

ABSTRACT
Trimethoprim (TMP)-sulfamethoxazole (SMX) is used to treat various types of infections, including community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) and Pneumocystis jirovecii infections in children. Pharmacokinetic (PK) data for infants and children are limited, and the optimal dosing is not known. We performed a multicenter, prospective PK study of TMP-SMX in infants and children. Separate population PK models were developed for TMP and SMX administered by the enteral route using nonlinear mixed-effects modeling. Optimal dosing was determined on the basis of the matching adult TMP exposure and attainment of the surrogate pharmacodynamic (PD) target for efficacy, a free TMP concentration above the MIC over 50% of the dosing interval. Data for a total of 153 subjects (240 samples for PK analysis) with a median postnatal age of 8 years (range, 0.1 to 20 years) contributed to the analysis for both drugs. A one-compartment model with first-order absorption and elimination characterized the TMP and SMX PK data well. Weight was included in the base model for clearance (CL/F) and volume of distribution (V/F). Both TMP and SMX CL/F increased with age. In addition, TMP and SMX CL/F were inversely related to the serum creatinine and albumin concentrations, respectively. The exposure achieved in children after oral administration of TMP-SMX at 8/40 mg/kg of body weight/day divided into administration every 12 h matched the exposure achieved in adults after administration of TMP-SMX at 320/1,600 mg/day divided into administration every 12 h and achieved the PD target for bacteria with an MIC of 0.5 mg/liter in >90% of infants and children. The exposure achieved in children after oral administration of TMP-SMX at 12/60 and 15/75 mg/kg/day divided into administration every 12 h matched the exposure achieved in adults after administration of TMP-SMX at 640/3,200 mg/day divided into administration every 12 h in subjects 6 to <21 years and 0 to <6 years of age, respectively, and was optimal for bacteria with an MIC of up to 1 mg/liter.
KEYWORDS: trimethoprim, sulfamethoxazole, infants, children, pharmacokinetics, methicillin-resistant Staphylococcus aureus
INTRODUCTION
Trimethoprim (TMP)-sulfamethoxazole (SMX) is a combination of TMP, which is a pyrimidine analogue, and SMX, which is from the sulfonamide family (1). Both components act sequentially in two successive steps in the biosynthesis of bacterial nucleic acids (1). TMP-SMX is commonly used in infants and children for the treatment and prevention of various clinical conditions, including urinary tract infections and Pneumocystis jirovecii pneumonia. Although TMP-SMX is not approved by the U.S. Food and Drug Administration (FDA) for the treatment of staphylococcal infections, it is recommended and used for the treatment of outpatients with skin and soft tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA) (2–4). Adult TMP-SMX dosing recommendations, based on the TMP component, are 320 to 640 mg/day, divided into administration every 12 h, orally for the treatment of bacterial infections and 15 to 20 mg/kg of body weight/day every 6 to 8 h intravenously and then orally for the treatment of P. jirovecii infections (1, 4).
Following oral administration in adults, TMP and SMX concentrations peak at about 1 to 4 h after dosing (1). Both components distribute well into tissues, and protein binding percentages are 45% and 70% for TMP and SMX, respectively (1). Oral adult volumes of distribution (V/F) are 1.4 to 1.8 liters/kg and 0.43 liters/kg for TMP and SMX, respectively (1). The primary route of TMP elimination is renal, with 75 to 85% of the parent drug being eliminated unchanged in urine (1, 5, 6). The remaining 15 to 25% is metabolized by the liver into inactive metabolites. SMX is metabolized predominantly by liver cytochrome P450 2C9 (CYP2C9) to multiple metabolites of unknown activity, with the most predominant being N4-acetylsulfamethoxazole (1). N4-Acetylsulfamethoxazole is mainly excreted in urine. In addition to undergoing biotransformation, 30% of SMX is excreted unchanged in the urine (5, 7). Both renal function and CYP2C9 metabolic activity undergo maturation during childhood, suggesting differences in TMP and SMX pharmacokinetics (PK) across the age continuum (8, 9).
TMP and SMX PK in infants and children are poorly described, especially following oral administration. A PK study of intravenous TMP-SMX in children (age range, 0 to 19 years; n = 17) and adults (n = 5) with P. jirovecii infection reported that the TMP and SMX weight-normalized volume of distribution decreases with age (6). Furthermore, TMP weight-normalized clearance in infants (<1 year of age) was similar to that in children >10 years of age and adults but was lower than that reported for children 1 to 9 years of age. However, that study (6) enrolled a limited number of infants (n = 2). For SMX, the weight-normalized clearance decreased with age (6).
The aim of the present analysis was to characterize the disposition of TMP and SMX across the age continuum from infants to adolescents using a population PK (PopPK) modeling approach and to optimize TMP-SMX dosing in this population.
RESULTS
Patient characteristics.
Data for a total of 153 subjects (240 samples) were included in the TMP and SMX PopPK analysis. Demographic and clinical variables are summarized in Table 1. Fifty-three subjects (35%) were obese. Ten subjects (7%) were ≤120 days of postnatal age (PNA) at the time of collection of samples for PK analysis (PK sampling); of these, all were ≥32 weeks gestational age (GA) and had a median PNA of 67 days (range, 20 to 114 days). The median serum creatinine concentration (SCR) was 0.5 mg/dl (range, 0.1 to 5.9 mg/dl), and 7 children had an SCR of >1.8 mg/dl. SCR was significantly correlated with PNA: for every 1-week increase in PNA, there was an increase in the SCR of 0.0005 mg/dl (P < 0.01) (see Fig. S1 in the supplemental material). This linear relationship was used to impute missing SCR values.
TABLE 1.
Clinical datae
| Characteristica | Values | No. (%) subjects with data available |
|---|---|---|
| Median (range) value for: | ||
| GA (wk)b | 38 (32–39) | 10 (7)b |
| PNA (yr) | 7.9 (0.1–20.2) | 153 (100) |
| WT (kg) | 30.8 (2.4–147.9) | 153 (100) |
| SCR (mg/dl) | 0.5 (0.1–5.9) | 117 (77) |
| AST (U/liter) | 31 (12–291) | 56 (37) |
| ALT (U/liter) | 42 (9–282) | 57 (37) |
| TBIL (mg/dl) | 0.5 (0.1–11.1) | 54 (35) |
| DBIL (mg/dl) | 0.2 (0–8.5) | 19 (12) |
| ALB (g/dl) | 3.4 (1.7–4.8) | 78 (51) |
| No. (%) of male subjects | 82 (54) | 153 (100) |
| No. (%) of subjects by race | 153 (100) | |
| White | 109 (71) | |
| Black | 29 (19) | |
| Unknown | 3 (2) | |
| Others | 12 (8) | |
| No. (%) of subjects by ethnicity | 153 (100) | |
| Hispanic | 26 (17) | |
| Not Hispanic | 123 (80) | |
| Unknown | 4 (3) | |
| No. (%) of obese subjectsc | 53 (35) | 153 (100) |
| No. (%) of subjects receiving the following drug formulation: | 153 (100) | |
| Oral suspension | 78 (51) | |
| Tablets | 75 (49) | |
| No. (%) of subjects receiving drug by the following route: | 153 (100) | |
| Oral | 125 (82) | |
| Gastrostomy | 17 (11) | |
| Othersd | 11 (7) |
Descriptive statistics are calculated on the basis of the values at the time of first recorded dose. GA, gestational age; PNA, postnatal age; WT, total body weight; SCR, serum creatinine concentration; AST, aspartate aminotransferase concentration; ALT, alanine aminotransferase concentration; TBIL, total bilirubin concentration; DBIL, direct bilirubin concentration; ALB, serum albumin concentration.
For infants of <120 days postnatal age (n = 10).
Body mass index, ≥95th percentile. The body mass index was not assessed for subjects <2 years of age.
Others include the nasogastric, orogastric, or nasojejunal route or administration by jejunostomy.
Data are for 153 subjects.
The median TMP and SMX doses were 2.5 mg/kg/dose (range, 0.5 to 12.1 mg/kg/dose) and 12.7 mg/kg/dose (range, 2.5 to 60.2 mg/kg/dose), respectively. The median daily TMP and SMX doses were 4.6 mg/kg/day (range, 0.5 to 12.1 mg/kg/day) and 23.0 mg/kg/day (range, 2.5 to 120.5 mg/kg/day), respectively. The median dosing interval was 12 h (range, 6 to 48 h). Seventy-eight subjects (51%) received an oral suspension (TMP-SMX at 8/40 mg/ml) at the time of the first recorded dose, while the remaining subjects received tablets (TMP-SMX at 80/400 mg or 160/800 mg) (Fig. 1). Three subjects received both drug formulations throughout the study period. Dosing was via the oral route in 125 subjects (82%), via a gastrostomy tube in 17 subjects (11%), and by other routes, including by the nasogastric, orogastric, and nasojejunal routes and via a jejunostomy tube, in the remaining 11 subjects (7%) at the time of the first recorded dose. Two subjects received TMP-SMX via two different routes throughout the study period.
FIG 1.
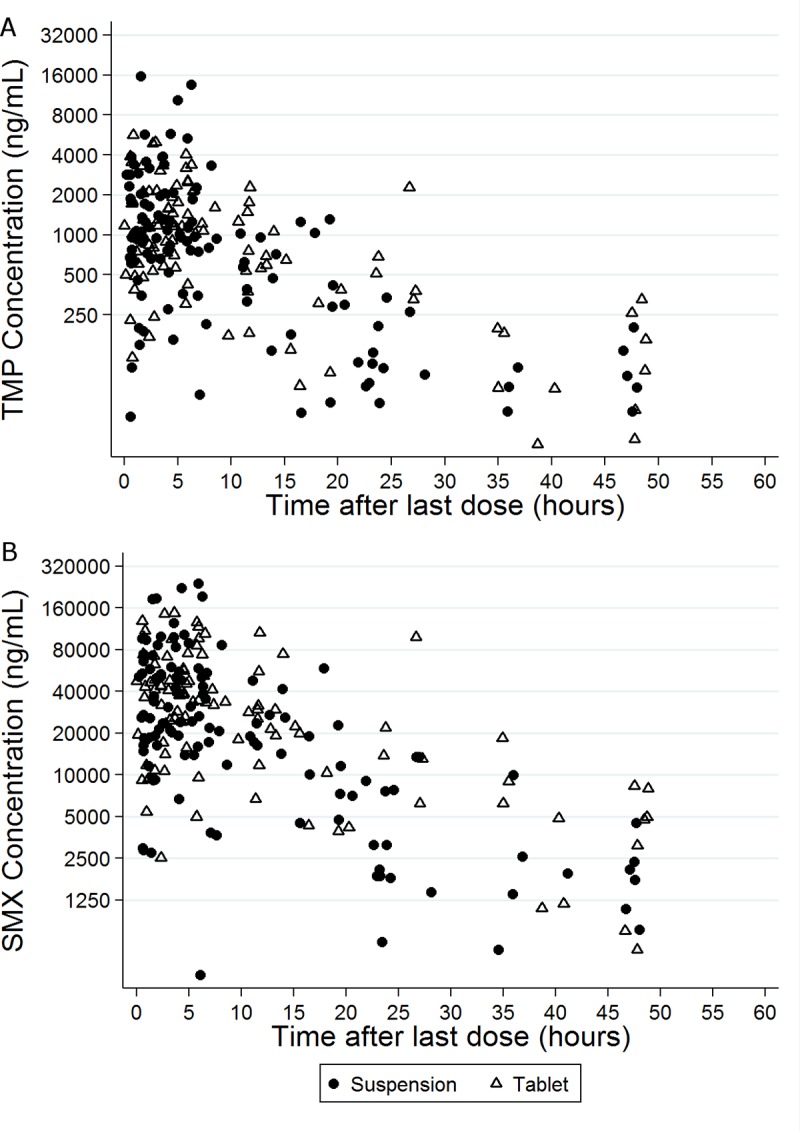
Trimethoprim (TMP) (A) and sulfamethoxazole (SMX) (B) concentrations versus time after the last dose.
PK specimens.
The median number of samples collected for PK analysis (PK samples) from each subject was 1 (range, 1 to 4). The median TMP and SMX concentrations were 0.94 mg/liter (range, 0.03 to 15.49 mg/liter) and 25.12 mg/liter (range, 0.36 to 238.25 mg/liter), respectively. No samples for analysis of TMP or SMX PK were excluded from the analysis. Among the 240 samples collected, 26 (10.8%) had TMP concentrations and 19 (7.9%) had SMX concentrations that were below the limit of quantification (BLQ). When the final PK model was run with a data set in which BLQ values were ignored, the final PK estimates were similar to the ones obtained with the method consisting of substitution of the BLQ for half of the lower limit of quantification (LLOQ) (within 12%; data not shown).
TMP population PK model development and evaluation.
A one-compartment PK model with first-order absorption and elimination fit the TMP concentration-versus-time data well (Fig. 2). For the base model, the use of total body weight (WT) to adjust for body size resulted in an objective function value (OFV) lower than that achieved with free fat mass (FFM), lean body weight (LBW), and body mass index (BMI). In the PK structural model, the use of a first-order absorption model with no lag time (Tlag) resulted in an OFV similar to that obtained in alternative absorption models. A transit compartment absorption model did not minimize successfully. Estimation of different absorption rate constant (Ka) values according to the oral formulation (tablet versus oral suspension) or route of administration (by oral versus other enteral routes) did not improve the fit (the models had the same OFV). The interindividual variability (IIV) in Ka could not be accurately estimated (relative standard error [RSE] > 200%), and therefore, this random effect parameter was fixed to zero. A covariance between the IIV in CL/F and the IIV in V/F was not estimated, as the inclusion of a block covariance matrix resulted in model instability with an unacceptably high proportion of runs with minimization problems (∼50%) during the bootstrap analysis.
FIG 2.

TMP diagnostic plots for the final model. (A) Observed versus population predictions; (B) observed versus individual predictions; (C) conditional weighted residuals versus time; (D) conditional weighted residuals versus population predictions. The dashed lines represent the loess curve. The solid grey lines represent conditional weighted residual values of 0 and ±2. Concentrations below the lower limit of quantifications (LLOQ) were imputed as LLOQ/2.
After accounting for body size, use of a maximum effect (Emax) maturation function using PNA on CL/F resulted in the largest OFV drop (−14.6 points; Table S1). However, the Hill coefficient (Hill) for the maturation model relationship between PNA and CL/F was fixed to 1 due to model instability, if it was estimated. Alternative models between PNA and CL/F (linear and power models) resulted in an OFV higher than that obtained with the Emax relationship (data not shown). Thereafter, SCR and the albumin concentration (ALB) on CL/F and obese status on V/F resulted in a significant reduction in the OFV (Table S1). However, only PNA and SCR on CL/F were retained in the model after the backward elimination steps. Thus, the final model included WT, an Emax maturation relationship between PNA and CL/F, and a power relationship with SCR on CL/F (Table 2), as follows: Ka = 1.27, CL/F = 10.0 · (WT/70)0.75 · [PNA1/(0.241 + PNA1)] · (0.5/SCR)0.40, and V/F = 148 · (WT/70), where Ka is in hours−1, CL/F is in liters per hour, WT is in kilograms, PNA is in years, SCR is in milligrams per deciliter, and V/F is in liters.
TABLE 2.
TMP PopPK parameter estimates from the final modela
| Parameter | Final model |
Bootstrap analysis (n = 1,000)b |
|||
|---|---|---|---|---|---|
| Estimate | RSE (%) | 2.5th percentile | Median | 97.5th percentile | |
| Ka (h−1) | 1.27 | 35.8 | 0.6 | 1.27 | 2.4 |
| CL/F70 kg (liters/h) | 10.0 | 5.5 | 8.8 | 9.9 | 11.0 |
| V/F70 kg (liters) | 148 | 6.8 | 129 | 148 | 173 |
| TM50 (yr) | 0.24 | 24.8 | 0.14 | 0.24 | 0.40 |
| Hill | 1 (fixed) | ||||
| Exponent for SCR effect on CL/F | 0.40 | 20.4 | 0.26 | 0.41 | 0.57 |
| IIV(CL/F) (% CV) | 33.8 | 36.8 | 10.0 | 31.6 | 44.7 |
| IIV(V/F) (% CV) | 20.6 | 89.2 | 4.7 | 22.3 | 50.1 |
| Proportional error (%) | 51.1 | 14.4 | 42.3 | 50.0 | 57.6 |
TMP, trimethoprim; PK, pharmacokinetic; RSE, relative standard error; Ka, population absorption rate constant; CL/F70 kg, apparent population clearance estimate scaled to a 70-kg adult; V/F70 kg, apparent population volume of distribution estimate scaled to a 70-kg adult; TM50, maturation half-life calculated as a function of postnatal age (in years); Hill, Hill coefficient in the Emax maturation function; SCR, serum creatinine concentration; IIV(CL/F), interindividual variability in drug clearance; IIV(V/F), interindividual variability in the volume of distribution; CV, coefficient of variation.
Altogether, 39 runs with minimization terminated were skipped when the bootstrap results were calculated; 249 runs with estimates near a boundary were skipped when the bootstrap results were calculated.
The PNA at which the value of CL/F reached 50% of the mature, adult value (i.e., maturation half-life [TM50]) was estimated to be 0.24 years, corresponding to ~13 weeks PNA. Estimation of the exponents in the relationship between WT and the PK parameters using the final model suggests an allometric relationship between CL/F and WT (exponent = 0.75) and an exponent close to 1 for the relationship between V/F and WT (exponent = 0.9). A proportional residual error model characterized unexplained residual variability and was estimated to be 51.1%. Shrinkage estimates for the IIV in the CL/F and V/F terms were 32% and 67%, respectively. Shrinkage for the proportional residual error parameter was 12%. The percent difference between the model and bootstrapped median parameter estimates was ≤8% for all parameters; 97% of the bootstrap runs minimized successfully, and the covariance step ran successfully in 82% of the bootstrap runs (Table 2). Diagnostic plots for the final model showed underprediction for TMP concentrations of >5,000 mg/liter (Fig. 2). Ten observed concentrations (4%) were outside the standardized visual predictive check (SVPC) 95% prediction interval, indicating the good predictive performance of the final model (Fig. S2).
Individual empirical Bayesian estimates for PK parameters were compared across age groups (Table 3). Following the Emax maturation function, the median CL/F increased from 9.6 liters/h/70 kg at <2 years of age to 11.4 liters/h/70 kg at 2 to <6 years of age. The median CL/F then decreased back to 9.3 liters/h/70 kg in the older age group (6 to <21 years of age). V/F normalized to a WT of 70 kg decreased in the older age group, although the individual empirical Bayesian estimates of V/F should be interpreted with caution, given the high shrinkage in the IIV parameter for V/F.
TABLE 3.
TMP individual empirical Bayesian post hoc parameter estimates stratified by age
| PK parametera | Median (range) value for the following age groups (yr)b: |
|||
|---|---|---|---|---|
| 0 to <2 (n = 46) | 2 to <6 (n = 25) | 6 to <21 (n = 82) | Total (n = 153) | |
| CL/F (liters/h/kg) | 0.25 (0.05–0.44) | 0.23 (0.14–0.43) | 0.14 (0.04–0.31) | 0.16 (0.04–0.44) |
| CL/F (liters/h/70 kg) | 9.6 (1.5–18.1) | 11.4 (6.2–21.4) | 9.3 (2.5–18.0) | 9.6 (1.5–21.4) |
| V/F (liters/kg) | 2.1 (1.8–2.5) | 2.1 (1.8–2.4) | 2.1 (1.4–2.4) | 2.1 (1.4–2.5) |
| V/F (liters/70 kg) | 149 (125–175) | 149 (127–171) | 147 (96–168) | 148 (96.2–175) |
| Half-life (h) | 5.9 (3.3–33.2) | 6.5 (3.1–11.3) | 11.1 (4.3–32.6) | 8.7 (3.1–33.2) |
CL/F, clearance; V/F, volume of distribution.
Values were calculated using age at the time that the first pharmacokinetic sample was collected.
SMX population PK model development and evaluation.
A one-compartment PK model with first-order absorption and elimination fit the SMX concentration-versus-time data well (Fig. 3). Similar to the results for TMP, the IIV in Ka could not be precisely estimated for SMX (RSE > 200%), and therefore, this random effect parameter was fixed to zero. After accounting for body size using WT, a sigmoidal Emax maturation function with PNA resulted in the largest OFV drop (−10.4 points) (Table S2). Thereafter, ALB and obese status accounted for a significant amount of the IIV in CL/F and V/F, respectively. Obese status was dropped from the model in the backward elimination step. Thus, the final model included WT, a sigmoidal maturation relationship between PNA and CL/F, and an exponential relationship between ALB and CL/F (Table 4), as follows: Ka = 0.58, CL/F = 1.46 · (WT/70)0.75 · [PNA2.13/(0.122.13 + PNA2.13)] · (ALB/3.4)−0.77, and V/F = 24 · (WT/70), where Ka is in hours−1, CL/F is in liters per hour, WT is in kilogram, PNA is in years, ALB is in grams per deciliter, and V/F is in liters.
FIG 3.

SMX diagnostic plots for the final model. (A) Observed versus population predictions; (B) observed versus individual predictions; (C) conditional weighted residuals versus time; (D) conditional weighted residuals versus population predictions. The dashed lines represent the loess curve. The solid grey lines represent conditional weighted residual values of 0 and ±2. Concentrations below the lower limit of quantifications (LLOQ) were imputed as LLOQ/2.
TABLE 4.
SMX PopPK parameter estimates from the final modela
| Parameter | Final model |
Bootstrap analysis (n = 1,000)b |
|||
|---|---|---|---|---|---|
| Estimate | RSE (%) | 2.5th percentile | Median | 97.5th percentile | |
| Ka (h−1) | 0.58 | 43.9 | 0.1 | 0.6 | 1.3 |
| CL/F70 kg (liters/h) | 1.46 | 5.1 | 1.30 | 1.45 | 1.76 |
| V/F70 kg (liters) | 24 | 10.0 | 6 | 23 | 29 |
| TM50 (years) | 0.12 | 16.4 | 0.05 | 0.13 | 0.17 |
| Hill | 2.13 | 59.6 | 0.3 | 2.3 | 11.4 |
| Exponent for ALB effect on CL/F | −0.77 | 34 | −1.5 | −0.76 | −0.20 |
| IIV(CL/F) (%) | 35.9 | 46.2 | 9.2 | 33.2 | 51.3 |
| IIV(V/F) (%) | 40.6 | 41.1 | 18.3 | 39.6 | 114.1 |
| ρ (CL/F − V/F) | 0.1 | 56.7 | −0.1 | 0.1 | 0.3 |
| Proportional error (%) | 46.9 | 16.7 | 34.7 | 45.8 | 53.4 |
| Additive error (mg/liters) | 5.1 | 38.0 | 1.8 | 5.5 | 322 |
SMX, sulfamethoxazole; PK, pharmacokinetic; RSE, relative standard error; Ka, population absorption rate constant; CL/F70 kg, apparent population clearance estimate scaled to a 70-kg adult; V/F70 kg, apparent population volume of distribution estimate scaled to a 70-kg adult; TM50, maturation half-life calculated as a function of postnatal age (in years); Hill, Hill coefficient in the Emax maturation function; ALB, albumin concentration; SCR, serum creatinine concentration; IIV(CL/F), interindividual variability in drug clearance; IIV(V/F), interindividual variability in the volume of distribution; ρ (CL/F − V/F), correlation between random-effect parameters for CL/F and V/F.
One hundred twenty-six runs were skipped when the bootstrap results were calculated due to minimization termination. One hundred six runs with estimates near a boundary were skipped when the bootstrap results were calculated.
The PNA at which the value of CL/F reached 50% of the mature, adult value (i.e., TM50) was estimated to be 0.12 years, corresponding to ~6 weeks PNA. A combined proportional and additive error model best characterized the residual variability (Table 4). Shrinkage estimates were 31% and 42% for the IIV in the CL/F and V/F terms, respectively. Shrinkage for both the proportional and the additive residual error parameters were 17%. The percent difference between the model and bootstrapped median parameter estimates was ≤8% for all parameters; 87% of the bootstrap runs minimized successfully, and in 87% the covariance step ran successfully (Table 4). Diagnostic plots for the final model showed underprediction for SMX concentrations of >100 mg/liter (Fig. 3). Fifteen (6%) observations were outside the SVPC 95% prediction interval, indicating the good predictive performance of the final model (Fig. S3).
Individual empirical Bayesian estimates were compared across age groups. As described using the sigmoidal maturation function, the median CL/F increased from 1.47 liters/h/70 kg at 0 to <2 years of age to 1.56 liters/h/70 kg at 2 to <6 years of age. CL/F then decreased back to 1.48 liters/h/70 kg in the older age group (6 to <21 years of age).
Dose-exposure relationship.
For all dosing regimens, the simulated total TMP steady-state area under the concentration-versus-time curve from time zero to τ (AUC0–τ,ss; where τ denotes the dosing interval) was similar in subjects 0 to <2 years and 2 to <6 years of age (within 20%; Table 5). However, the value of AUC0–τ,ss for children in the older age group (6 to <21 years of age) was up to 29% higher than that simulated for infants 0 to <2 years of age (Table 5). In general, with all dosing regimens the simulated AUC0–τ,ss for infants and children was lower than that for 70-kg adults, except for the regimen of 8 mg/kg/day divided into administration every 12 h, for which AUC0–τ,ss values were similar in children and adults (Table 5).
TABLE 5.
Simulated trimethoprim exposure at steady state
| Age group (yr) | No. of subjects | AUC0–τ,ss (mg · h/liter)a |
|||||
|---|---|---|---|---|---|---|---|
| Oral dosing every 12 h |
Oral dosing every 8 h |
Oral dosing every 6 h |
|||||
| 8 mg/kg/dayb | 12 mg/kg/dayc | 15 mg/kg/dayd | 20 mg/kg/daye | 15 mg/kg/dayf | 20 mg/kg/dayg | ||
| 0 to <2 | 500 | 19.2 (9.2, 59.1) | 28.7 (13.8, 88.7) | 23.9 (11.5, 73.9) | 32.1 (15.4, 99.0) | 17.9 (8.6, 55.4) | 23.9 (11.5, 73.9) |
| 2 to <6 | 500 | 19.0 (9.8, 35.9) | 28.5 (14.7, 53.8) | 23.8 (12.2, 44.9) | 31.8 (16.4, 60.1) | 17.8 (9.2, 33.6) | 23.8 (12.2, 44.9) |
| 6 to <21 | 1,500 | 22.8 (11.4, 45.7) | 36.2 (18.0, 76.5) | 30.7 (15.7, 63.7) | 41.0 (21.0, 85.4) | 23.1 (11.8, 47.8) | 30.7 (15.7, 63.7) |
| 18 to 21 (70 kg) | 500 | 19.5 (10.2, 39.5) | 39.1 (20.3, 79.0) | 42.8 (22.2, 86.4) | 57.3 (29.7, 115.7) | 32.1 (16.6, 64.8) | 42.8 (22.2, 86.4) |
Data are presented as the median (2.5th, 97.5th percentile). AUC0–τ,ss, steady-state area under the concentration-versus-time curve from time zero to τ (where τ denotes the dosing interval).
Maximum daily dose, 320 mg (1 double-strength tablet every 12 h) achieved at a WT of 40 kg.
Maximum daily dose, 640 mg (2 double-strength tablets every 12 h) achieved at a WT of 53 kg.
Maximum daily dose, 1,200 mg (2 double-strength tablets + 1 single-strength tablet every 8 h) achieved at a WT of 60 kg.
Maximum daily dose, 1,440 mg (3 double-strength tablets every 8 h) achieved at a WT of 72 kg.
Maximum daily dose, 1,280 mg (2 double-strength tablets every 6 h) achieved at a WT of 85 kg.
Maximum daily dose, 1,600 mg (2 double-strength tablets + 1 single-strength tablet every 6h) achieved at a WT of 80 kg.
Pharmacodynamic (PD) target attainment parameters. (i) Antibacterial target of a free TMP C50,ss greater than the MIC.
A simulated dosing regimen of 8 mg/kg/day of the TMP component administered every 12 h was associated with a free total concentration at 50% of the dosing interval at steady state (C50,ss) greater than the MIC of 0.5 mg/liter in 91%, 94%, and 97% of subjects ages 0 to <2 years, 2 to <6 years, and 6 to <21 years, respectively (Fig. 4). For an MIC of 1 mg/liter, the target attainment rate ranged from 38 to 49% in all age groups. Therefore, administration of 8 mg/kg/day of the TMP component every 12 h appears to be adequate to treat infections caused by bacteria with an MIC of ≤0.5 mg/liter in children from all age groups.
FIG 4.
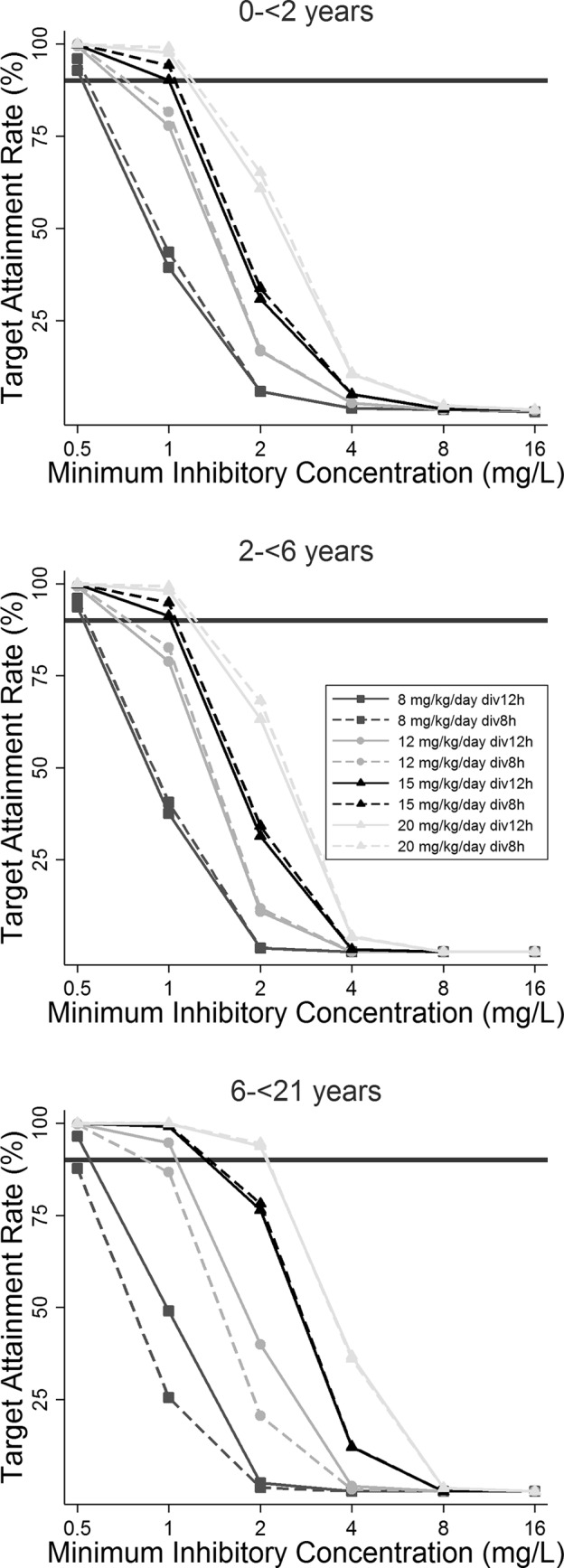
Target attainment rates by MIC for free trimethoprim concentrations at 50% of the dosing interval in 1,000 simulated children by age group. The solid horizontal black lines represent the 90% target attainment rate. div, the dose was divided by administration by the indicated times.
As shown in Fig. 4, the administration of 12 mg/kg/day every 12 h achieved target attainment in 99%, 99%, and 100% of subjects ages 0 to <2 years, 2 to <6 years, and 6 to <21 years, respectively, against bacteria with an MIC of 0.5 mg/liter. For an MIC of 1 mg/liter, target attainment was 78 to 79% for subjects 0 to <6 years of age and was 95% for subjects 6 to <21 years of age. Therefore, the administration of 12 mg/kg/day every 12 h appears to be adequate to treat infections caused by bacteria with an MIC of ≤0.5 mg/liter in all age groups and bacteria with an MIC of 1 mg/liter in those who are ≥6 years of age. A higher dose of 15 mg/kg/day divided into administration every 12 h was necessary for an optimal target attainment rate (90 to 91%) against bacteria with an MIC of 1 mg/liter in younger children (age, 0 to <6 years) (Fig. 4).
The only simulated dosing regimen associated with a free C50,ss of >2 mg/liter was 20 mg/kg/day divided into administration every 6 to 8 h for children 6 to <21 years of age (Fig. 4). For children in younger age groups, none of the simulated dosing regimens reached the surrogate PD target for bacteria with MICs of ≥2 mg/liter. These results suggest that TMP-SMX should not be recommended for the treatment of infections caused by bacteria with MICs of ≥2 mg/liter in children <6 years of age, for whom alternative antibiotic agents should be used.
(ii) Antifungal target of a total TMP Cmax,ss of ≥3 mg/liter.
In all age groups, a dose of the TMP component of 15 mg/kg/day divided into administration every 12 h was the lowest daily dose associated with the surrogate PD target for P. jirovecii of a total maximal drug concentration at steady state (Cmax,ss) of ≥3 mg/liter in 94%, 95%, and 99% of simulated subjects 0 to <2 years, 2 to <6 years, and 6 to <21 years of age, respectively (Fig. 5).
FIG 5.
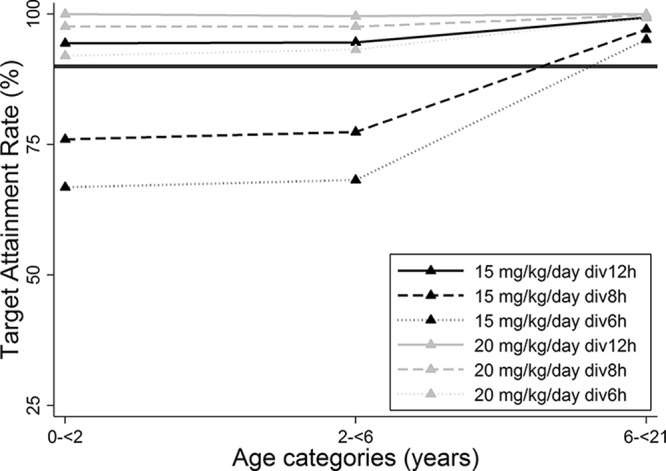
Target attainment rate for Pneumocystis jirovecii by age group. The solid horizontal black line represents the 90% target attainment rate. The target was defined as a total maximum concentration at steady state (Cmax,ss) of ≥3 mg/liter.
Safety threshold of a total TMP Cmax,ss of >8 mg/liter.
Simulated dosing of the TMP component at 8 to 12 mg/kg/day divided into administration every 8 to 12 h was associated with a Cmax,ss of >8 mg/liter in ≤3% of virtual subjects. Simulated dosing of 15 mg/kg/day divided into administration every 12 h exceeded the safety threshold in 1% of subjects 2 to <6 years of age but in up to 11% of subjects in the other age groups. Finally, administration of the TMP component at 20 mg/kg/day every 6 to 12 h was associated with achievement of a Cmax,ss of >8 mg/liter in a high proportion of subjects (up to 48%).
DISCUSSION
This is the first PopPK analysis of TMP-SMX administered by the enteral route in infants and children with dose-exposure simulations to support dosing recommendations for various indications. Similar to a published TMP and SMX PK model developed using data for adults, one-compartment models best characterized the data for both components (10–14). Population estimates for the TMP and SMX Ka were similar to values previously reported in adults (1.6 h−1 and 0.4 h−1, respectively) (10, 15). However, as suggested by our difficulty with estimation of the IIV in Ka, we were limited in our ability to characterize the absorption phase, even with alternative absorption models. The reasons for this include the high variability in the concentrations associated with oral administration, sparse PK sampling, the use of an opportunistic study design, and the questionable reliability of parental reports of outpatient dosing information.
Population estimates for TMP and SMX V/F values standardized to a 70-kg adult weight were 17 to 51% higher than the V/F values previously reported in HIV-infected adults (98 to 126 liters and 17.5 to 18.7 liters/h, respectively) (16–19). This difference may be explained by a difference in population characteristics. In the literature, the mean TMP volume of distribution was also higher in infants (139 liters/70 kg) than in adults (95 liters/70 kg) after intravenous administration of TMP-SMX for the treatment of P. jirovecii infections (6). However, in our cohort, after accounting for size, no other covariates further explained the IIV in V/F.
Population estimates for TMP and SMX CL/F in children standardized to a 70-kg adult weight were comparable (within 21%) to estimates previously reported in adults (8.93 liters/h and 1.72 liters/h, respectively) (16). In this analysis, TMP and SMX CL/F increased with PNA following an Emax model (20). Consistent with a predominant renal elimination, the TMP maturation half-life (i.e., TM50) estimate (13 weeks of PNA) is similar to previously reported estimates of TM50 for glomerular filtration rate maturation (47.7 weeks of postmenstrual age [PMA]), assuming a GA of 40 weeks (20). Even though glomerular filtration rate maturation was previously reported with PMA, we retained PNA because of the improved model fit. In our cohort, only 10/153 infants were <120 days of age, and the GA for these infants was then recorded. For the remaining infants and children, GA was not collected per study protocol and was imputed as 40 weeks for this analysis. Hence, our population estimate for TM50 is comparable to the TM50 reported with PMA, but our data should not be extrapolated to preterm infants. SMX is predominantly metabolized by CYP2C9, which has been shown to be expressed at birth at levels 20 to 30% of those for mature adults. Consistent with our results, the CL of indomethacin, which is also predominantly metabolized by CYP2C9, was shown to increase with PNA (21). In the previous study, the relationship between indomethacin CL and PNA in a cohort of young infants (PNA, 1 to 77 days) was characterized using an exponential function. In our study, we retained an Emax model, allowing a plateau to be achieved when adult CL values were reached. This difference is probably explained by the wide range of PNAs of our study population, with the study subjects being up to 21 years of age. CYP2C9 expression levels in the first week of life were reported to be half of those in adults, reaching mature values at approximately 5 months of age (8, 22). In our study, we estimated an SMX TM50 of 6 weeks of age. This small discrepancy may be explained by the high degree of variability in expression levels between birth and 5 months of age (8).
In addition to PNA, we found that TMP CL/F was inversely related to SCR, which is a surrogate endpoint for evaluating renal function. This relationship is consistent with the primary route of TMP elimination and with previous information in the literature reporting a correlation between SCR and TMP plasma levels in children and adults (23). These results suggest the need to adjust TMP dosing when renal function is impaired, as recommended in the product monograph (1).
An inverse exponential relationship between ALB and CL/F has not previously been reported in the literature. These findings are consistent with SMX's predominant elimination through metabolism (1). Given that the metabolic SMX CL/F is low (∼1 liter/h/70 kg) (1), SMX likely behaves as a low-extraction-ratio drug (24). This means that CL is primarily driven by the metabolizing capacity of the liver and by the free-drug fraction (24). A lower ALB will likely result in a higher free-drug fraction and, therefore, a higher CL.
Goodness-of-fit plots for the final TMP and SMX models suggested a certain degree of misspecification in the model (Fig. 2 and 3). For TMP and SMX concentrations greater than 5 mg/liter and 100 mg/liter, respectively, our model appears to underpredict plasma concentrations. This underprediction of concentrations at the upper end of the concentration range may be a result of the variability in drug absorption, which is inadequately accounted for in our model, including the differences between the absorption achieved with oral formulations and that achieved with enteral routes of administration. Thus, the peak concentrations predicted using our model should be interpreted cautiously. TMP-SMX's indications are broad, and dosing varies according to the indication (1). The opportunistic design of our study resulted in a wide spectrum of indications and dosing regimens. Despite these limitations, we believe that our PopPK model is useful in optimizing dosing for the treatment of bacterial infection because the surrogate PD parameter is the time that the plasma concentration exceeds the MIC and not Cmax,ss. However, dosing recommendations for the treatment of P. jirovecii infections are based on a Cmax,ss target and therefore need further validation, ideally, in children who receive high doses of TMP-SMX for this specific indication.
Another limitation of the TMP and SMX models is the high proportional residual error component (∼50%). Oral drug PK features are associated with high variability due to several factors, including food intake and maturation of absorption processes. Moreover, our study population was heterogeneous in terms of diagnoses and disease states. Another factor contributing to the high residual error is the potential imprecision regarding the time of drug administration associated with oral dosing. Lastly, the subjects received two types of oral formulations (tablets and suspension), and although this covariate was not statistically significant, this might have increased the variability. More specifically, the TMP-SMX oral liquid formulation is a suspension, meaning that parents must shake the bottle well before administering the dose. Failure to do so may have introduced variability in the dose administered.
Our TMP dose-exposure simulations indicate that the level of exposure in children in all age groups receiving 8 mg/kg/day of the TMP component divided into administration every 12 h matches the level of exposure in adults receiving 160 mg every 12 h (1 double-strength tablet twice daily), which is currently recommended for the treatment of urinary tract infections, shigellosis, acute otitis media, acute exacerbations of chronic bronchitis, and traveler's diarrhea in adults (1). This dosing is also recommended for the treatment of urinary tract infections and acute otitis media in children (1). However, recent literature suggests that common pathogens causing urinary tract infections and acute otitis media (Escherichia coli and Streptococcus pneumoniae, respectively) have high MIC values (MIC90, >2 mg/liter) (25, 26). Given that 8 mg/kg/day did not achieve the optimal target attainment rate for bacteria with MICs of 2 mg/liter, we propose a higher dosing regimen as empirical therapy when targeting E. coli and S. pneumoniae. Regarding acute otitis media, although TMP-SMX is labeled for its treatment, this drug is not recommended in clinical guidelines, which is consistent with our findings (27). For patients with nonfebrile urinary tract infections, TMP-SMX at 8 mg/kg/day is commonly used in clinical practice, and this dosing regimen may be adequate, given the renal excretion of TMP, resulting in high urinary TMP concentrations. However, for pyelonephritis, in which TMP tissue penetration is required, a higher dosing regimen may be necessary for adequate treatment.
On the contrary, 8 mg/kg/day every 12 h was associated with an optimal target attainment rate for bacteria with an MIC of ≤0.5 mg/liter. Given that the MIC90 for CA-MRSA is 0.5 mg/liter, 8 mg/kg/day every 12 h is optimal for oral therapy of CA-MRSA infections (28). These results are in line with those of a randomized controlled trial of clindamycin versus TMP-SMX for the treatment of uncomplicated small (≤5-cm) skin abscesses in 786 subjects, including 281 children (29). This trial showed that TMP-SMX was associated with cure rates (83.1%) similar to those achieved with clindamycin (81.7%; P = 0.73). Adverse events were less frequent with TMP-SMX (11.1%) than with clindamycin (21.9%). In this efficacy trial, children received TMP-SMX doses of 8 to 10/40 to 50 mg/kg/day divided into administration every 12 h, with the maximum adult dose being 160/800 mg of TMP-SMX twice a day. TMP dosing varied between 8 and 10 mg/kg/day to allow convenient dosing using the oral suspension of TMP-SMX at 8/40 mg/ml. Our recommendations are also in line with the Infectious Diseases Society of America guidelines suggesting TMP dosing of 8 to 10 mg/kg/day divided into administration every 12 h for the treatment of uncomplicated CA-MRSA cellulitis in children (4).
Our simulations indicate that TMP at 15 and 12 mg/kg/day every 12 h in children 0 to <6 and 6 to <21 years of age, respectively, matched the adult dose of 320 mg every 12 h (2 double-strength tablets twice daily), which is recommended for the same indications as 160 mg every 12 h (1). Although this dosing regimen is not indicated for use in children in the product monograph, our results suggest that this dosing regimen may be appropriate when treating infections caused by bacteria with an MIC of 1 mg/liter.
Assuming that the surrogate PD target for P. jirovecii is a TMP Cmax,ss of ≥3 mg/liter (30), we recommend a dosing regimen of 15 mg/kg/day of the TMP component every 12 h across age groups. Although this dosing regimen is not currently indicated in the product label, it had better target attainment rates than 15 mg/kg/day every 6 to 8 h, which is recommended in the label (Fig. 5) (1). The target attainment rates achieved with 20 mg/kg/day were also optimal; however, this higher daily dose was more frequently associated with a TMP Cmax,ss above the safety threshold than 15 mg/kg/day every 12 h was (up to 48% versus 7%, respectively), making the latter dosing regimen the preferred option. Before consideration of a change in current practice, the PD target for P. jirovecii should be confirmed, and the TMP PopPK model should be further validated for TMP concentrations ≥5 mg/liter, as discussed above.
The safety of TMP-SMX was not assessed in this study. Therefore, dosing recommendations in term infants 0 to 2 months of age are based on PK simulations. Additional studies are needed to better determine safety.
The PopPK of oral TMP-SMX in infants and children showed that PNA is associated with the CL/F of both components. In addition, the TMP CL/F and SMX CL/F were inversely related to SCR and ALB, respectively. For children 0 to 21 years of age, the exposure achieved by enteral administration of TMP-SMX at 8/40 mg/kg/day divided into administration every 12 h matched the exposure achieved in adults receiving TMP-SMX at 320/1,600 mg/day divided into administration every 12 h, and this dose is recommended for the treatment of infections caused by bacteria with an MIC ≤0.5 mg/liter in children. The exposure achieved by enteral administration of TMP-SMX at 12/60 mg/kg/day divided into administration every 12 h matched the exposure achieved in adults for a TMP-SMX dose of 640/3,200 mg/day divided into administration every 12 h and was optimal for the treatment of infections caused by bacteria with an MIC of 1 mg/liter in children 6 to <21 years of age. In younger children aged 0 to <6 years, the exposure achieved with TMP-SMX at 15/75 mg/kg/day divided into administration every 12 h matched that achieved with 640/3,200 mg/day divided into administration every 12 h in adults and was optimal for the treatment of infections caused by bacteria with an MIC of 1 mg/liter.
MATERIALS AND METHODS
Patient population.
PK samples used to develop the PopPK model were collected through the Pediatric Trials Network's (PTN′s) Pharmacokinetics of Understudied Drugs Administered to Children Per Standard of Care trial (PTN_POPS; ClinicalTrials.gov number NCT01431326; protocol NICHD-2011-POP01), a multicenter, prospective study of the PK and safety of understudied drugs administered to children (<21 years of age) per the standard of care. Subjects who received TMP-SMX per the standard of care as administered by their treating caregiver were eligible for enrollment. Only subjects who received oral TMP-SMX were included in this PopPK analysis. Exclusion criteria included failure to obtain consent or assent or a known pregnancy, as determined by interview or testing, if available. PK samples were collected optimally from standard-of-care laboratory collections or at different times from standard-of-care collections if allowed per the consent. Because this was a standard-of-care study, dosing and PK sample collection times varied between subjects. Standard-of-care laboratory assessments (e.g., basic metabolic panels) were recorded if samples were collected within 72 h of a sampling dose (the last dose prior to biological sample collection) of the drug. GA was collected for infants <120 days of PNA. Subjects were enrolled in the study for up to 90 days. This study protocol was reviewed and approved by the institutional review board of each participating institution (Ann and Robert H. Lurie Children's Hospital of Chicago, University of Louisville Kosair Children's Hospital, Medical University of South Carolina Children's Hospital, Oregon Health and Science University, University of Maryland, Riley Hospital for Children at Indiana University, Wesley Medical Center, Seattle Children's Hospital, University of California San Diego Medical Center, Children's National Medical Center, University of Utah Hospitals and Clinics, Axis Clinical Trials, Duke University Medical Center, Alfred I. DuPont Hospital for Children, Arkansas Children's Hospital Research Institute, University of North Carolina at Chapel Hill, and Yale University School of Medicine.
Drug dosing and sample collection.
Dosing information was collected for up to 8 doses prior to the sampling dose. As this study used an opportunistic study design, the timing of blood sample collection was dependent on standard-of-care laboratory assessments. However, parents and/or guardians were also given the option to allow sample collection at times different from those for the standard-of-care laboratory assessments. In this case, PK samples were collected at time points according to the frequency of administration of the study drug (see Table S2 in the supplemental material).
Analytical methods.
Blood was collected (0.2 ml from subjects <1 year of age and 2 ml from subjects ≥1 year of age) in a Microtainer tube with K2 EDTA and was processed into plasma immediately before it was frozen at the study sites. PK samples were sent to the PTN central laboratory (OpAns, LLC, Durham, NC, USA) for storage and analysis. TMP and SMX concentrations were quantified using validated liquid chromatography-tandem mass spectrometry assays (31). The chromatography system and mass spectrometer used for sample analysis were an Agilent 1200 series high-performance liquid chromatography system and an Agilent 6410 series triple-quadrupole system, respectively. An Agilent Zorbax XDB-C8 column (2.1 by 30 μm; inside diameter, 3.5 μm; Agilent) and a gradient mobile phase (water containing 0.1% [vol/vol] formic acid, methanol containing 0.1% [vol/vol] formic acid) were used. The TMP validation range for the assay was 25 to 50,000 ng/ml. The TMP LLOQ was 25 ng/ml. The SMX validation range for the assay was 250 to 500,000 ng/ml. The SMX LLOQ was 250 ng/ml. For both the TMP and the SMX assays, the accuracy and precision assessed were within FDA bioanalytical assay validation criteria (e.g., ± 15%).
Statistical analysis.
Using the value at the time of the first recorded dose, the median and range were calculated for demographic and dosing variables. With the exception of the PK modeling, all statistical analyses were performed using Stata (version 13.1) software (StataCorp, College Station, TX, USA).
Population PK model development.
TMP and SMX plasma PK data collected following oral administration were analyzed with a nonlinear mixed-effects modeling approach using the software NONMEM (version 7.2; Icon Development Solutions, Ellicott City, MD, USA). Given that BLQ approximately represented ∼≤10% of the data set, all TMP and SMX concentrations for samples with concentrations BLQ were imputed using LLOQ/2 (12.5 ng/ml and 125 ng/ml for TMP and SMX, respectively) (32). A separate PopPK analysis for each drug was performed. The first-order conditional estimation method with an eta-epsilon interaction was used for all model runs. Run management was performed using Pirana (version 2.8.2) software (33). Bootstrap analyses were performed with Perl-speaks-NONMEM (version 3.7.6) software (34). Data manipulation and visualization were performed using the packages Xpose in the software R (version 3.0.3; R Foundation for Statistical Computing, Vienna, Austria) and RStudio (version 0.97.551; RStudio, Boston, MA, USA) (35–37).
One- and two-compartment PK models with assumed linear PK were explored (10). Different absorption models—including a first-order absorption model, a Tlag model, a model with a sigmoidal Emax function (to describe the time dependency in the absorption rate constant), a model with a Weibull function, and a transit compartment model—were assessed (38, 39). The impact of the drug formulation (tablet versus oral suspension) and route of administration (by oral versus other enteral routes) on the first-order Ka was also explored using the following equations:
| (1) |
| (2) |
where θPop,j is the population value for Ka, and θDFRM and θDRTE are the parameters representing the effect of the oral formulation and the route of administration, respectively. DFRM is equal to 0 if the formulation was a liquid suspension and to 1 if the formulation was a tablet; and DRTE is equal to 0 if the drug was administered orally and to 1 if the drug was administered by other enteral routes (by the nasogastric, orogastric, or nasojejunal route, by jejunostomy, etc.).
Between-subject variability in the PK model parameters was assessed using an exponential relationship (equation 3):
| (3) |
where PARij denotes the estimate of parameter j in the ith individual, θPop,j is the population value for parameter j, and ηij denotes the deviation from the average population value for parameter j in the ith individual with a mean of zero and a variance of ω2.
The correlation between random-effect parameters (ρ) was calculated according to equation 4, where ωCL,V denotes the off-diagonal element of the variance covariance matrix of CL/F and V/F parameters and ω2CL and ω2V are the variance estimates for the IIV in CL/F and V/F, respectively.
| (4) |
Proportional, additive, and combined (additive plus proportional) residual error models were explored.
Covariate analysis.
Total body weight (WT) was assumed to be a significant covariate for CL/F and V/F and was included in the base model. The relationship between WT and PK parameters was characterized using a fixed-exponent (0.75 and 1) allometric and linear relationship for the parameters CL and V (scaled to a 70-kg standardized WT), respectively:
| (5) |
| (6) |
where CL/Fstd and V/Fstd represent population estimates of CL/F and V/F in a 70-kg adult, respectively, and WTi denotes the body weight for the ith subject. Once the final model was developed, the exponent of the relationship between WT and the PK parameters was estimated.
To assess whether the use of other indirect measures of body size—namely, BMI, FFM, and LBW—resulted in superior model performance, we tested these in place of WT before the covariate analysis (40–42). If the height measurement was missing, the 50th percentile value of height for weight and sex was imputed (43).
Other covariates were tested for model inclusion. Determination of which covariates to test for model inclusion was assessed on the basis of physiological relevance and by visual inspection of scatter and box plots (continuous and categorical variables, respectively) of the individual deviations from the values of the PK parameters typical for the population (eta) against covariates. The following covariates were explored: postmenstrual age (PMA), PNA, alanine aminotransferase concentration, aspartate aminotransferase concentration, SCR, bilirubin (total and direct) concentration, ALB, obese status (defined as a BMI ≥95th percentile in children ≥2 years of age), ethnicity, and sex (44).
The relationship between age and CL/F was characterized using a sigmoidal Emax maturation function, as shown in equation 7. As a measure of age, PNA and PMA were explored.
| (7) |
where Fage denotes clearance as a fraction of the adult clearance value, TM50 represents the age (in years for PNA and weeks for PMA) when 50% of the clearance achieved in adults is reached, and Hill is a slope parameter for the sigmoidal maturation model. This relationship was first tested by estimating TM50 and Hill and then by fixing the value of Hill to 1.
With the exception of WT and age, other continuous covariates were normalized to the population median value (equations 8 and 9), whereas for dichotomous categorical covariates, the relationship shown in equation 10 was used.
| (8) |
| (9) |
| (10) |
where PARij denotes the estimate of parameter j in the ith individual, θPop,j is the population value for parameter j, covi denotes the individual covariate value, covm is the population median covariate value, θcov is a parameter that represents the covariate effect, and categorical is a dichotomous categorical variable that can take a value of 0 or 1. For all continuous covariates except SCR, missing data values were imputed using the median value for the sample by group age (0 to 30 days, 31 days to <2 years, 2 to <13 years, 13 to <16 years, or 16 to <21 years). To compensate for missing SCR data, a univariable linear regression between PNA (the predictor) and SCR (the dependent variable) was performed for all subjects with available data and for subjects with an SCR of <1.8 mg/dl. For subjects with more than one SCR value, we used the median SCR for that subject through the study period in the regression. For each subject with a missing SCR, we imputed the predicted SCR based on their PNA.
A forward inclusion (P < 0.05 and change in OFV [ΔOFV] > 3.8) and backward elimination (P < 0.01 and ΔOFV >6.6) approach was used to evaluate statistical significance in the covariate analysis.
Population PK model evaluation.
Standard model diagnostic methods were used and included successful minimization, diagnostic plots, plausibility, and the precision of parameter estimates, as well as OFV and shrinkage values. The generated diagnostic plots included individual predictions and population predictions (PRED) versus observations and conditional weighted residuals (CWRES) versus PRED and time.
Parameter precision for the final PopPK model was evaluated using nonparametric bootstrapping (1,000 replicates) to generate the 95% confidence intervals for parameter estimates. SVPCs were performed, whereby the base and final models were used to generate 1,000 Monte Carlo simulation replicates of TMP and SMX concentration measurements per time point, and the results observed in the simulation were compared with those observed in the study (45). An SVPC was preferred to the commonly used visual predictive check because of the opportunistic design of the study, resulting in variable dosages and times of PK sampling across subjects (45). The percentile of each subject's observation in the marginal distribution of model-simulated endpoints (Pi,j) as a function of time and dosing was estimated, using the subject's individual time and dosing. Observations outside the 95% prediction interval were then estimated.
| (11) |
where Pij is the percentile of the jth observation for the ith subject and δij,n is equal to 1 if C′obs,ij is greater than C′obs,ij,n (otherwise, δij,n is equal to 0), where C′obs,ij is the jth observed concentration for the ith individual and C′obs,ij,n is the nth simulated concentration corresponding to C′obs,ij.
The dosing and covariate values used to generate the simulations in the SVPC were the same as those used in the study population.
Dosing simulations.
Virtual pediatric populations were created using the population module of the PK-Sim (version 5.5) program (Bayer Technology Services, Leverkusen, Germany). Five groups according to PNA (ages, 0 to <2 years, 2 to <6 years, 6 to <13 years, 13 to <16 years, and 16 to <21 years) were selected, with each group consisting of 500 subjects. The weight, race, and sex of the simulated subjects were generated using the distribution of weight, race, and sex of the subjects in the observed data. SCR was predicted on the basis of PNA using the linear regression equation derived from the original data set used for model development. ALB was set to the median ALB in each age group in the original data set.
TMP-SMX dosing is based on the TMP component because SMX concentrations have not been associated with efficacy or toxicity (46). The TMP PopPK model was therefore used to evaluate the dose-exposure relationship. Parameter estimates generated for virtual subjects using the final TMP PopPK model were used to perform the dosing simulations. The pediatric dosing regimens recommended in the package insert and those recommended in clinical practice guidelines were simulated (1, 4). For the dosing regimens of 8 and 12 mg/kg/day for the TMP component, the maximum dose was set as the corresponding dose recommended for adults (Table S3) (1, 4, 47). For the regimens of 15 to 20 mg/kg/day for the TMP component, the maximum dosing regimen was set as the dose reached in adults (53 to 64 kg), given the available tablet strengths (80 and 160 mg).
The following TMP parameters were calculated for each virtual subject: total drug steady-state area under the concentration-versus-time curve from time zero to the end of τ (AUC0–τ,ss; where τ denotes the dosing interval), the total maximal drug concentration at steady state (Cmax,ss), the total minimum concentration at steady state (Cmin,ss), and the total concentration at 50% of the dosing interval at steady state (C50,ss). The model equations are shown in equations 12 to 16:
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
where Ke denotes the first-order elimination rate constant, calculated as (CL/F)/(V/F); Ka denotes the absorption rate constant; Tmax,ss is the time of the maximal steady-state concentration; and τ is the dosing interval (6 to 12 h in dosing simulations). F denotes bioavailability and was assumed to be complete (i.e., 100%) (16).
Simulation results were compared to (i) the exposures achieved in adult and pediatric populations with various infections and published in the literature and (ii) simulated exposures in adult subjects (age, 18 to <21 years) weighing 70 kg. Given that the antibacterial activity of TMP is time dependent, we compared the free C50,ss to a range of MIC values for the relevant bacteria (S. pneumoniae, E. coli, and CA-MRSA) (25, 26, 28, 48). Free (unbound fraction) TMP C50,ss values were calculated by assuming an unbound fraction of 55% (1). No MIC value for P. jirovecii is available in the literature because this fungus cannot be cultured in vitro. A target TMP Cmax,ss of ≥3 mg/liter was used, and target attainment was calculated (30).
The TMP or SMX dose that either is associated with the symptoms of an overdosage or is likely to be life-threatening has not been reported. However, the safety target was defined to be a TMP Cmax,ss of 8 mg/liter because concentrations exceeding this threshold have been associated with an increased risk of anemia and neutropenia (30, 49). Other adverse events (gastrointestinal disorders, rash, fever, and liver function abnormalities) have been reported to be independent of the plasma drug concentration (49).
Supplementary Material
ACKNOWLEDGMENTS
The assay measuring trimethoprim and sulfamethoxazole concentrations was performed at OpAns, LLC, Durham, NC, by Alan Cunningham (analyst), Christine Grosse (responsible scientist), and Kenneth Lewis (chief executive officer).
The PTN Publications Committee consists of Gary Furda, Duke Clinical Research Institute, Durham, NC; Danny Benjamin, Duke Clinical Research Institute, Durham, NC; Edmund Capparelli, University of California San Diego, San Diego, CA; Gregory L. Kearns, Arkansas Children's Hospital Research Institute, Little Rock, AR; Ian M. Paul, Penn State College of Medicine, Hershey, PA; Christoph Hornik, Duke Clinical Research Institute, Durham, NC; and Kelly Wade, Children's Hospital of Philadelphia, Philadelphia, PA. The Eunice Kennedy Shriver National Institute of Child Health and Human Development was represented by David Siegel and Anne Zajicek. The Emmes Corporation (the data coordinating center) was represented by Ravinder Anand and Gina Simone. The Pediatric Trials Network trimethoprim-sulfamethoxazole study team, principal investigators (PIs), and study coordinators (SCs) are as follows: at the Duke Clinical Research Institute, Tammy Day (CRA); at the Université de Montréal, Julie Autmizguine (PI); at The University of North Carolina at Chapel Hill, Daniel Gonzalez; at Ann and Robert H. Lurie Children's Hospital, Ram Yogev (site PI), Laura Fearn (SC), Rohit Kalra (SC), Mayra Gomez (SC), and Bemajin Traisman (SC); at the University of Louisville, Norton Children's Hospital, and Kosair Charities Pediatric Clinical Research Unit, Janice Sulliver (site PI), Jen Comings (SC), Jackie Perry (SC), and Michelle Wiseheart (SC); at the Medical University of South Carolina Children's Hospital, Andrew Atz (site PI) and Layla Al Sarraf (SC); at the Oregon Health and Science University, Amira Al-Uzri (site PI), Kira Clark (SC), Carrie Farrar (SC), and Connie Swanson (SC); at the University of Maryland, Susan Mendley (site PI) and Donna Cannonier (SC); at the Riley Hospital for Children at Indiana University, Brenda Poindexter (site PI), Susan Gunn (SC), and Dianne Wilson (SC); at the Wesley Medical Center, Paula Delmore (site PI and SC); at Seattle Children's Hospital, Joseph Flynn (site PI), Shannon Granillo (SC), Rob Johnson (SC), and Megan Kelton-Rehkopf (SC); at the University of California San Diego Medical Center, Adriana Tremoulet (site PI) and Baharin Abdullah (SC); at Children's Mercy Hospital, Kathy Neville (site PI) and Jaylene Weigel (SC); at Children's National Medical Center, John Van Den Anker (site PI) and Elaine Williams (SC); at the University of Utah Hospitals and Clinics, Catherine Sherwin (site PI), Fumiko Alger (SC), Sharada Dixit (SC), JoAnn Narus (SC), Rebbecca Perez (SC), Priscilla Rosen (SC), and Yakub Salman (SC); at Axis Clinical Trials, Lydie Hazan (site PI) and Angelica Covarrubias (SC); at the Duke University Medical Center, Kevin Watt (site PI), Christie Milleson (SC), and Samantha Wrenn (SC); at the Alfred I. DuPont Hospital for Children, Marisa Meyer (site PI) and Ramany John (SC); at the Arkansas Children's Hospital Research Institute, Laura James (site PI), Lee Howard (SC), D. Ann Pierce (SC), and Kristin Richmond (SC); at the University of North Carolina at Chapel Hill, Matthew Laughon (site PI), Janice Bernhardt (SC), and Ashley Mariconti (SC); and at the Yale University School of Medicine, Matthew Bizarro (site PI).
This work was funded under National Institute of Child Health and Human Development (NICHD) contract HHSN275201000003I for the Pediatric Trials Network (PI, Danny Benjamin).
S.D. receives salary support from NIH/NIGMS (T32GM086330). M.C.-W. receives support for research from the NIH (1R01-HD076676-01A1), NIAID (HHSN272201500006I and HHSN272201300017I), NICHD (HHSN275201000003I), the Biomedical Advanced Research and Development Authority (HHSO100201300009C), and industry for drug development in adults and children (all disclosures for M.C.-W. are available at www.dcri.duke.edu/research/coi.jsp). D.G. receives support for research from NICHD (K23HD083465), the nonprofit organization Thrasher Research Fund (www.thrasherresearch.org), and industry (Cempra, Inc., and Jacobus Pharmaceutical Company, Inc.) for drug development in adults and children. The remaining authors have no relevant conflicts to disclose. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH.
Julie Autmizguine and Daniel Gonzalez wrote the manuscript; Michael Cohen-Wolkowiez designed the research; Julie Autmizguine (primary author), Chiara Melloni (study PI), Christoph P. Hornik (PTN PI), Samantha Dallefield (fellow), Barrie Harper (project leader), Ram Yogev (site PI), Janice E. Sullivan (site PI), Andrew M. Atz (site PI), Amira Al-Uzri (site PI), Susan Mendley (site PI), Brenda Poindexter (site PI), Jeff Mitchell (clinical trial manager), Andrew Lewandowski (statistician), Paula Delmore (site PI), and Michael Cohen-Wolkowiez (protocol PI) performed the research; and Julie Autmizguine, Daniel Gonzalez, and Michael Cohen-Wolkowiez analyzed the data.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01813-17.
REFERENCES
- 1.Scientific Inc AR. 2013. Bactrim (sulfamethoxazole and trimethoprim) package insert. Scientific Inc AR, Philadelphia, PA. [Google Scholar]
- 2.Gerber JS, Coffin SE, Smathers SA, Zaoutis TE. 2009. Trends in the incidence of methicillin-resistant Staphylococcus aureus infection in children's hospitals in the United States. Clin Infect Dis 49:65–71. doi: 10.1086/599348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iwamoto M, Mu Y, Lynfield R, Bulens SN, Nadle J, Aragon D, Petit S, Ray SM, Harrison LH, Dumyati G, Townes JM, Schaffner W, Gorwitz RJ, Lessa FC. 2013. Trends in invasive methicillin-resistant Staphylococcus aureus infections. Pediatrics 132:e817–e824. doi: 10.1542/peds.2013-1112. [DOI] [PubMed] [Google Scholar]
- 4.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, Kaplan SL, Karchmer AW, Levine DP, Murray BE, Rybak JM, Talan DA, Chambers HF. 2011. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis 52:285–292. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 5.Kremers P, Duvivier J, Heusghem C. 1974. Pharmacokinetic studies of co-trimoxazole in man after single and repeated doses. J Clin Pharmacol 14:112–117. doi: 10.1002/j.1552-4604.1974.tb02300.x. [DOI] [PubMed] [Google Scholar]
- 6.Siber GR, Gorham CC, Ericson JF, Smith AL. 1982. Pharmacokinetics of intravenous trimethoprim-sulfamethoxazole in children and adults with normal and impaired renal function. Rev Infect Dis 4:566–578. doi: 10.1093/clinids/4.2.566. [DOI] [PubMed] [Google Scholar]
- 7.Kagaya H, Miura M, Niioka T, Saito M, Numakura K, Habuchi T, Satoh S. 2012. Influence of NAT2 polymorphisms on sulfamethoxazole pharmacokinetics in renal transplant recipients. Antimicrob Agents Chemother 56:825–829. doi: 10.1128/AAC.05037-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koukouritaki SB, Manro JR, Marsh SA, Stevens JC, Rettie AE, McCarver DG, Hines RN. 2004. Developmental expression of human hepatic CYP2C9 and CYP2C19. J Pharmacol Exp Ther 308:965–974. [DOI] [PubMed] [Google Scholar]
- 9.Rhodin MM, Anderson BJ, Peters AM, Coulthard MG, Wilkins B, Cole M, Chatelut E, Grubb A, Veal GJ, Keir MJ, Holford NH. 2009. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol 24:67–76. doi: 10.1007/s00467-008-0997-5. [DOI] [PubMed] [Google Scholar]
- 10.Jelliffe RW, Gomis P, Tahani B, Ruskin J, Sattler FR. 1997. A population pharmacokinetic model of trimethoprim in patients with pneumocystis pneumonia, made with parametric and nonparametric methods. Ther Drug Monit 19:450–459. doi: 10.1097/00007691-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 11.Nolte H, Buttner H. 1973. Pharmacokinetics of trimethoprim and its combination with sulfamethoxazole in man after single and chronic oral administration. Chemotherapy 18:274–284. doi: 10.1159/000221272. [DOI] [PubMed] [Google Scholar]
- 12.Welling PG, Craig WA, Amidon GL, Kunin CM. 1973. Pharmacokinetics of trimethoprim and sulfamethoxazole in normal subjects and in patients with renal failure. J Infect Dis 128(Suppl):556–566. [DOI] [PubMed] [Google Scholar]
- 13.Hess MM, Boucher BA, Laizure SC, Stevens RC, Sanders PL, Janning SW, Croce MA, Fabian TC. 1993. Trimethoprim-sulfamethoxazole pharmacokinetics in trauma patients. Pharmacotherapy 13:602–606. [PubMed] [Google Scholar]
- 14.Lares-Asseff I, Villegas F, Perez G, Toledo A, Camacho A, Lopez DC. 1996. Kinetic effects of trimethoprim-sulfamethoxazole in children with biliary atresia: a new dosing regimen. Arch Med Res 27:183–190. [PubMed] [Google Scholar]
- 15.Alsaad N, van Altena R, Pranger AD, van Soolingen D, de Lange WC, van der Werf TS, Kosterink JG, Alffenaar JW. 2013. Evaluation of co-trimoxazole in the treatment of multidrug-resistant tuberculosis. Eur Respir J 42:504–512. doi: 10.1183/09031936.00114812. [DOI] [PubMed] [Google Scholar]
- 16.Klepser ME, Zhu Z, Nicolau DP, Banevicius MA, Belliveau PP, Ross JW, Broisman L, Quintiliani R, Nightingale CH. 1996. Oral absorption of trimethoprim-sulfamethoxazole in patients with AIDS. Pharmacotherapy 16:656–662. [PubMed] [Google Scholar]
- 17.Stevens RC, Laizure SC, Sanders PL, Stein DS. 1993. Multiple-dose pharmacokinetics of 12 milligrams of trimethoprim and 60 milligrams of sulfamethoxazole per kilogram of body weight per day in healthy volunteers. Antimicrob Agents Chemother 37:448–452. doi: 10.1128/AAC.37.3.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varoquaux O, Lajoie D, Gobert C, Cordonnier P, Ducreuzet C, Pays M, Advenier C. 1985. Pharmacokinetics of the trimethoprim-sulphamethoxazole combination in the elderly. Br J Clin Pharmacol 20:575–581. doi: 10.1111/j.1365-2125.1985.tb05114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stevens RC, Laizure SC, Williams CL, Stein DS. 1991. Pharmacokinetics and adverse effects of 20-mg/kg/day trimethoprim and 100-mg/kg/day sulfamethoxazole in healthy adult subjects. Antimicrob Agents Chemother 35:1884–1890. doi: 10.1128/AAC.35.9.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson BJ, Holford NH. 2008. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 48:303–332. doi: 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 21.Smyth JM, Collier PS, Darwish M, Millership JS, Halliday HL, Petersen S, McElnay JC. 2004. Intravenous indometacin in preterm infants with symptomatic patent ductus arteriosus. A population pharmacokinetic study. Br J Clin Pharmacol 58:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson TN, Rostami-Hodjegan A, Tucker GT. 2006. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet 45:931–956. doi: 10.2165/00003088-200645090-00005. [DOI] [PubMed] [Google Scholar]
- 23.Klinker H, Langmann P, Zilly M, Richter E. 1998. Drug monitoring during the treatment of AIDS-associated Pneumocystis carinii pneumonia with trimethoprim-sulfamethoxazole. J Clin Pharm Ther 23:149–154. doi: 10.1046/j.1365-2710.1998.00152.x. [DOI] [PubMed] [Google Scholar]
- 24.Rowland M. 1984. Protein binding and drug clearance. Clin Pharmacokinet 9(Suppl 1):S10–S17. [DOI] [PubMed] [Google Scholar]
- 25.Chin TL, MacGowan AP, Bowker KE, Elder F, Beck CR, McNulty C. 2015. Prevalence of antibiotic resistance in Escherichia coli isolated from urine samples routinely referred by general practitioners in a large urban centre in south-west England. J Antimicrob Chemother 70:2167–2169. doi: 10.1093/jac/dkv050. [DOI] [PubMed] [Google Scholar]
- 26.Pfaller MA, Farrell DJ, Sader HS, Jones RN. 2012. AWARE ceftaroline surveillance program (2008-2010): trends in resistance patterns among Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis in the United States. Clin Infect Dis 55(Suppl 3):S187–S193. doi: 10.1093/cid/cis561. [DOI] [PubMed] [Google Scholar]
- 27.Lieberthal AS, Carroll AE, Chonmaitree T, Ganiats TG, Hoberman A, Jackson MA, Joffe MD, Miller DT, Rosenfeld RM, Sevilla XD, Schwartz RH, Thomas PA, Tunkel DE. 2013. The diagnosis and management of acute otitis media. Pediatrics 131:e964–e999. doi: 10.1542/peds.2012-3488. [DOI] [PubMed] [Google Scholar]
- 28.Mendes RE, Moet GJ, Janechek MJ, Jones RN. 2010. In vitro activity of telavancin against a contemporary worldwide collection of Staphylococcus aureus isolates. Antimicrob Agents Chemother 54:2704–2706. doi: 10.1128/AAC.00301-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daum RS, Miller LG, Immergluck L, Fritz S, Creech CB, Young D, Kumar N, Downing M, Pettibone S, Hoagland R, Eells SJ, Boyle MG, Parker TC, Chambers HF. 2017. A placebo-controlled trial of antibiotics for smaller skin abscesses. N Engl J Med 376:2545–2555. doi: 10.1056/NEJMoa1607033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kovacs JA, Masur H. 1988. Pneumocystis carinii pneumonia: therapy and prophylaxis. J Infect Dis 158:254–259. doi: 10.1093/infdis/158.1.254. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez D, Melloni C, Poindexter BB, Yogev R, Atz AM, Sullivan JE, Mendley SR, Delmore P, Delinsky A, Zimmerman K, Lewandowski A, Harper B, Lewis KC, Benjamin DK Jr, Cohen-Wolkowiez M. 2015. Simultaneous determination of trimethoprim and sulfamethoxazole in dried plasma and urine spots. Bioanalysis 7:1137–1149. doi: 10.4155/bio.15.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keizer RJ, Jansen RS, Rosing H, Thijssen B, Beijnen JH, Schellens JH, Huitema AD. 2015. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol Res Perspect 3:e00131. doi: 10.1002/prp2.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keizer RJ, van Benten M, Beijnen JH, Schellens JH, Huitema AD. 2011. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed 101:72–79. doi: 10.1016/j.cmpb.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 34.Lindbom L, Pihlgren P, Jonsson EN. 2005. PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79:241–257. doi: 10.1016/j.cmpb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Sarkar D. 2008. Lattice: multivariate data visualization with R, 1st ed Springer-Verlag New York, New York, NY. [Google Scholar]
- 36.Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer-Verlag New York, New York, NY. [Google Scholar]
- 37.Jonsson EN, Karlsson MO. 1999. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64. [DOI] [PubMed] [Google Scholar]
- 38.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. 2007. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn 34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 39.Zhou H. 2003. Pharmacokinetic strategies in deciphering atypical drug absorption profiles. J Clin Pharmacol 43:211–227. doi: 10.1177/0091270002250613. [DOI] [PubMed] [Google Scholar]
- 40.Janmahasatian S, Duffull SB, Ash S, Ward LC, Byrne NM, Green B. 2005. Quantification of lean bodyweight. Clin Pharmacokinet 44:1051–1065. doi: 10.2165/00003088-200544100-00004. [DOI] [PubMed] [Google Scholar]
- 41.Peters AM, Snelling HL, Glass DM, Bird NJ. 2011. Estimation of lean body mass in children. Br J Anaesth 106:719–723. doi: 10.1093/bja/aer057. [DOI] [PubMed] [Google Scholar]
- 42.Holford N. 2010. Dosing in children. Clin Pharmacol Ther 87:367–370. doi: 10.1038/clpt.2009.262. [DOI] [PubMed] [Google Scholar]
- 43.Centers for Disease Control and Prevention, National Center for Health Statistics. 2001. Clinical growth charts. Centers for Disease Control and Prevention, National Center for Health Statistics, Atlanta, GA: https://www.cdc.gov/growthcharts/clinical_charts.htm Accessed 3 April 2017. [Google Scholar]
- 44.Barlow SE. 2007. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 120(Suppl 4):S164–S192. doi: 10.1542/peds.2007-2329C. [DOI] [PubMed] [Google Scholar]
- 45.Wang DD, Zhang S. 2012. Standardized visual predictive check versus visual predictive check for model evaluation. J Clin Pharmacol 52:39–54. doi: 10.1177/0091270010390040. [DOI] [PubMed] [Google Scholar]
- 46.Joos B, Blaser J, Opravil M, Chave JP, Luthy R. 1995. Monitoring of co-trimoxazole concentrations in serum during treatment of Pneumocystis carinii pneumonia. Antimicrob Agents Chemother 39:2661–2666. doi: 10.1128/AAC.39.12.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller LG, Daum RS, Creech CB, Young D, Downing MD, Eells SJ, Pettibone S, Hoagland RJ, Chambers HF. 2015. Clindamycin versus trimethoprim-sulfamethoxazole for uncomplicated skin infections. N Engl J Med 372:1093–1103. doi: 10.1056/NEJMoa1403789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clinical and Laboratory Standards Institute. 2015. Performance standards for antimicrobial susceptibility testing, 26th ed Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 49.Hughes WT, LaFon SW, Scott JD, Masur H. 1995. Adverse events associated with trimethoprim-sulfamethoxazole and atovaquone during the treatment of AIDS-related Pneumocystis carinii pneumonia. J Infect Dis 171:1295–1301. doi: 10.1093/infdis/171.5.1295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.