
Glomerular cell death rate was assessed by IHC for active caspase‐3 using a rabbit polyclonal antibody. Positive cells were counted and expressed as % of total cells (Iacobini et al., 2004; Menini et al., 2007). A custom‐made, C‐language macro was written to count the number of cells within each glomerular tuft profile by means of the Optimas™6.5 image analysis system. At least 60 glomerular tuft profiles per sample were measured.
Serum and renal markers of oxidative and carbonyl stress and FL‐926‐16 levels
Serum levels of isoprostane 8‐epi‐PGF2α, an index of systemic oxidative stress, were determined by elisa using a commercial kit (Iacobini et al., 2009a,b). Total carbonylated proteins (PCOs) were measured by slot blot immunoassay using an anti‐dinitrophenylhydrazone antibody, after carbonyl derivatization with dinitrophenylhydrazine (Vistoli et al., 2009; Orioli et al., 2011); serum AGE levels by a competitive elisa technique (Menini et al., 2007; Iacobini et al., 2009a); serum pentosidine levels by HPLC (Odetti et al., 1992); and FL‐926‐16 concentrations in serum and urine by LC‐ESI‐MS/MS (MRM mode) (Anderson EJ et al., unpublished observations). The tissue content of CML, a glyoxal‐derived protein adduct, HNE histidine adduct (Michael adduct), AGER and the renal isoform of NAD(P)H oxidase (Nox4) were assessed by IHC (see above), using a biotinylated mouse monoclonal antibody against CML, a rabbit antiserum against the HNE protein adduct, a goat polyclonal antibody to AGER and a rabbit monoclonal antibody to Nox4 respectively (Iacobini et al., 2009a,b).
Renal inflammation
The mRNA expression of the genes for TNF‐α (Tnfa), monocyte chemoattractant protein‐1 (MCP‐1, Mcp1; also known as CCL2), the monocyte/macrophage cell marker Cell surface glycoprotein F4/80 or adhesion GPCR E1 (Adgre1) and of the NLRP3 inflammasome (Nlrp3) were assessed by qRT‐PCR (Iacobini et al., 2009a,b; Solini et al., 2013).
Protein content and distribution of F4/80, MCP‐1 and NLRP3 were assessed by IHC (see above), using a rat monoclonal antibody to mouse F4/80 and rabbit polyclonal antibodies to MCP‐1 and NLRP3 (Solini et al., 2013). For F4/80 and MCP‐1, positive staining was measured in 20 random fields of the renal cortex examined at a final magnification of 400× and expressed as the mean percentage of field area occupied by the specific stain.
Finally, protein levels of F4/80 in homogenates of renal cortex were also assessed by Western blot using the same antibody utilized for IHC.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results are expressed as mean ± SD and % change versus controls.
Statistical significance was evaluated by Student's t‐test or one‐way ANOVA followed by the Bonferroni test for multiple comparisons. For variables not normally distributed, the Kruskal–Wallis test was applied, followed by the Mann–Whitney U‐test for pairwise comparisons. The post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity.
A P‐value <0.05 was considered significant. All statistical tests were performed on raw data using the SPSS version 13.0 (SPSS Inc., Chicago, IL, USA).
Materials
The db/db, db/m and C57BL6 mice were purchased from Charles River (Calco, Italy). The study drug FL‐926‐16 was obtained from Flamma S.p.A. (Chignolo d'Isola, Italy).
DMEM, mouse serum albumin, glyoxylic acid, sodium cyanoborohydrate, glyoxal and antibodies to human fibronectin and NLRP3 (HPA012878) were purchased from Sigma (St. Louis, MO, USA); FBS, L‐glutamine and antibiotics from Flow Laboratories (Irvine, Scotland, UK); the elisa kit for insulin from Mercodia AB (Uppsala, Sweden); the enzymatic colorimetric kit for total cholesterol and triglycerides from Roche Diagnostics (Milan, Italy); the Bradford dye‐binding protein assay kit from Pierce (Rockford, IL, USA); the Mouse Albumin elisa Quantification Kit from Bethyl (Montgomery, TX, USA); the RNAeasy mini kit from Qiagen (Milan, Italy); HNE and the elisa kit for 8‐epi‐ PGF2α from Cayman (Ann Arbor, MI, USA); the High Capacity cDNA Reverse Transcription Kit and TaqMan Gene Expression Assays from Applied Biosystems (Monza, Italy); the antibodies to mouse collagen IV α1 chain (IHC and western blot), Nox4 (UOTR1B492), F4/80 (IHC and western blot) and MCP‐1 from Abcam (Cambridge, UK); the Anti‐ACTIVE® caspase‐3 antibody from Promega Italia (Milan, Italy); the antibody against CML from Wako (Neuss, Germany); the antibody against the HNE protein adduct from Alpha Diagnostic International (San Antonio, TX, USA); and the antibody to AGER (ab7764) from Novus Biologicals (Littleton, CO, USA).
As previously reported (Iacobini et al., 2011), CML was prepared by incubating 175 mg mouse serum albumin in 1 mL of 0.2 M phosphate buffer, pH 7.8, containing 0.15 M glyoxylic acid and 0.45 M sodium cyanoborohydrate for 24 h at 37°C.
The instrument for blood glucose measurement Glucocard™ SM was from A.Menarini Diagnostics (Florence, Italy); the image analysis system Optimas 6.5 was from Bioscan (Washington DC, USA); the StepOne™ RT‐PCR instrument was from Thermo Fisher Scientific (Monza, Italy); and the SDS 2.1 software was from Applied Biosystem.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
In vivo studies
Baseline body weights and blood glucose levels in Ctrl and Diab mice were 20.2 ± 1.4 and 23.3 ± 2.7 g and 6.57 ± 1.02 and 9.50 ± 2.67 mmol·L−1 respectively.
Prevention protocol
Metabolic parameters
Growth impairment and metabolic derangement were similar in Diab versus Ctrl mice and were not affected by FL‐926‐16 treatment (Table 1).
Table 1.
Metabolic parameters, kidney weight and renal function
| Ctrl | Ctrl‐FL‐926‐16 | Diab | Diab‐FL‐926‐16 | |
|---|---|---|---|---|
| Body weight, g | 30.2 ± 1.2 | 30.0 ± 2.1 | 46.2 ± 4.8* | 46.1 ± 3.9* |
| Glucose, mmol·L−1 | 7.85 ± 1.02 | 7.80 ± 1.40 | 25.18 ± 4.78* | 25.24 ± 5.11* |
| Insulin, pmol·L−1 | 77.6 ± 17.2 | 78.2 ± 9.5 | 287.6 ± 30.3* | 278.4 ± 31.2* |
| HOMA‐IR | 3.80 ± 0.77 | 3.78 ± 0.76 | 46.19 ± 9.59* | 41.60 ± 9.25* |
| Cholesterol, mmol·L−1 | 1.88 ± 0.43 | 1.86 ± 0.43 | 3.27 ± 0.30* | 3.23 ± 0.37* |
| Triglycerides, mmol·L−1 | 0.69 ± 0.13 | 0.70 ± 0.12 | 1.04 ± 0.11* | 1.01 ± 0.15* |
| 8‐epi‐PGF2α, pg·mL−1 | 65.0 ± 6.52 | 62.4 ± 12.5 | 146.8 ± 14.4* | 75.8 ± 10.8† |
| PCOs, pmol·mg·prot−1 | 1.50 ± 0.27 | 1.47 ± 0.24 | 2.90 ± 0.61* | 1.55 ± 0.16† |
| AGEs, U·mL−1 | 3.98 ± 1.14 | 3.78 ± 0.82 | 9.27 ± 1.23* | 5.31 ± 0.84† |
| Pentosidine, pg·mL−1 | 53.1 ± 2.8 | 51.8 ± 5.3 | 117.4 ± 10.4* | 71.4 ± 8.1† |
| P‐FL‐926‐16, μmol·L−1 | 0.00 ± 0.00 | 0.21 ± 0.07† | 0.00 ± 0.00 | 0.22 ± 0.04† |
| U‐FL‐926‐16, μmol·L−1 | 0.00 ± 0.00 | 37.09 ± 6.46† | 0.00 ± 0.00 | 16.83 ± 2.50*, † |
| Kidney weight, mg | 217.7 ± 35.9 | 213.8 ± 33.1 | 253.8 ± 18.5* | 241.5 ± 20.8 |
| Creatinine, μmol·L−1 | 30.5 ± 2.4 | 30.1 ± 1.7 | 51.3 ± 7.1* | 34.7 ± 2.3† |
| Urinary P/C, mg·g−1 | 1.86 ± 0.51 | 1.89 ± 0.25 | 35.68 ± 7.88* | 10.26 ± 1.93*, † |
| Urinary A/C, mg·g−1 | 1.28 ± 0.34 | 1.24 ± 0.25 | 25.72 ± 7.52* | 6.90 ± 1.68*, † |
Values of untreated and FL‐926‐16‐treated db/m and db/db mice at 20 weeks of age (prevention protocol; n = 10 per group).
Kidney weight refers to both kidneys. HOMA‐IR, Homeostasis Model of Assessment – Insulin Resistance; N/A, not assessed; Post hoc multiple comparison:
P < 0.05 versus Ctrl;
P < 0.05 versus Diab.
Renal function
Serum creatinine increased significantly in Diab mice, and these increases were significantly reduced in Diab‐FL animals; values from Diab‐FL mice did not differ from those of Ctrl mice. Likewise, A/C and P/C ratios increased significantly in Diab mice, and these increments were markedly attenuated in Diab‐FL animals, although values remained significantly higher than in Ctrl mice (Table 1).
Renal structure
Kidney weights increased significantly versus Ctrl mice in untreated but not in FL‐926‐16‐treated Diab animals (Table 1). Moreover, mGA, mMA and fMA were significantly higher in Diab than in Ctrl mice, and increases were significantly reduced, but not normalized, in Diab‐FL animals (Figure 1), indicating that treatment largely prevented glomerular lesions. Consistent with previous observations in db/db mice (Sharma et al., 2003), no expansion of the tubule‐interstitium was observed in Diab animals.
Figure 1.
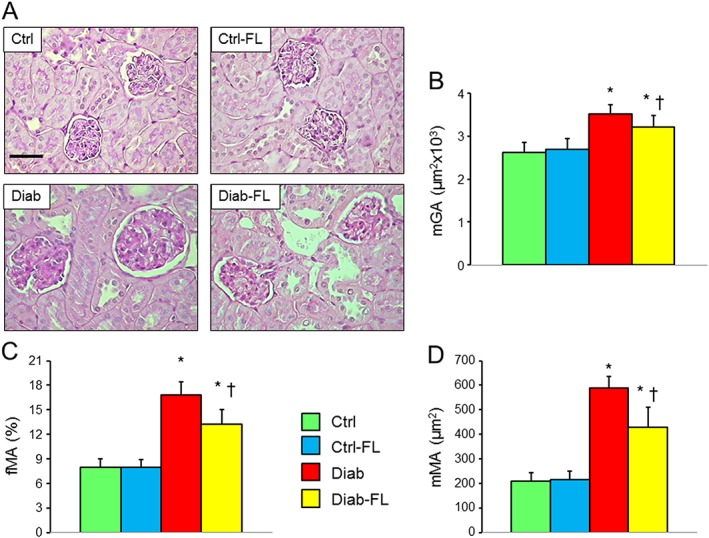
Prevention protocol: renal structure. PAS staining of kidney sections from representative animals (A) and quantification of mGA (B), fMA (C) and mMA (D) in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 10 per group). Scale bar = 50 μm. Post hoc multiple comparison: *P < 0.05 versus Ctrl; †P < 0.05 versus Diab.
Glomerular extracellular matrix and cell apoptosis
At IHC, the glomerular concentration of the extracellular matrix proteins fibronectin and collagen IV was significantly higher in Diab versus Ctrl mice and was significantly reduced, but not normalized, in Diab‐FL versus Diab mice (Figure 2A, B). Similar findings were observed for kidney cortex transcripts of Fn1, Col1a1 and Tgfb (Figure 2C–E) and for kidney cortex protein levels of collagen IV, as assessed by Western blot (Supporting Information Figure S1). The number of glomerular cells positive for active caspase‐3 was very low in Ctrl animals and increased significantly in Diab animals, with a predominant involvement of podocytes, although mesangial and endothelial cells were also affected. Once again, these increases in apoptosis were significantly attenuated, but not prevented, in Diab‐FL mice (Figure 2F).
Figure 2.
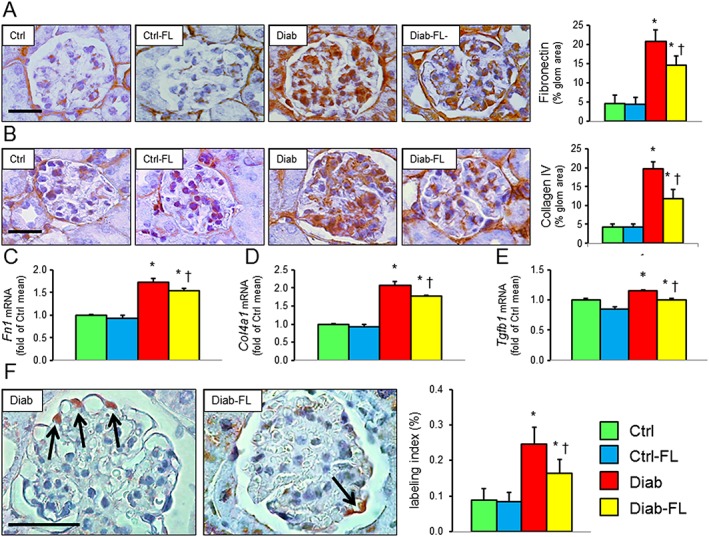
Prevention protocol: glomerular extracellular matrix and cell apoptosis. IHC of kidney sections from representative animals and quantification of fibronectin (A) and collagen IV α1 chain (B); kidney mRNA expression of FN1 (C), Col4a1 (D) and Tgfb (E); IHC of kidney sections (original quantification 1000×) from representative animals and quantification of active caspase‐3 (F, arrows indicate positive cells) in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). Scale bar = 50 μm. Post hoc multiple comparison: *P < 0.05 versus Ctrl; †P < 0.05 versus Diab.
Systemic and renal oxidative and carbonyl stress and FL‐926‐16 levels
Circulating levels of isoprostane 8‐epi‐PGF2α, PCOs and AGEs were increased in Diab versus Ctrl mice and reduced by FL‐926‐16 treatment to values that did not differ significantly from those of Ctrl animals. A similar trend was observed for pentosidine (Table 1). Glomerular staining for CML and HNE adducts was slightly positive in Ctrl mice and increased significantly in Diab mice. Increases were significantly attenuated in Diab‐FL animals, which showed values that did not differ from those of Ctrl mice for CML and HNE adducts (Figure 3A, B). Also immunostaining for AGER and Nox4, which was minimal in glomeruli from Ctrl animals, increased significantly in Diab, and these increases were markedly reduced in Diab‐FL mice (Figure 3C, D). FL‐926‐16 was detected only in the plasma and urine from treated animals, with lower urinary levels in Diab versus Ctrl mice, probably due to the reduced renal function and/or increased FL‐926‐16‐RCS formation (Table 1).
Figure 3.
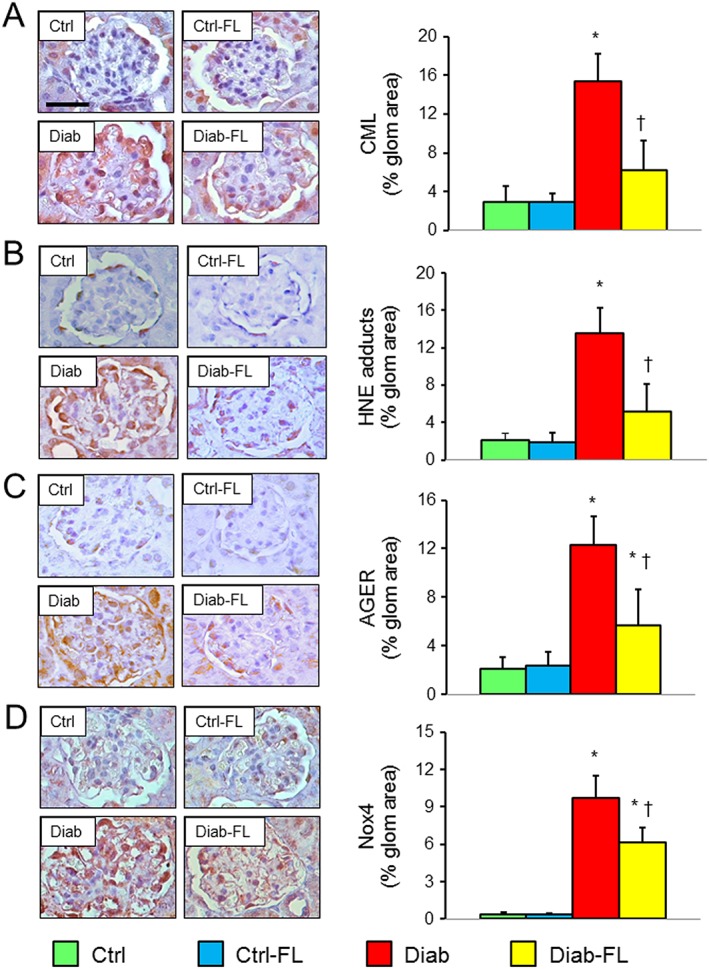
Prevention protocol: renal oxidative and carbonyl stress. IHC of kidney sections from representative animals and quantification of CML (A), HNE (B), AGER (C) and Nox4 (D) in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). Scale bar = 50 μm. Post hoc multiple comparison: *P < 0.05 versus Ctrl; †P < 0.05 versus Diab.
Renal inflammation
At IHC, the renal concentrations of the inflammatory markers F4/80 and MCP‐1 were markedly increased in Diab mice and these increases were significantly attenuated in Diab‐FL animals, which showed a level of positivity similar to that of Ctrl mice (Figure 4A, B). Similar findings were observed for protein levels of F4/80 in renal cortex homogenates, as evaluated by Western blot analysis (Supporting Information Figure S1). Likewise, tubular staining for NLRP3, which was insignificant in both Ctrl and Ctrl‐FL mice, increased significantly in Diab animals and also showed a strong positivity of glomerular cells but not in Diab‐FL mice (Figure 5A). Renal cortex mRNA levels of Adgre1, Mcp1, Tnfa (Figure 4C–E) and Nlrp3 (Figure 5B) were significantly increased in Diab mice, and these changes were significantly attenuated by FL‐926‐16 treatment, although Diab‐FL animals still exhibited higher values than Ctrl mice.
Figure 4.
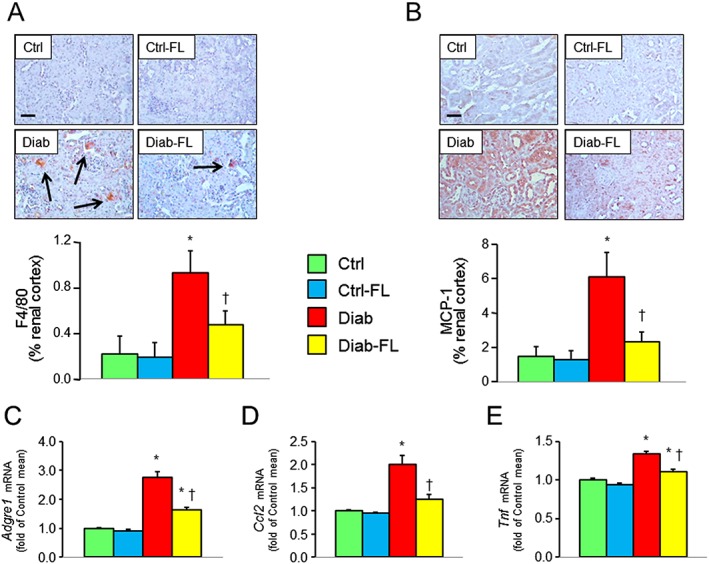
Prevention protocol: renal inflammation. IHC of kidney sections from representative animals and quantification of F4/80 (A) and MCP‐1 (B) and kidney mRNA expression of Adgre1 (C), Mcp1 (D) and Tnfa (E) in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). Scale bar = 50 μm. Post hoc multiple comparison: *P < 0.05 versus Ctrl, †P < 0.05 versus Diab.
Figure 5.
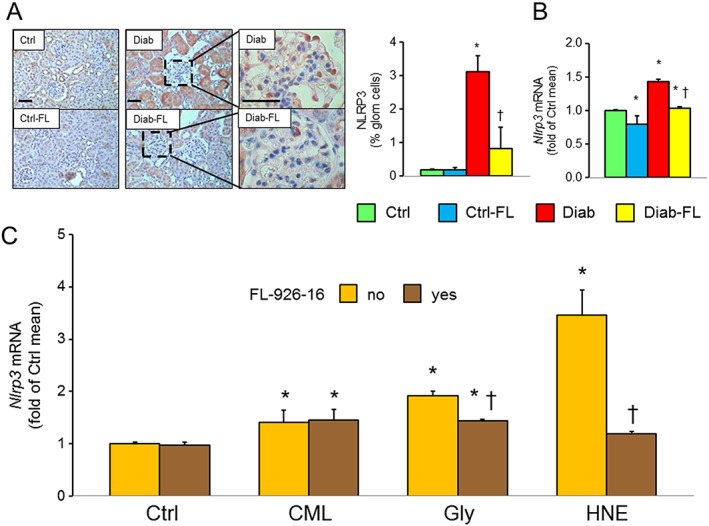
In vivo and in vitro studies: NLRP3 expression. IHC of kidney sections from representative animals and quantification of NLRP3 (A) and kidney mRNA expression of Nlrp3 (B) in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). Scale bar = 50 μm. Post hoc multiple comparison: *P < 0.05 versus Ctrl; †P < 0.001 versus Diab. Expression levels of Nlrp3 in mouse mesangial cells incubated with vehicle (Ctrl), 100 μg·mL−1 CML, 100 μM glyoxal (Gly) or 5 μM HNE, without and with FL‐926‐16 20 mM (C) (mean ± SD; n = 5 experiments in triplicate). Pairwise comparison: *P < 0.05 versus Ctrl; †P < 0.05 versus untreated.
Regression protocol
Metabolic parameters
As in the prevention protocol, FL‐926‐16 treatment did not affect the growth impairments and metabolic derangement in Diab mice (Table 2).
Table 2.
Metabolic parameters, kidney weight and renal function
| Ctrl | Ctrl‐FL‐926‐16 | Diab | Diab‐FL‐926‐16 | |
|---|---|---|---|---|
| Body weight, g | 34.5 ± 3.0 | 33.8 ± 1.7 | 45.5 ± 2.0* | 45.6 ± 7.6* |
| Glucose, mmol·L−1 | 9.18 ± 0.43 | 9.11 ± 0.63 | 29.74 ± 2.43* | 28.95 ± 2.38* |
| Insulin, pmol·L−1 | 72.6 ± 8.1 | 73.0 ± 10.5 | 254.4 ± 18.8* | 251.3 ± 15.6* |
| HOMA‐IR | 4.27 ± 0.51 | 4.23 ± 0.49 | 48.20 ± 1.81* | 46.39 ± 1.91* |
| Cholesterol, mmol·L−1 | 1.98 ± 0.24 | 1.98 ± 0.15 | 3.80 ± 0.41* | 3.81 ± 0.40* |
| Triglycerides, mmol·L−1 | 0.76 ± 0.10 | 0.77 ± 0.07 | 1.09 ± 0.10* | 1.06 ± 0.10* |
| 8‐epi‐PGF2α, pg·mL−1 | 71.0 ± 4.7 | 70.4 ± 2.4 | 165.6 ± 8.8* | 77.6 ± 13.3† |
| PCOs, pmol·mg·prot−1 | 1.57 ± 0.06 | 1.56 ± 0.13 | 3.12 ± 0.34* | 1.94 ± 0.16† |
| AGEs, U·mL−1 | 5.37 ± 0.64 | 4.94 ± 0.83 | 10.57 ± 1.30* | 6.67 ± 1.09† |
| Pentosidine, pg·mL−1 | N/A | N/A | N/A | N/A |
| P‐FL‐926‐16, μmol·L−1 | N/A | N/A | N/A | N/A |
| U‐FL‐926‐16, μmol·L1 | N/A | N/A | N/A | N/A |
| Kidney weight, mg | 234.0 ± 19.5 | 232.0 ± 14.8 | 278.0 ± 14.8* | 256.0 ± 11.4 |
| Creatinine, μmol·L−1 | 31.5 ± 1.3 | 31.3 ± 1.6 | 62.1 ± 6.4* | 50.7 ± 5.9*, † |
| Urinary P/C, mg·g−1 | 2.03 ± 0.32 | 2.08 ± 0.09 | 44.65 ± 7.89* | 28.04 ± 4.84*, † |
| Urinary A/C, mg·g−1 | 1.42 ± 0.23 | 1.43 ± 0.22 | 33.48 ± 4.96* | 19.65 ± 2.63*, † |
Values of untreated and FL‐926‐16‐treated db/m and db/db mice at 34 weeks of age (regression protocol; n = 5 per group).
Kidney weight refers to both kidneys. HOMA‐IR, Homeostasis Model of Assessment – Insulin Resistance; N/A, not assessed; Post hoc multiple comparison:
P < 0.05 versus Ctrl;
P < 0.05 versus Diab.
Renal function
In the regression protocol, the increases in serum creatinine as well as in A/C and P/C ratios in Diab mice were also significantly reduced by FL‐926‐16 treatment, although values remained significantly higher than in Ctrl mice. Interestingly, as compared with 20‐week‐old Diab mice, levels were higher in 34‐week‐old Diab (P < 0.05), but not Diab‐FL mice, indicating that the progression of renal dysfunction was blocked by initiation of FL‐926‐16 treatment (Table 2).
Renal structure
Kidney weights increased significantly versus Ctrl mice in untreated but not in FL‐926‐16‐treated Diab animals also in the regression protocol (Table 2). The increases in mGA, mMA and fMA (Figure 6) observed in Diab animals were significantly reduced but not normalized in Diab‐FL animals. When compared with those of 20‐week‐old untreated Diab animals, changes in the structural parameters were significantly higher in untreated Diab mice but not in FL‐926‐16‐treated at 34 weeks of age, thus indicating that progression was halted by FL‐926‐16 treatment, although it did not cause significant regression (Figure 7).
Figure 6.
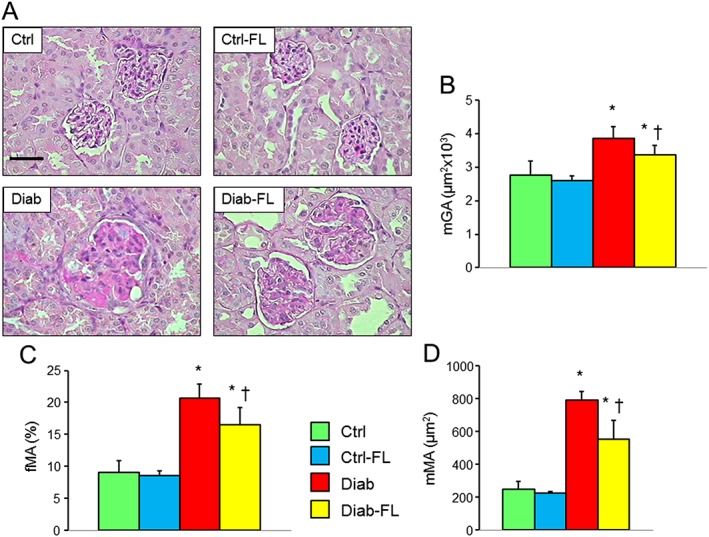
Regression protocol: renal structure. PAS staining of kidney sections from representative animals (A) and quantification of mGA (B), fMA (C) and mMA (D) in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). Scale bar = 50 μm. Post hoc multiple comparison: *P < 0.05 versus Ctrl, †P < 0.05 versus Diab.
Figure 7.
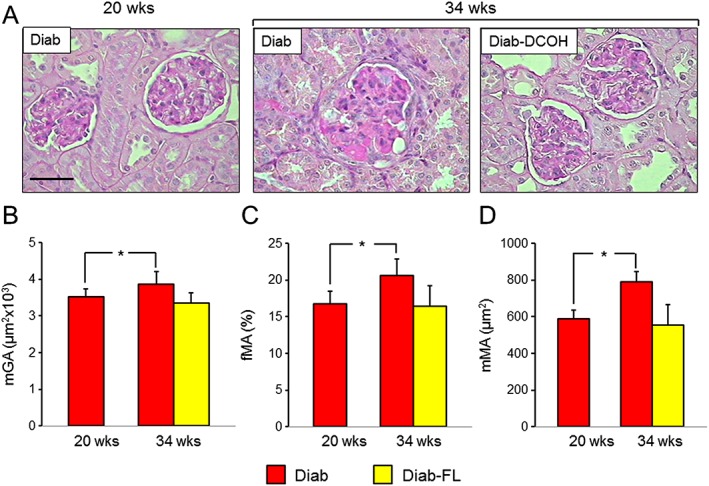
Prevention and regression protocols: renal structure. PAS staining of kidney sections from representative animals (A) and quantification of mGA (B), fMA (C) and mMA (D) in untreated and FL‐926‐16 (FL)‐treated db/db diabetic mice (Diab and Diab‐FL) of 34 weeks of age versus untreated Diab mice of 20 weeks of age (mean ± SD; n = 10 for 20‐week‐old mice and 5 for 34‐week‐old mice). Scale bar = 50 μm. Pairwise comparison: *P < 0.05 versus Diab‐20 weeks.
Systemic oxidative and carbonyl stress
Also in the regression protocol, the increased levels of isoprostane 8‐epi‐PGF2α, PCOs and AGEs observed in Diab mice were reduced by FL‐926‐16 treatment to values similar to those of Ctrl animals (Table 2).
In vitro studies
The RCSs glyoxal and HNE and the glyoxal‐derived protein adduct CML induced the mRNA expression of Nlrp3 in mesangial cells. FL‐926‐16 completely prevented HNE‐induced Nlrp3 up‐regulation and significantly attenuated glyoxal‐induced Nlrp3 up‐regulation but was not able to inhibit Nlrp3 expression induced by CML (Figure 5C).
Discussion
In view of the key role of AGE accumulation in the pathogenesis of diabetic nephropathy (Wendt et al., 2003; Cooper, 2004), therapeutic strategies aimed at reducing AGE‐induced tissue injury have been proposed and tested in experimental animals (Bakris et al., 2004; Reddy and Beyaz, 2006). Some of these agents have also been investigated in humans with inconclusive results, due to safety concerns and inconsistent efficacy, possibly due to the advanced degree of disease progression in patients enrolled in these studies (Cooper, 2004). The AGE‐reducing strategies inhibit the formation of AGEs by quenching their precursors, RCS, with carbonyl scavengers, which include histidine‐containing dipeptides (Ellis, 2007).
In the present study, we evaluated a novel carnosine peptidomimetic, FL‐926‐16, in the prevention/attenuation of the onset of diabetic nephropathy and the halting/regression of established diabetic nephropathy in db/db mice, a model of type 2 diabetes. These mice carry a G‐to‐T point mutation of the leptin receptor that causes polyphagia, obesity and insulin resistance followed by frank hyperglycaemia (by 4–6 weeks of age) and renal features resembling those of human diabetic nephropathy (Sharma et al., 2003). The main finding of our study was that treatment with FL‐926‐16 significantly reduced the changes in renal function and structure occurring in db/db mice. Importantly, FL‐926‐16 treatment was effective both in attenuating the onset of diabetic nephropathy and in stopping its progression. This was demonstrated by the markedly reduced increment in serum creatinine, albuminuria, total proteinuria, glomerular hypertrophy and mesangial expansion, as compared with age‐matched, untreated db/db mice. Of note, these differences in the severity of renal involvement between the two diabetic groups occurred despite similar degrees of metabolic derangement. This observation is at variance with previous studies showing an improvement in glucose homeostasis in db/db mice treated with L‐carnosine (Block et al., 2009; Gorin et al., 2015).
The glomeruli of db/db mice underwent significant remodelling, as indicated by fibronectin and collagen IV accumulation and increased glomerular cell apoptosis. These changes were associated with increased serum levels of isoprostane 8‐epi‐PGF2α, PCOs and AGEs, augmented glomerular content of AGEs and their receptor AGER and markedly increased Nox4 expression, the major NADPH oxidase renal isoform involved in ROS production and diabetes‐associated kidney damage (Block et al., 2009; Gorin et al., 2015). In addition, kidneys of db/db mice showed signs of inflammation, as demonstrated by the increased protein levels of F4/80 and transcripts for Adgre1, Mcp1 and Tnfa. Interestingly, the NLRP3 inflammasome, which has been recently shown to serve as a central mechanism in the pathogenesis of type 2 diabetes‐associated renal inflammation and injury (Anders and Muruve, 2011; Solini et al., 2013), was also markedly increased in renal tissue of db/db mice, at both the protein and mRNA level. The elevation of all these parameters was markedly blunted in db/db mice treated with FL‐926‐16, in which a reduction in systemic and renal carbonyl and oxidative stress was associated with attenuated renal/glomerular fibrosis, apoptosis and inflammation.
These data provide further insights into the mechanisms underlying the injurious effect of AGEs in diabetic nephropathy as well as the favourable action of carnosine derivatives on this disorder. In fact, FL‐926‐16 was very effective in blunting the diabetes‐induced up‐regulation of the NLRP3 inflammasome, thus suggesting that the pro‐inflammatory response of the innate immune system triggered by AGEs involves the activation of this danger recognition platform, possibly via ROS generation (Anders and Muruve, 2011; Solini et al., 2013). In addition, mesangial cells treated with FL‐926‐16 showed no or significantly attenuated RCS‐induced Nlrp3 expression, thus supporting the concept that the protective effect exerted by L‐carnosine derivatives is due to their ability to scavenge RCS and inhibit AGE formation. As evidence of this, FL‐926‐16 was not able to reduce Nlrp3 overexpression induced by CML, the most abundant AGE found in vivo (Fu et al., 1996). Moreover, our previous finding that D‐carnosine octylester is effective in preventing cytotoxicity induced by HNE, but not by H2O2 in cultured mesangial cells, ruled out the possibility that carnosine derivatives provide protection through direct antioxidant effects (Menini et al., 2012).
Taken together, the results from this study support the concept that extensive AGE formation by chronic hyperglycaemia and dyslipidaemia plays a pivotal role in the pathogenesis of diabetic nephropathy, by favouring inflammation and oxidative stress through pathways involving the activation of the NLRP3 inflammasome. In turn, these pathways trigger extracellular matrix remodelling and cell loss through apoptosis, which eventually end up causing a deterioration of renal function. This conclusion is consistent with previous studies indicating that a reduction in oxidative stress (Melhem et al., 2002; DeRubertis et al., 2004) or inflammation (Okada et al., 2003; Tarabra et al., 2009) is effective in preventing or attenuating experimental diabetic glomerulopathy. In addition, our data indicate that a therapeutic strategy aimed at reducing AGE formation and accumulation is effective in attenuating the inflammatory and oxidative pathways triggered by these by‐products, thus blunting the onset of diabetes‐induced glomerular lesions and stopping the progression of established diabetic nephropathy. As a corollary, treatment with a derivative of the endogenous histidine‐containing dipeptide L‐carnosine would raise less safety concerns than those associated with the use of pharmacological agents acting on the inflammatory and oxidative stress pathways.
The strengths of this study include the use of a well‐established animal model of type 2 diabetes and nephropathy as well as of a novel, rationally designed carnosine derivative with a favourable pharmacokinetic profile, which might be suitable for testing in human subjects. The main limitation is the preliminary nature of these observations, which cannot be readily translated to human diabetic nephropathy.
In conclusion, these data show that treatment with the L‐carnosine derivative FL‐926‐16 provides protection from the development of diabetic glomerulopathy and halts the progression of established diabetic nephropathy. This protection appears to be dependent on a reduced carbonyl stress due to the RCS scavenging activity of FL‐926‐16, with consequent inhibition of the formation and accumulation of AGEs, which is associated with reduced fibrosis, apoptosis and inflammation, including the activation of the NLRP3 inflammasome. These results suggest that FL‐926‐16 might be a suitable strategy for the prevention and treatment of diabetic nephropathy in humans with type 2 diabetes.
Author contributions
C.I. contributed to the conception and design, acquisition of data, analysis and interpretation of data, critical revision of the article for important intellectual content and final approval of the version to be published. C.B.F., C.M.P., A.G., E.S., A.L., M.O. and G.A. contributed to the acquisition of data, critical revision of the article for important intellectual content and final approval of the version to be published. S.M. and G.P. contributed to the conception and design, analysis and interpretation of data, drafting the article and final approval of the version to be published. All Authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of interest
G.A. and M.O. are co‐authors of the patent application no. WO/2011/080139 on ‘Amino alcohol derivatives and their therapeutic activities’, which represents the rationale for using FL‐926‐16 in this work. G.P. is a recipient of an unconditional grant from Flamma S.p.A., Chignolo d'Isola, Italy.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Prevention protocol: western blot analysis for collagen IV and F4/80. Western blot analysis for collagen IV a1 chain (Panel A) and F4/80 (Panel B) protein levels in representative animals and relative band densitometry analysis in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). The Y axis label “Relative level” indicates fold of mean value of Ctr (db/m control) mice. Post hoc multiple comparison: *P < 0.05 vs Ctrl, †P < 0.05 vs Diab.
Table S1 Assays used for qRT‐PCR analysis.
Acknowledgements
The Authors thank Cinzia Cataldo for technical assistance in sample collection, coding and histology processing.
This work was supported by a research grant of the Research Foundation of the Italian Society of Diabetology (Associazione Diabete Ricerca ONLUS) to G.P.; Sapienza Università di Roma‐Progetti Ateneo 2016 to S.M.; and by an unconditional grant from Flamma S.p.A., Chignolo d'Isola, Italy, that also provided the tested compound FL‐926‐16 to GP. The sponsors had no role in the design and conduct of the study; collection, management and interpretation of the data; or preparation, review and approval of the manuscript.
Iacobini, C. , Menini, S. , Blasetti Fantauzzi, C. , Pesce, C. M. , Giaccari, A. , Salomone, E. , Lapolla, A. , Orioli, M. , Aldini, G. , and Pugliese, G. (2018) FL‐926‐16, a novel bioavailable carnosinase‐resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy in db/db mice. British Journal of Pharmacology, 175: 53–66. doi: 10.1111/bph.14070.
References
- Ahluwalia TS, Lindholm E, Groop LC (2011). Common variants in CNDP1 and CNDP2, and risk of nephropathy in type 2 diabetes. Diabetologia 54: 2295–2302. [DOI] [PubMed] [Google Scholar]
- Ahmed N (2005). Advanced glycation endproducts – role in pathology of diabetic complications. Diabetes Res Clin Pract 67: 3–21. [DOI] [PubMed] [Google Scholar]
- Aldini G, Facino RM, Beretta G, Carini M (2005). Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. Biofactors 24: 77–87. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders HJ, Muruve DA (2011). The inflammasomes in kidney disease. J Am Soc Nephrol 22: 1007–1018. [DOI] [PubMed] [Google Scholar]
- Bakris GL, Bank AJ, Kass DA, Neutel JM, Preston RA, Oparil S (2004). Advanced glycation end‐product cross‐link breakers. A novel approach to cardiovascular pathologies related to the aging process. Am J Hypertens 17: 23S–30S. [DOI] [PubMed] [Google Scholar]
- Block K, Gorin Y, Abboud HE (2009). Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A 106: 14385–14390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlender JM, Franke S, Stein G, Wolf G (2005). Advanced glycation end products and the kidney. Am J Physiol Renal Physiol 289: F645–F659. [DOI] [PubMed] [Google Scholar]
- Cooper ME (2004). Importance of advanced glycation end products in diabetes‐associated cardiovascular and renal disease. Am J Hypertens 17: 31S–38S. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzzocrea S, Genovese T, Failla M, Vecchio G, Fruciano M, Mazzon E et al (2007). Protective effect of orally administered carnosine on bleomycin‐induced lung injury. Am J Physiol Lung Cell Mol Physiol 292: L1095–L1104. [DOI] [PubMed] [Google Scholar]
- DeRubertis FR, Craven PA, Melhem MF, Salah EM (2004). Attenuation of renal injury in db/db mice overexpressing superoxide dismutase: evidence for reduced superoxide–nitric oxide interaction. Diabetes 53: 762–768. [DOI] [PubMed] [Google Scholar]
- Ellis EM (2007). Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther 115: 13–24. [DOI] [PubMed] [Google Scholar]
- Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR (1996). The advanced glycation end product, Nε‐(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 271: 9982–9986. [DOI] [PubMed] [Google Scholar]
- Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S et al (2015). Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol 308: F1276–F1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjutsalo V, Groop PH (2014). Epidemiology and risk factors for diabetic kidney disease. Adv Chronic Kidney Dis 21: 260–266. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Menini S, Oddi G, Ricci C, Amadio L, Pricci F et al (2004). Galectin‐3/AGE‐receptor 3 knockout mice show accelerated AGE‐induced glomerular injury evidence for a protective role of galectin‐3 as an AGE‐receptor. FASEB J 18: 1773–1775. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Mazzitelli G et al (2009a). Advanced lipoxidation end‐products mediate lipid‐induced glomerular injury: role of receptor‐mediated mechanisms. J Pathol 218: 360–369. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Cordone S et al (2009b). Accelerated lipid‐induced atherogenesis in galectin‐3 deficient mice: role of lipoxidation via receptor‐mediated mechanisms. Arterioscler Thromb Vasc Biol 29: 831–836. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Menini S, Ricci C, Blasetti Fantauzzi C, Scipioni A, Salvi L et al (2011). Galectin‐3 ablation protects mice from diet‐induced nonalcoholic steatohepatitis. Major scavenging role of galectin‐3 in liver. J Hepatol 54: 975–983. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köppel H, Riedl E, Braunagel M, Sauerhoefer S, Ehnert S, Godoy P et al (2011). L‐carnosine inhibits high‐glucose‐mediated matrix accumulation in human mesangial cells by interfering with TGF‐β production and signalling. Nephrol Dial Transplant 26: 3852–3858. [DOI] [PubMed] [Google Scholar]
- Kurata H, Fujii T, Tsutsui H, Katayama T, Ohkita M, Takaoka M et al (2006). Renoprotective effects of l‐carnosine on ischemia/reperfusion‐induced renal injury in rats. J Pharmacol Exp Ther 319: 640–647. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melhem MF, Craven PA, Liachenko J, DeRubertis FR (2002). α‐Lipoic acid attenuates hyperglycemia and prevents glomerular mesangial matrix expansion in diabetes. J Am Soc Nephrol 13: 108–116. [DOI] [PubMed] [Google Scholar]
- Menini S, Iacobini C, Ricci C, Oddi G, Pesce C, Pugliese F et al (2007). Ablation of the gene encoding p66Shc protects mice against AGE‐induced glomerulopathy by preventing oxidant‐dependent tissue injury and further AGE accumulation. Diabetologia 50: 1997–2007. [DOI] [PubMed] [Google Scholar]
- Menini S, Iacobini C, Ricci C, Scipioni A, Blasetti Fantauzzi C, Giaccari A et al (2012). D‐Carnosine octylester attenuates atherosclerosis and renal disease in ApoE null mice fed a Western diet through reduction of carbonyl stress and inflammation. Br J Pharmacol 166: 1344–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menini S, Iacobini C, Ricci C, Blasetti Fantauzzi C, Pugliese G (2015). Protection from diabetes‐induced atherosclerosis and renal disease by D‐carnosine‐octylester: effects of early vs late inhibition of advanced glycation end‐products in Apoe‐null mice. Diabetologia 58: 845–853. [DOI] [PubMed] [Google Scholar]
- Molitch ME, DeFronzo RA, Franz MJ, Keane WF, Mogensen CE, Parving HH et al (2004). Nephropathy in diabetes. Diabetes Care 27 (S1): S79–S83. [DOI] [PubMed] [Google Scholar]
- Negre‐Salvayre A, Coatrieux C, Ingueneau C, Salvayre R (2008). Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol 153: 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrisoli G, Canevotti R, Previtali M, Aldini G, Carini M, Orioli M et al (2011) Amino alcohol derivatives and their therapeutic activities. Patent application No.: WO/2011/080139; International Application No.: PCT/EP2010/070238, Jun 7.
- Odetti P, Fogarty J, Sell DR, Monnier VM (1992). Chromatographic quantitation of plasma and erythrocyte pentosidine in diabetic and uremic subjects. Diabetes 41: 153–159. [DOI] [PubMed] [Google Scholar]
- Okada S, Shikata K, Matsuda M, Ogawa D, Usui H, Kido Y et al (2003). Intercellular adhesion molecule‐1‐deficient mice are resistant against renal injury after induction of diabetes. Diabetes 52: 2586–2593. [DOI] [PubMed] [Google Scholar]
- Orioli M, Vistoli G, Regazzoni L, Pedretti A, Lapolla A, Rossoni G et al (2011). Design, synthesis, ADME Properties, and pharmacological activities of β‐alanyl‐D‐histidine (D‐Carnosine) prodrugs with improved bioavailability. ChemMedChem 6: 1269–1282. [DOI] [PubMed] [Google Scholar]
- Peters V, Lanthaler B, Amberger A, Fleming T, Forsberg E, Hecker M et al (2015). Carnosine metabolism in diabetes is altered by reactive metabolites. Amino Acids 47: 2367–2376. [DOI] [PubMed] [Google Scholar]
- Pugliese G, Pricci F, Leto G, Amadio L, Iacobini C, Romeo G et al (2000). The diabetic milieu modulates the advanced glycation end product‐receptor complex in the mesangium by inducing or upregulating galectin‐3 expression. Diabetes 49: 1249–1257. [DOI] [PubMed] [Google Scholar]
- Rajanikant GK, Zemke D, Senut MC, Frenkel MB, Chen AF, Gupta R et al (2007). Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke 38: 3023–3031. [DOI] [PubMed] [Google Scholar]
- Reddy VP, Beyaz A (2006). Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discov Today 11: 646–654. [DOI] [PubMed] [Google Scholar]
- Sharma K, McCue P, Dunn SR (2003). Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol 284: F1138–F1144. [DOI] [PubMed] [Google Scholar]
- Soliman KM, Abdul‐Hamid M, Othman AI (2007). Effect of carnosine on gentamicin‐induced nephrotoxicity. Med Sci Monit 13: BR73–BR83. [PubMed] [Google Scholar]
- Solini A, Menini S, Rossi C, Ricci C, Santini E, Blasetti Fantauzzi C et al (2013). The purinergic 2X7 receptor participates in renal inflammation and injury induced by high‐fat diet: possible role of NLRP3 inflammasome activation. J Pathol 231: 342–353. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Cutler RG, Jo DG, Magnus T, Chan SL et al (2007). Neuroprotective actions of a histidine analogue in models of ischemic stroke. J Neurochem 101: 729–736. [DOI] [PubMed] [Google Scholar]
- Tarabra E, Giunti S, Barutta F, Salvidio G, Burt D, Deferrari G et al (2009). Effect of the monocyte chemoattractant protein‐1/CC chemokine receptor 2 system on nephrin expression in streptozotocin‐treated mice and human cultured podocytes. Diabetes 58: 2109–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle KR, Bakris GL, Bilous RW, Chiang JL, de Boer IH, Goldstein‐Fuchs J et al (2014). Diabetic kidney disease: a report from an ADA Consensus Conference. Am J Kidney Dis 64: 510–533. [DOI] [PubMed] [Google Scholar]
- Vistoli G, Orioli M, Pedretti A, Regazzoni L, Canevotti R, Negrisoli G et al (2009). Design, synthesis, and evaluation of carnosine derivatives as selective and efficient sequestering agents of cytotoxic reactive carbonyl species. Chem Med Chem 4: 967–975. [DOI] [PubMed] [Google Scholar]
- Wendt T, Tanji N, Guo J, Hudson BI, Bierhaus A, Ramasamy R et al (2003). Glucose, glycation, and RAGE: implications for amplification of cellular dysfunction in diabetic nephropathy. J Am Soc Nephrol 14: 1383–1395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Prevention protocol: western blot analysis for collagen IV and F4/80. Western blot analysis for collagen IV a1 chain (Panel A) and F4/80 (Panel B) protein levels in representative animals and relative band densitometry analysis in untreated and FL‐926‐16 (FL)‐treated db/m control (Ctrl) and db/db diabetic (Diab) mice (mean ± SD; n = 5 per group). The Y axis label “Relative level” indicates fold of mean value of Ctr (db/m control) mice. Post hoc multiple comparison: *P < 0.05 vs Ctrl, †P < 0.05 vs Diab.
Table S1 Assays used for qRT‐PCR analysis.