

ABSTRACT
Vancomycin-resistant Enterococcus faecium strains (VREfm) are critical public health concerns because they are among the leading causes of hospital-acquired bloodstream infections. Chlorhexidine (CHX) is a bisbiguanide cationic antiseptic that is routinely used for patient bathing and other infection control practices. VREfm are likely frequently exposed to CHX; however, the long-term effects of CHX exposure have not been studied in enterococci. In this study, we serially exposed VREfm to increasing concentrations of CHX for a period of 21 days in two independent experimental evolution trials. Reduced CHX susceptibility emerged (4-fold shift in CHX MIC). Subpopulations with reduced daptomycin (DAP) susceptibility were detected, which were further analyzed by genome sequencing and lipidomic analysis. Across the trials, we identified adaptive changes in genes with predicted or experimentally confirmed roles in chlorhexidine susceptibility (efrE), global nutritional stress response (relA), nucleotide metabolism (cmk), phosphate acquisition (phoU), and glycolipid biosynthesis (bgsB), among others. Moreover, significant alterations in membrane phospholipids were identified for some populations with reduced DAP susceptibility. Our results are clinically significant because they identify a link between serial subinhibitory CHX exposure and reduced DAP susceptibility. In addition, the CHX-induced genetic and lipidomic changes described in this study offer new insights into the mechanisms underlying the emergence of antibiotic resistance in VREfm.
KEYWORDS: Enterococcus, chlorhexidine, daptomycin
INTRODUCTION
Enterococcus faecium is a Gram-positive bacterium that normally colonizes the human gastrointestinal tract and is an opportunistic pathogen associated with bacteremia, urinary tract infections, endocarditis, and wound infections (1–3). Vancomycin-resistant E. faecium strains (VREfm) are of particular concern for infection treatment. VREfm are among the primary etiological agents of central line-associated bloodstream infections (CLABSIs), a type of health care-associated infection (HAI) that arises from central venous catheter use and is associated with high mortality in the United States (4, 5). E. faecium contamination on indwelling venous catheters, surgical instruments, and hospital surfaces is challenging to eradicate (2, 6–8). In hospital and clinical settings, improper infection control practices, contaminated surfaces, and indiscriminate use of antibiotics contribute to the persistence of E. faecium (6, 9).
Chlorhexidine (CHX) is a cationic antiseptic and membrane-active antimicrobial (10–12). The primary mechanism of action of CHX is to disrupt the bacterial cell membrane and cause leakage of cytoplasmic contents and precipitation of cytoplasm (13–15). CHX is recommended by the Society for Healthcare Epidemiology of America to reduce CLABSI occurrence in acute care hospitals (16). Specifically, CHX bathing and CHX-impregnated cardiovascular catheters are used for CLABSI control (16–18). Clinical reports have raised concerns about the long-term effects of CHX bathing on hospital-associated pathogens (19–22). The CHX concentrations on patient skin can fall below the MIC for VREfm between bathings (22). Frequent exposure to subinhibitory CHX could select for VREfm mutants with reduced susceptibility to CHX and other antimicrobials that also interact with the bacterial cell surface. It was recently reported that colistin resistance emerged in the Gram-negative pathogen Klebsiella pneumoniae after exposure to CHX (23).
In a previous study, we used RNA sequencing to study the global transcriptomic responses of a VanA-type VREfm strain to CHX (24). We found that CHX exposure elicited expression of genes associated with antibiotic resistance and extracytoplasmic stress, including genes associated with vancomycin resistance (vanHAX) and reduced daptomycin (DAP) susceptibility (liaXYZ) (24). In the present study, we test the hypothesis that serial exposure to sub-MIC CHX selects for VREfm mutants with reduced susceptibilities to CHX and other membrane and cell wall-targeting antimicrobials, with particular focus on DAP.
RESULTS
In vitro evolution of reduced CHX susceptibility in VREfm.
Previous RNA sequencing analysis identified up to 118-fold upregulation of liaXYZ in CHX-treated E. faecium 1,231,410 (E. faecium 410) (24), a VanA-type vancomycin- and ampicillin-resistant blood isolate and member of the hospital-adapted clade A1 (25, 26). This was of interest because liaXYZ is protective against DAP (27–32). We confirmed the CHX-stimulated upregulation of liaX in E. faecium 410 and a commensal clade B strain, E. faecium 1,141,733 (26), using reverse transcription-quantitative PCR (RT-qPCR; see Fig. S1 in the supplemental material).
We hypothesized that repeat exposure to subinhibitory CHX could select for mutants with reduced susceptibility to CHX and concomitant reduced susceptibility to other antimicrobials. To test this, we performed in vitro serial passaging of E. faecium 410 with CHX for a period of 21 days, starting with a sub-MIC of 2.9 μg/ml (Fig. 1). Similar patterns of MIC shifts were observed for two independent trials over the course of 21 days. The CHX MICs of the evolved populations (referred to as populations A and B) (Table 1) recovered after one drug-free passage were confirmed to be increased (19.6 μg/ml) compared to the parental strain (Table 2). The CHX MIC was not altered in E. faecium 410 passaged for 21 days in medium without CHX. We conclude that reduced CHX susceptibility emerges in VREfm after serial in vitro CHX exposure.
FIG 1.
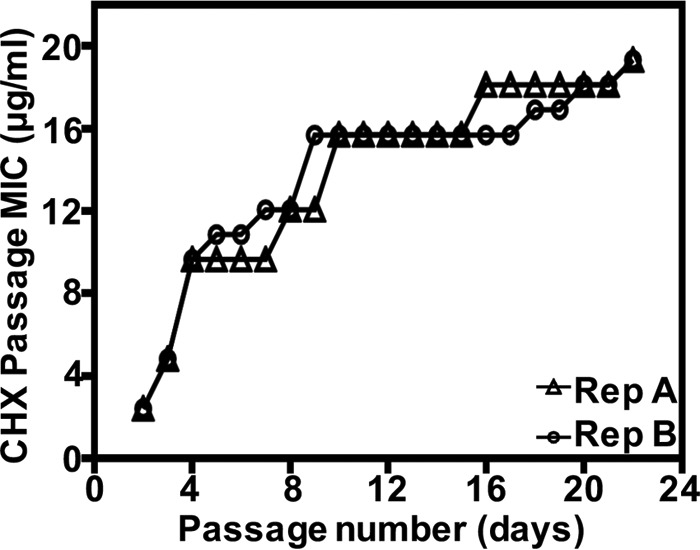
E. faecium adapts to CHX. In vitro evolution of reduced CHX susceptibility in E. faecium 1,231,410 (E. faecium 410) by serial passaging in increasing concentrations of CHX for a period of 21 days. CHX passage MIC (y axis) for each day of passage (x axis) is shown for two independent experiments (A and B).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Source or reference |
|---|---|---|
| Bacterial strains | ||
| E. faecium | ||
| 1,231,410 | Clade A skin and soft tissue infection isolate; VanA-type VRE; DAPs | 25, 26 |
| 1,141,733 | Clade B clinical isolate; VANs | 25, 26 |
| Population A | In vitro-evolved population with reduced CHX susceptibility from expt A, stocked from day 22 | This study |
| Population B | In vitro-evolved population with reduced CHX susceptibility from expt B, stocked from day 22 | This study |
| 410-P1 | E. faecium 410 in vitro-evolved population in BHI from day 21, trial 1 | This study |
| 410-P2 | E. faecium 410 in vitro-evolved population in BHI from day 21, trial 2 | This study |
| DAP-A1 | Mutants with reduced DAP susceptibility from population A | This study |
| DAP-A2 | Mutants with reduced DAP susceptibility from population A | This study |
| DAP-B1 | Mutants with reduced DAP susceptibility from population B | This study |
| DAP-B2 | Mutants with reduced DAP susceptibility from population B | This study |
| PB301 | E. faecium 410 EFTG_02287-88 deletion mutant | This study |
| PB302 | E. faecium DAP-A2 EFTG_02287-88 deletion mutant | This study |
| AH101 | PB301 transformed with empty pLZ12 plasmid | This study |
| AH102 | PB301 transformed with pAH201 | This study |
| PB303 | PB301 transformed with pPB202 | This study |
| E. coli EC1000 | E. coli cloning host; provides repA in trans; F− araD139(ara ABC-leu)7679 galU galK lacX74 rspL thi; repA of pWV01 in glgB; KANr | 74 |
| Plasmids | ||
| pLZ12 | E. coli and Streptococcus shuttle vector | 69 |
| pHA101 | Markerless counterselectable exchange plasmid | 24 |
| pAH201 | pLZ12 containing a 4.186-kb BamHI/BamHI-digested fragment with EFTG_02287-88 and predicted promoter | This study |
| pPB202 | pLZ12 containing a 4.186-kb BamHI/BamHI-digested fragment with EFTG_02287-88 and predicted promoter from strain DAP-A2 | This study |
| pPB301 | pHA101 containing a 1.998-kb EcoRI/BamHI-digested fragment with upstream and downstream regions flanking EFTG_02287-88 | This study |
VANs, vancomycin susceptible; DAPs, daptomycin susceptible; Km, kanamycin.
TABLE 2.
MIC values for cell wall-targeting antibiotics
| Strain or population | MIC (μg/ml)a |
|||
|---|---|---|---|---|
| CHX | DAP range, median (P) | AMP | VAN | |
| E. faecium 410 | 4.9 | 2, 2 (NA) | 195 | 250 |
| Population A | 19.6 | 2–6, 3 (0.0085) | 390 | 500 |
| Population B | 19.6 | 3–4, 3.5 (0.0006) | 195 | 500 |
| DAP-A1 | 19.6 | 3–6, 4 (0.0043) | 195 | 500 |
| DAP-A2 | 19.6 | 3–6, 4 (0.0015) | 390 | 1,000 |
| DAP-B1 | 19.6 | 3–4, 3.5 (0.0006) | 195 | 1,000 |
| DAP-B2 | 19.6 | 3–4, 3.5 (0.0006) | 195 | 1,000 |
| PB301 (E. faecium 410 ΔefrEF) | 1.2 | 2–3, 2 (0.211) | ND | ND |
| PB302 (DAP-A2 ΔefrEF) | 2.4 | 2–4, 3 (0.00188) | ND | ND |
DAP MICs were determined independently either seven times (E. faecium 1,231,410 and population A) or six times (all other determinations). A one-tailed unpaired Student t test was used to assess the significance (P) of the difference in MIC versus E. faecium 1,231,410. E. faecium 410, E. faecium 1,231,410; AMP, ampicillin; VAN, vancomycin; NA, not applicable; ND, not determined.
Populations A and B had variable but significantly higher DAP MICs relative to the wild-type strain (Table 2), with the DAP MIC in some experimental trials meeting the ≥4 μg/ml breakpoint for DAP resistance (33). The vancomycin MIC was 2-fold higher for both populations relative to the wild type, and ampicillin MIC was 2-fold higher for population A.
Reduced DAP susceptibility emerges during serial CHX exposure.
We sought to further quantify and investigate the basis for the elevated DAP MIC in the CHX-passaged populations. The CHX-passaged populations (A and B) were cultured on agar with or without 10 μg/ml DAP to quantify the CFU (Fig. 2). E. faecium 410 wild-type stock cultures (410-1 and 410-2) and E. faecium 410 passaged for 21 days in the absence of CHX (410-P1 and 410-P2) were used as controls. Subpopulations with reduced DAP susceptibility were detected in the CHX-passaged populations (Fig. 2).
FIG 2.
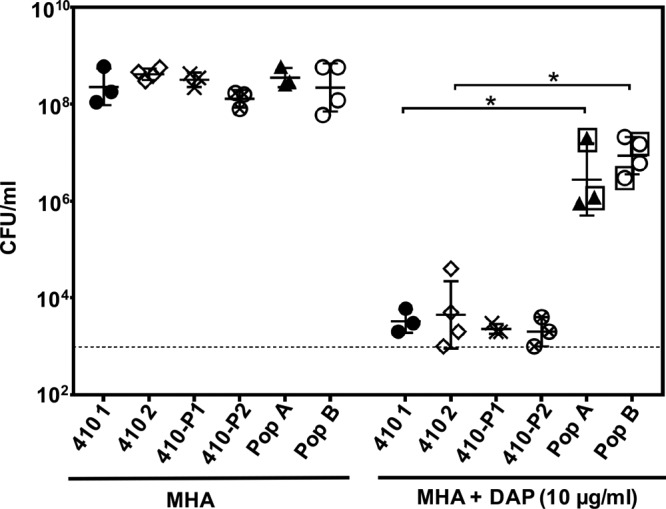
Reduced DAP susceptibility in CHX-passaged E. faecium populations A and B. The geometric mean and the geometric standard deviation of the CFU/ml count for n = 3 or 4 independent trials are shown. Strains and populations are described in the text. The dashed line represents limit of detection (103 CFU/ml). The boxed populations were analyzed by whole-genome sequencing. *, P < 0.05 (one-tailed Student t test).
Next, isolated colonies arising on DAP plates from each of two independent DAP plating trials were pooled and stocked for further analysis. These mutants are referred to here as DAP-A1 and DAP-A2 (from population A; 14 and 11 colonies were pooled, respectively) and DAP-B1 and DAP-B2 (from population B; 18 and 5 colonies were pooled, respectively). The DAP MICs of these mutants after two DAP-free passages were found to be variable but were significantly higher than the wild-type E. faecium 410 (Table 2). The DAP mutants, with the exception of DAP-B2, have significantly longer generation times than E. faecium 410 wild-type in BHI broth (see Table S2 in the supplemental material). No significant differences in spontaneous rifampin resistance frequencies were observed between the DAP mutants and wild-type E. faecium 410 (see Table S2 in the supplemental material), indicating that these strains are not hypermutators. RT-qPCR analysis identified a small but statistically significant increase in liaX expression in the DAP-A1 and DAP-A2 mutants but not the DAP-B1 and DAP-B2 mutants relative to the wild type (see Fig. S2 in the supplemental material). Recently, a two-component system that contributes to CHX tolerance, referred to as ChtRS, was identified in E. faecium (34). The ChtRS regulon is currently undefined. We identified a small (1.5-fold) but statistically significant increase in chtS transcript levels in the DAP-A2 mutant compared to wild-type E. faecium 410 (see Fig. S2 in the supplemental material).
Mutations in DAP strains.
Genome sequencing identified mutations occurring in the DAP strains relative to wild-type E. faecium 410 (Table 3). All four share a mutation in efrE (EFTG_02287), which encodes one subunit of the heterodimeric ABC transporter EfrEF (35). The mutation results in an Ala290Val substitution within the sixth transmembrane helix of EfrE, as predicted by TMHMM 2.0 (36). efrE is upregulated 22-fold by E. faecium 410 in response to CHX (24), and deletion of efrE from E. faecalis OG1RF confers increased susceptibility to CHX (37, 38). No mutations other than in efrE were common to all strains.
TABLE 3.
Mutations identified by whole-genome sequencing in the DAP-resistant mutants
| Mutant | Gene description | Nucleotide variation (frequency [%])a | Amino acid changeb | E. faecalis V583 ortholog |
|---|---|---|---|---|
| DAP-A1 | Eftg_02287 ABC transporter | C67008T (98.47) | Ala290Val | EF2226 |
| Eftg_01724 RelA/SpoT (relA) | CTAGGATTTAC30479Del (98.19) | Leu505fs | EF1974 | |
| Eftg_01135 alpha/beta hydrolase | G137748A (55.64) | Trp150* | EF1505 | |
| Eftg_00577 cytidylate kinase (cmk) promoter | A618699IS1251 | IS1251 insertion | EF1547 | |
| DAP-A2 | Eftg_02287 ABC transporter | C67008T (99.77) | Ala290Val | EF2226 |
| Eftg_01724 RelA/SpoT (relA) | CA29139AG (95.74) | Ala58Glu | EF1974 | |
| Eftg_01135 alpha/beta hydrolase | 137847G insertion (88.17) | Ile184fs | EF1505 | |
| Eftg_00577 cytidylate kinase (cmk) promoter | A618699IS1251 | IS1251 insertion | EF1547 | |
| DAP-B1 | Eftg_02287 ABC transporter | C67008T (98.962) | Ala290Val | EF2226 |
| Eftg_01175 PhoU regulator | G173845A (99.61) | Glu61Lys | EF1754 | |
| Eftg_02534 transposase | C71844A (99.47) | Lys206Asn | ||
| DAP-B2 | Eftg_02287 ABC transporter | C67008T (99.36) | Ala290Val | EF2226 |
| Eftg_02162 glycosyltransferase (bgsB) | C50223T (67.09) | Val57Met | EF2890 |
The mutation frequency in the read assembly was determined by variant detection in CLC Genomics Workbench where applicable. The precise location of insertion for variation A618699IS1251 was mapped by Sanger sequencing.
Asterisk indicates stop codon.
DAP-A1 and DAP-A2 share IS1251 insertions in the promoter region of cmk, which encodes cytidylate kinase (Table 3). Population heterogeneity in regard to IS1251 insertion at the cmk promoter was observed in the DAP-A1 and DAP-A2 mutants by gel electrophoresis analysis of cmk promoter amplicons (see Fig. S3 in the supplemental material). RT-qPCR confirmed significant downregulation of cmk transcript levels in the DAP-A mutants in three independent trials (see Fig. S3 in the supplemental material).
DAP-A1 and DAP-A2 each have different mutations in relA and in a gene encoding a predicted alpha/beta hydrolase (Table 3). In DAP-A1, the deletion of 11 bases (CTAGGATTTAC) from relA results in synthesis of a truncated, 505-amino-acid protein that lacks the C-terminal ACT regulatory domain. The suggested function of the ACT domain is regulation of the catalytic activities of the amino-terminal domain to ensure that synthesis and degradation of ppGpp are not costimulated (39, 40). In DAP-A2, the A58E substitution is predicted by EMBOSS (41) to convert a beta-strand fold into an alpha helix in the HD4 metal-dependent phosphohydrolase domain of RelA. DAP-A1 and DAP-A2 also possess nonsense and frameshift mutations, respectively, in a gene encoding a predicted alpha/beta hydrolase. To our knowledge, this gene has not previously been linked with antimicrobial susceptibility.
Different mutations were identified in DAP-B1 and DAP-B2. In DAP-B1, an E61K substitution occurs in PhoU, a regulator of phosphate transport. We confirmed that inorganic phosphate concentrations were significantly lower in the DAP-B1 mutant relative to the wild type for two of four time points assayed (see Fig. S4 in the supplemental material).
In DAP-B2, a V57M substitution occurs in BgsB (42). BgsB, a glycosyltransferase (GT), catalyzes the transfer of glucose from UDP-glucose to 1,2-diacylglycerol (DAG) forming 3-d-glucosyl-1,2-diacylglycerol (MGlcDAG), which is converted to diglucosyl-DAG (DGlcDAG) by BgsA (43).
Distinct alternations of membrane phospholipid compositions in DAP mutants.
We next compared lipid profiles between the DAP mutants and wild-type E. faecium 410 in three independent trials. Ion intensities of phospholipid and glycolipids detected were normalized by wet pellet weight for each lipid extraction, and the normalized levels were compared for wild-type and mutant strains (see Data Set S1 in the supplemental material). Significant changes, as assessed by the one-tailed Student t test, are presented in Table 4. Representative negative-ion chromatogram data are shown in Fig. S5 in the supplemental material.
TABLE 4.
Average fold ratios of significant phospholipid changes in DAP-A1 and DAP-A2 strains
| Lipid | Avg fold ratio for lipids detected in DAP strainsa |
|
|---|---|---|
| DAP-A1 | DAP-A2 | |
| Phosphatidylglycerol | −1.41 | −1.17 |
| Cardiolipin | −11.28 | −6.97 |
| MHDAG (monohexosyldiacylglycerol) | −1.89 | |
| DHDAG (dihexosyldiacylglycerol) | 1.89 | |
| GPDD (glycerolphosphate diglucosyl diacylglycerol) | 2.07 | |
The average fold ratios of differences between normalized ion intensities of the DAP mutants were compared to wild-type E. faecium 410 in three independent trials. The ion intensities were normalized by wet pellet weight. A one-tailed Student t test was applied to calculate the P value.
Cardiolipin (bisphosphatidylglycerol; CL), an anionic phospholipid, was significantly reduced the in DAP-A1 and DAP-A2 mutants compared to E. faecium 410 (Table 4). The expression levels of two predicted cardiolipin synthase (cls) genes (cls-1 [EFTG_00614] and cls-2 [EFTG_01168]), which mediate the reversible transphosphatidylation of PG molecules to synthesize cardiolipin, were not significantly altered compared to E. faecium 410 (see Fig. S2 in the supplemental material). We infer that E. faecium possesses posttranscriptional mechanisms to regulate cardiolipin synthesis and turnover. In the DAP-A1 mutant, the levels of the glycolipids MHDAG and DHDAG were also significantly altered compared to the E. faecium 410 parental strain (Table 4). In the DAP-A2 mutant, the levels of GPDD (glycerolphosphate diglucosyl-diacylglycerol), a lipoteichoic acid precursor, were 2-fold higher (Table 4).
In contrast to DAP-A mutants, lipid profiles of the DAP-B mutants were not significantly altered from the parental strains (see Data Set S1 in the supplemental material), indicating that DAP-A and DAP-B mutants do not share identical molecular mechanisms for reduced DAP susceptibility. We note that CL levels were reduced in every trial for DAP-B2, although this did not achieve statistical significance (P = 0.0503; our cutoff for significance is P < 0.05; see Data Set S1 in the supplemental material).
efrE impacts CHX but not DAP susceptibility.
Because efrE mutations were shared by all DAP strains, we deleted the EfrEF transport system from E. faecium 410 and the DAP-A2 mutant to assess its role in CHX and DAP susceptibility. The deletion of efrEF from both E. faecium 410 and the DAP-A2 mutant resulted in increased CHX susceptibility, as determined by broth microdilution assays (Table 2). We also utilized a spot-plating assay to assess CHX susceptibility in the presence of different concentrations of CHX (Fig. 3). As expected, the efrEF deletion mutants were significantly more susceptible to CHX than their parent strains.
FIG 3.

Agar CHX susceptibility assay. The geometric means and the geometric standard deviations of the CFU/ml are shown for wild-type E. faecium 410 and the ΔefrEF deletion mutant (A), the DAP-A2 mutant and the DAP-A2 ΔefrEF deletion mutant (PB302) (B), and the ΔefrEF deletion mutant transformed with empty pLZ12 vector or the complementation vectors pAH201 or pPB202 (C). The CFU/ml value was determined using BHI or BHI-chloramphenicol agars with or without CHX supplementation at 1/8×, 1/4×, or 1/2× MIC CHX. Data from three independent trials are shown for each condition. The dashed line represents the limit of detection (103 CFU/ml). *, P < 0.05 value (one-tailed Student t test). Cam, chloramphenicol.
To complement the efrEF deletion in trans, we first cloned efrEF from wild-type E. faecium 410, along with its predicted native promoter, into plasmid pLZ12 and then transformed it into the efrEF deletion mutant, generating strain AH102. As a control, empty pLZ12 vector was also transformed into the efrEF deletion mutant, generating strain AH101. We utilized the spot-plating assay to quantify CHX susceptibility differences between strains AH101 and AH102 in the presence of chloramphenicol selection for pLZ12 (Fig. 3). Complementation with the wild-type efrEF genes in trans significantly reduced the CHX susceptibility of the efrEF mutant (Fig. 3).
Next, to assess the contribution of the EfrE Ala290Val substitution in CHX susceptibility, we cloned efrEF with its predicted native promoter from the DAP-A2 mutant into the plasmid pLZ12 to generate a second complementation construct, pPB202. This plasmid was transformed into the efrEF deletion mutant to generate strain PB303. This construct significantly reduced the CHX susceptibility of the efrEF deletion mutant (Fig. 3) and provided greater protection from CHX than the wild-type efrEF genes (see data for 1/2× MIC CHX cultures in Fig. 3).
We assessed DAP MICs for the efrEF deletion mutants. The DAP MIC of the E. faecium 410 ΔefrEF mutant was not significantly different from the wild-type parent (Table 2). For the DAP-A2 ΔefrEF mutant, the range and median of the DAP MICs were lower than for DAP-A2, but this change was not significant (n = 6 trials; P = 0.07 in a one-tailed unpaired Student t test).
We performed comparative lipidomic analysis of E. faecium 410 and the efrEF deletion mutant in two independent trials. Data for anionic membrane phospholipid and glycolipid levels are shown in Data Set S2 in the supplemental material; note that we did not perform a statistical analysis because only two trials were performed. The most striking differences observed were for two species whose positive ion [M+H]+ signals are detected at m/z 288 and m/z 316 by electrospray ionization/mass spectrometry (ESI/MS). These two species are present in wild-type E. faecium 410 but undetected in the efrEF deletion mutant (see Fig. S6 in the supplemental material). High-resolution mass measurement and tandem MS analysis identified these species as ethoxylated fatty amines previously identified as components of the anti-static additive Atmer-163 (44), which is commonly used in consumer products such as plastics. The m/z 288 and 316 ions correspond to the [M+H]+ ions of Atmer-163 containing C13 and C15 fatty alkyl chains, respectively. The absence of Atmer-163 in the efrEF mutant suggests that this ABC transporter is involved in transporting the Atmer-163 species, with lipid-like structures, from media into the cells.
DISCUSSION
The goal of this study was to test the hypothesis that serial exposure to sub-MIC CHX selects for VREfm mutants with reduced susceptibilities to CHX, with concomitant effects on susceptibility to other membrane- and cell wall-targeting antimicrobials. Our serial passage experiments demonstrate that reduced CHX susceptibility emerges in VREfm after repeat subinhibitory exposure. Moreover, our DAP plating experiments demonstrate that reduced DAP susceptibility concomitantly emerges in a subpopulation of CHX-passaged cells. Using genomics and lipidomics, we identified genetic and physiological changes occurring in these subpopulations with reduced DAP susceptibility. That the strains arising on DAP plates are not hypermutators indicates that these mutants emerged under CHX selection. It remains to be determined at what point in the CHX serial passage experiments reduced DAP susceptibility emerged, since only the beginning and endpoints of the evolution experiments were assessed in this study. Deep sequencing of populations at the beginning, middle, and end of the CHX passage experiments could be used in future studies to further examine the diversity and frequency of genetic variations arising as a result of serial subinhibitory CHX exposure. Moreover, it remains to be determined whether susceptibility to host-associated cationic antimicrobial peptides is also altered as a result of serial CHX exposure.
CHX, a cationic antiseptic, interacts with the bacterial cell membrane (12). Various mechanisms of reduced CHX susceptibility in Gram-negative and -positive bacteria have been reported. The two main mechanisms include CHX efflux (23, 35, 37, 38, 45–48) and changes in outer membrane content (49, 50). In this study, we have confirmed a role for the heterodimeric ABC transporter in CHX susceptibility in VREfm. Deletion of efrEF increases susceptibility of E. faecium 410 to CHX, and an amino acid substitution in EfrE is associated with decreased susceptibility to CHX. By lipidomic analysis, we found that the presence of two ethoxylated fatty amine compounds was abolished in the efrEF deletion mutant relative to the wild type. It remains to be determined whether and how these compounds are protective against CHX.
Daptomycin (DAP) is a cyclic lipopeptide antibiotic used to treat infections caused by multidrug-resistant Gram-positive pathogens, including VREfm (51–53). DAP resistance arises by mutation, leading to treatment failure (54). DAP is a negatively charged molecule that requires calcium ions for activity. Interaction of the cationic DAP-calcium complex with the membrane induces daptomycin oligomerization, membrane phospholipid remodeling, and other physiological alterations, ultimately leading to cell death (55–60). Broadly speaking, alterations in cell surface composition and in cellular stress responses are associated with reduced DAP susceptibility in Gram-positive bacteria (61, 62). In this study, we identified adaptive changes in genes with predicted or experimentally confirmed roles in chlorhexidine susceptibility (efrE), global nutritional stress response (relA), nucleotide metabolism (cmk), phosphate acquisition (phoU), and glycolipid biosynthesis (bgsB) occurring in the CHX-passaged mutants with reduced DAP susceptibilities. Of these, relA has been directly implicated in DAP resistance. Production of the alarmone (p)ppGpp is controlled by RelA. Changes in ppGpp concentration modulate the stringent stress response and impact antibiotic tolerance and virulence in Enterococcus (63, 64). Mutations in relA have previously been associated with in vitro (55) and in vivo (65) emergence of DAP resistance in Bacillus subtilis and E. faecium, respectively.
We identified other phenotypes occurring in CHX-evolved VREfm with reduced DAP susceptibility. The DAP-A1, DAP-A2, and DAP-B1 populations each had significantly lower growth rates than did the wild-type parent, indicating that CHX adaptation comes with a fitness cost. However, the growth rate of DAP-B1 was not significantly different than the parent strain. Moreover, significant changes in cellular membrane phospholipid and glycolipid content, particularly for CL, occurred in the DAP-A1 and DAP-A2 populations, but not the DAP-B populations. It is likely that these different phenotypes reflect the different genotypes of the CHX-evolved populations, since different mutations emerged in the two independent evolution experiments. A weakness of our study is that we did not assess each mutation by introducing them back to the parent strain in controlled genetic experiments, in order to assess the contribution of each to DAP susceptibility and cellular lipid content. This will be a focus of future work.
Our work has clinical implications. If subinhibitory CHX exposure selects for VREfm mutants with enhanced abilities to tolerate or resist DAP, these mutants could contribute to treatment failures with DAP. Frequent improper use of CHX (i.e., the presence of subinhibitory concentrations on patient skin) may favor the emergence and persistence of these VREfm mutants in health care settings. Surveillance of VREfm from hospital wards utilizing CHX bathing would be useful for monitoring the long-term impact of CHX bathing on these organisms. Routine subinhibitory CHX exposure may be a contributing factor to the clinical emergence of DAP resistance in VREfm.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains used in this study are listed in Table 1. E. faecium was cultured at 37°C on brain heart infusion (BHI) agar or in BHI broth without agitation unless otherwise stated. The CHX product used for experiments was Hibiclens (4% [wt/vol] chlorhexidine gluconate with 4% isopropyl alcohol). Chloramphenicol was used at 15 μg/ml for E. coli and E. faecium.
Routine molecular biology techniques.
E. faecium genomic DNA was isolated according to a previously published protocol (66). DNA fragments were purified using the Purelink PCR purification kit (Thermo Fisher). Taq polymerase (New England BioLabs) was used for routine PCR. Routine DNA sequencing was performed by the Massachusetts General Hospital DNA core facility (Boston, MA). The primers used are shown in Table S1 in the supplemental material.
MIC determinations.
Vancomycin, ampicillin, and CHX MICs were determined by broth microdilution in BHI as described previously (24). CHX MICs were recorded at 48 h postinoculation. DAP MICs were measured using Etest (bioMérieux, Inc.) strips on Mueller-Hinton agar (MHA) plates according to the manufacturer's instructions. MICs were independently assessed at least three times.
RT-qPCR.
E. faecium 1,231,410 (E. faecium 410) and 1,141,733 cultures treated for 15 min with 0× (control) and 1× MIC CHX were harvested and mixed with 2 volumes of RNAprotect Bacteria Reagent (Qiagen) according to the manufacturer's recommendations and a previously published protocol (24). Then, 100 ng RNA was used to synthesize cDNA with Superscript II (Life Technologies), and 5 ng cDNA was used as the template in RT-qPCR with primers to amplify internal regions of liaX or clpX. Threshold cycle (CT) values were used to calculate the fold change of liaX gene expression (normalized by clpX expression) between 1× MIC CHX-treated cultures and control cultures (n = 2 independent trials).
Serial passage experiments.
Wild-type E. faecium 410 was used for in vitro serial passage according to a previously published protocol (27). The CHX 1× MIC value for E. faecium 410 was 4.9 μg/ml on day 1. For serial passage, overnight culture was adjusted to an optical density at 600 nm (OD600) of 0.1 and exposed to CHX below, at, and above the MIC in BHI broth. The cultures were incubated at 37°C, and after 24 h, the cultures with visible growth in the highest drug concentration were used as an inoculum for the next round of passaging on day 2. Again, the inoculum OD600 was adjusted to 0.1, and cultures were exposed to CHX as described above. Passages were performed for 21 days. The serial passage experiment was performed independently twice, referred to as experiments A and B. Cultures from the 22nd day of experiment A (referred to as population A) and experiment B (referred to as population B) were analyzed in this study. E. faecium 410 cultures were also passaged in BHI without CHX for 21 days in two independent trials. Populations from day 21 of these trials are referred to as 410-P1 and 410-P2 in this study.
Agar daptomycin susceptibility assay.
Cultures of populations A and B were inoculated directly from glycerol stocks into BHI broth and incubated overnight. Cultures were serially diluted in 1× phosphate-buffered saline (PBS) and spotted on MHA plates supplemented with calcium (50 μg/ml) with or without 10 μg/ml DAP. Plates were incubated for 24 to 36 h at 37°C prior to colony counting. Three (for population A) or four (for population B) independent trials were performed. A one-tailed Student t test was used to assess significance. E. faecium 410 unpassaged (seven trials) and 410-P1 and 410-P2 populations (three trials each) were assayed as controls. Colonies arising on DAP plates from two independent trials each for populations A and B were pooled, inoculated in BHI broth, and cryopreserved.
Genome sequencing and analysis.
Genomic DNA was isolated from overnight broth cultures. The DAP MIC was confirmed for these cultures by an Etest assay. Library preparation and 2×150 paired-end Illumina sequencing was performed by Molecular Research LP (Shallowater, TX). The wild-type E. faecium 410 strain was sequenced as a control. Sequence reads were first assembled to the E. faecium 410 draft genome sequence (NZ_ACBA00000000.1) using default parameters for local alignment in CLC Genomics Workbench (Qiagen). Polymorphisms in the read assemblies were detected using the basic variant mapping tool for sites with ≥10-fold coverage. Variations occurring with ≥50% frequency were compared to the wild-type E. faecium 410 read assembly to find mutant-specific SNPs. To detect putative transposon hops, the read-mapping parameters were changed to global instead of local alignment, and regions of interest were manually analyzed. ≥98.8% of the nucleotide positions in the E. faecium 410 reference were covered ≥10-fold and included in variation analyses. Variants unique to the DAP-resistant mutants were confirmed by Sanger sequencing.
Phosphate assay.
A commercially available phosphate colorimetric kit (Sigma-Aldrich; MAK030) and a previously published protocol (67) were utilized to measure intracellular inorganic phosphate (Pi) levels in wild-type and DAP-B1 cultures. Overnight cultures were diluted to an OD600 of 0.01 in 50 ml of prewarmed BHI, followed by incubation at 37°C with shaking at 100 rpm. The phosphate levels were measured at four time points: 1, OD600 = 0.4 to 0.5; 2, OD600 = 0.5 to 0.6; 3, OD600 = 0.6 to 0.7; and 4, OD600 = 0.7 to 0.8. For each time point, 100-μl culture was serially diluted in 0.9% sterile NaCl and spotted on BHI plates for CFU determination. Also, 1 ml of the culture was incubated on ice for 5 min and then pelleted at 13,300 × g for 2 min at 4°C. The pellet was washed twice in 1 ml of double-distilled water, resuspended in 0.5 ml of double-distilled water, and then disrupted three times (Fast-Prep-24; MPBio) at 6.5 m/s for 30 s. The homogenized samples were centrifuged at 13,300 × g for 15 min at 4°C. Twenty-five μl of the supernatant was diluted with 25 μl double-distilled water, and the Pi levels were measured per the manufacturer's instructions. The phosphate levels were normalized by determining the CFU. A one-tailed Student t test was used to assess significance from three independent trials.
Bacterial growth curves.
Overnight broth cultures inoculated from glycerol stocks were diluted to an OD600 of 0.01 in 50 ml of prewarmed BHI and incubated at 37°C with shaking at 100 rpm. The OD600 values were measured for 6 h. The experiment was performed independently three times, and a one-tailed Student t test was used to assess significance.
Rifampin resistance frequency.
Overnight cultures of E. faecium 410 wild-type and DAP-resistant mutant strains were serially diluted in 1× PBS and spot plated on BHI agar to obtain the total CFU counts. Three milliliters of the cultures were pelleted and spread onto BHI agar plates supplemented with 50 μg/ml rifampin to obtain rifampin-resistant CFU counts. Plates were incubated for 24 to 48 h. Four independent trials were performed, and the significance value was calculated using a one-tailed Student t test.
Gene deletion and agar CHX susceptibility assay.
Loci (EFTG_02287-02288) encoding the predicted ABC transport system EfrEF were deleted in-frame utilizing plasmid pHA101 in wild-type E. faecium 410 and the DAP-A2 mutant. Briefly, 999-bp flanking upstream and downstream regions of the genes of interest were amplified using the primers in Table S1 in the supplemental material, ligated with the plasmid pHA101, and propagated in E. coli EC1000. The construct, pPB301, was sequence verified and electroporated into E. faecium 410 according to a previously published protocol (24). Temperature shift at a nonpermissible temperature of 42°C and counterselection with p-chlorophenylalanine was monitored according to a previously published protocol (68). Deletion of EFTG_02287-02288 in the mutant strains (E. faecium PB301 and PB302) was confirmed by Sanger sequencing. Next, overnight cultures of wild-type E. faecium 410, PB301, DAP-A2, and PB302 strains were serially diluted and spot plated onto BHI plates supplemented with or without 1/8×, 1/4×, and 1/2× MIC CHX. The plates were incubated at 37°C for 24 to 36 h, and the CFU/ml values from three independent trials were quantified. A one-tailed Student t test was used to assess significance.
Complementation assay for CHX susceptibility.
To complement the transporter deletion in trans, the genes EFTG_02287-02288 with their putative native promoter were amplified from wild-type E. faecium 410 or the DAP-A2 mutant and ligated with pLZ12 (69) to generate constructs pAH201 and pPB202. The complementation constructs were sequence verified and electroporated into the efrEF deletion mutant (PB301) to generate strains AH102 and PB303. The empty pLZ12 plasmid was also transformed into the deletion mutant as a control (strain AH101). To assay complementation, overnight cultures were serially diluted and spot plated on BHI-chloramphenicol (15 μg/ml) plates supplemented with or without 1/8×, 1/4×, and 1/2× MIC CHX. The plates were incubated at 37°C for 24 to 36 h. The CFU/ml values for three independent trials were quantified, and a one-tailed Student t test was used to assess significance.
Lipidomic analysis.
Cultures were inoculated from glycerol stock into 10 ml of BHI broth, followed by incubation at 37°C overnight. The 10-ml cultures were added to prewarmed 250 ml of BHI and incubated until reaching an OD600 of ∼0.6. Then, 100 μl was removed for DAP Etest testing on MHA plates. Also, 15 ml of the cultures was added to 2 volumes of RNAprotect Bacteria Reagent according to the manufacturer's recommendations and a previously published protocol (24), and samples were used to assess cls, cmk, clpX, or liaX expression by RT-qPCR as described above. The remaining culture was pelleted at 10,000 rpm at 4°C. Cell pellets were stored at −80°C prior to lipid extraction by the Bligh and Dyer method (70). Lipid analysis by normal-phase liquid chromatography ESI/MS (LC-ESI/MS) was performed using an Agilent 1200 Quaternary LC system coupled to a high-resolution TripleTOF5600 mass spectrometer (Sciex, Framingham, MA), as previously described (71, 72). Normal-phase LC was performed on an Agilent 1200 Quaternary LC system using an Ascentis Silica HPLC column, (5 μm; 25 cm by 2.1 mm; Sigma-Aldrich). Mobile phase A and B solvents, flow conditions, and instrumental settings for ESI/MS and MS/MS were as previously published (73). Data analysis was performed using Analyst TF1.5 software (Sciex). To compare wild-type and DAP mutant strains, three independent lipidomics trials were performed. To compare wild-type and ΔefrEF strains, two independent trials were performed.
Accession number(s).
Raw Illumina sequence reads generated in this study are available in the Sequence Read Archive under accession number SRP108331.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ronda Akins for providing daptomycin.
This study was supported by start-up funds from the University of Texas at Dallas to K.L.P. The MS facility in the Department of Biochemistry at Duke University Medical Center and Z.G. were partially supported by grants GM-069338 and EY023666 from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01235-17.
REFERENCES
- 1.Agudelo Higuita NI, Huycke MM. 2014. Enterococcal disease, epidemiology, and implications for treatment, p 45–70. In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug-resistant infection. Massachusetts Eye and Ear Infirmary, Boston, MA. [Google Scholar]
- 2.Arias CA, Murray BE. 2012. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol 10:266–278. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilmore MS, Lebreton F, van Schaik W. 2013. Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol 16:10–16. doi: 10.1016/j.mib.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, National Healthcare Safety Network. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. 2011. Vital signs: central line-associated blood stream infections–United States, 2001, 2008, and 2009. MMWR Morb Mortal Wkly Rep 60:243–248. [PubMed] [Google Scholar]
- 6.Muto CA, Jernigan JA, Ostrowsky BE, Richet HM, Jarvis WR, Boyce JM, Farr BM, Shea. 2003. SHEA guideline for preventing nosocomial transmission of multidrug-resistant strains of Staphylococcus aureus and enterococcus. Infect Control Hosp Epidemiol 24:362–386. doi: 10.1017/S0195941700083375. [DOI] [PubMed] [Google Scholar]
- 7.Boyce JM. 2007. Environmental contamination makes an important contribution to hospital infection. J Hosp Infect 65(Suppl 2):S50–S54. doi: 10.1016/S0195-6701(07)60015-2. [DOI] [PubMed] [Google Scholar]
- 8.Weber DJ, Anderson D, Rutala WA. 2013. The role of the surface environment in healthcare-associated infections. Curr Opin Infect Dis 26:338–344. doi: 10.1097/QCO.0b013e3283630f04. [DOI] [PubMed] [Google Scholar]
- 9.Hota B. 2004. Contamination, disinfection, and cross-colonization: are hospital surfaces reservoirs for nosocomial infection? Clin Infect Dis 39:1182–1189. doi: 10.1086/424667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies A. 1973. The mode of action of chlorhexidine. J Periodont Res Suppl 12:68–75. doi: 10.1111/j.1600-0765.1973.tb02167.x. [DOI] [PubMed] [Google Scholar]
- 11.Davies GE, Francis J, Martin AR, Rose FL, Swain G. 1954. 1:6-Di-4′-chlorophenyldiguanidohexane (hibitane): laboratory investigation of a new antibacterial agent of high potency. Br J Pharmacol Chemother 9:192–196. doi: 10.1111/j.1476-5381.1954.tb00840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koontongkaew S, Jitpukdeebodintra S. 1995. Interaction of chlorhexidine with cytoplasmic membranes of Streptococcus mutans GS-5. Caries Res 29:413–417. doi: 10.1159/000262101. [DOI] [PubMed] [Google Scholar]
- 13.Hugo WB, Longworth AR. 1966. The effect of chlorhexidine on the electrophoretic mobility, cytoplasmic constituents, dehydrogenase activity, and cell walls of Escherichia coli and Staphylococcus aureus. J Pharm Pharmacol 18:569–578. doi: 10.1111/j.2042-7158.1966.tb07935.x. [DOI] [PubMed] [Google Scholar]
- 14.Hugo WB, Longworth AR. 1964. Some aspects of the mode of action of chlorhexidine. J Pharm Pharmacol 16:655–662. doi: 10.1111/j.2042-7158.1964.tb07384.x. [DOI] [PubMed] [Google Scholar]
- 15.Hugo WB, Longworth AR. 1965. Cytological aspects of the mode of action of chlorhexidine diacetate. J Pharm Pharmacol 17:28–32. doi: 10.1111/j.2042-7158.1965.tb07562.x. [DOI] [PubMed] [Google Scholar]
- 16.Marschall J, Mermel LA, Fakih M, Hadaway L, Kallen A, O'Grady NP, Pettis AM, Rupp ME, Sandora T, Maragakis LL, Yokoe DS, Society for Healthcare Epidemiology of America. 2014. Strategies to prevent central line-associated bloodstream infections in acute care hospitals: 2014 update. Infect Control Hosp Epidemiol 35:753–771. doi: 10.1086/676533. [DOI] [PubMed] [Google Scholar]
- 17.Supple L, Kumaraswami M, Kundrapu S, Sunkesula V, Cadnum JL, Nerandzic MM, Tomas M, Donskey CJ. 2015. Chlorhexidine only works if applied correctly: use of a simple colorimetric assay to provide monitoring and feedback on effectiveness of chlorhexidine application. Infect Control Hosp Epidemiol 36:1095–1097. doi: 10.1017/ice.2015.124. [DOI] [PubMed] [Google Scholar]
- 18.Rupp ME, Lisco SJ, Lipsett PA, Perl TM, Keating K, Civetta JM, Mermel LA, Lee D, Dellinger EP, Donahoe M, Giles D, Pfaller MA, Maki DG, Sherertz R. 2005. Effect of a second-generation venous catheter impregnated with chlorhexidine and silver sulfadiazine on central catheter-related infections: a randomized, controlled trial. Ann Intern Med 143:570–580. doi: 10.7326/0003-4819-143-8-200510180-00007. [DOI] [PubMed] [Google Scholar]
- 19.Noto MJ, Wheeler AP. 2015. Understanding chlorhexidine decolonization strategies. Intensive Care Med 41:1351–1354. doi: 10.1007/s00134-015-3846-6. [DOI] [PubMed] [Google Scholar]
- 20.Noto MJ, Domenico HJ, Byrne DW, Talbot T, Rice TW, Bernard GR, Wheeler AP. 2015. Chlorhexidine bathing and health care-associated infections: a randomized clinical trial. JAMA 313:369–378. doi: 10.1001/jama.2014.18400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pittet D, Angus DC. 2015. Daily chlorhexidine bathing for critically ill patients: a note of caution. JAMA 313:365–366. doi: 10.1001/jama.2014.18482. [DOI] [PubMed] [Google Scholar]
- 22.Popovich KJ, Lyles R, Hayes R, Hota B, Trick W, Weinstein RA, Hayden MK. 2012. Relationship between chlorhexidine gluconate skin concentration and microbial density on the skin of critically ill patients bathed daily with chlorhexidine gluconate. Infect Control Hosp Epidemiol 33:889–896. doi: 10.1086/667371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wand ME, Bock LJ, Bonney LC, Sutton JM. 2017. Mechanisms of increased resistance to chlorhexidine and cross-resistance to colistin following exposure of Klebsiella pneumoniae clinical isolates to chlorhexidine. Antimicrob Agents Chemother 61:e01162-16. doi: 10.1128/AAC.01162-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhardwaj P, Ziegler E, Palmer KL. 2016. Chlorhexidine induces VanA-type vancomycin resistance genes in enterococci. Antimicrob Agents Chemother 60:2209–2221. doi: 10.1128/AAC.02595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer KL, Gilmore MS. 2010. Multidrug-resistant enterococci lack CRISPR-cas. mBio 1:e00227-10. doi: 10.1128/mBio.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J, Feldgarden M, Haas B, Birren B, Gilmore MS. 2012. Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 3:e00318-11. doi: 10.1128/mBio.00318-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer KL, Daniel A, Hardy C, Silverman J, Gilmore MS. 2011. Genetic basis for daptomycin resistance in enterococci. Antimicrob Agents Chemother 55:3345–3356. doi: 10.1128/AAC.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arias CA, Panesso D, McGrath DM, Qin X, Mojica MF, Miller C, Diaz L, Tran TT, Rincon S, Barbu EM, Reyes J, Roh JH, Lobos E, Sodergren E, Pasqualini R, Arap W, Quinn JP, Shamoo Y, Murray BE, Weinstock GM. 2011. Genetic basis for in vivo daptomycin resistance in enterococci. N Engl J Med 365:892–900. doi: 10.1056/NEJMoa1011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davlieva M, Shi Y, Leonard PG, Johnson TA, Zianni MR, Arias CA, Ladbury JE, Shamoo Y. 2015. A variable DNA recognition site organization establishes the LiaR-mediated cell envelope stress response of enterococci to daptomycin. Nucleic Acids Res 43:4758–4773. doi: 10.1093/nar/gkv321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davlieva M, Tovar-Yanez A, DeBruler K, Leonard PG, Zianni MR, Arias CA, Shamoo Y. 2016. An adaptive mutation in Enterococcus faecium LiaR associated with antimicrobial peptide resistance mimics phosphorylation and stabilizes LiaR in an activated state. J Mol Biol 428:4503–4519. doi: 10.1016/j.jmb.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diaz L, Tran TT, Munita JM, Miller WR, Rincon S, Carvajal LP, Wollam A, Reyes J, Panesso D, Rojas NL, Shamoo Y, Murray BE, Weinstock GM, Arias CA. 2014. Whole-genome analyses of Enterococcus faecium isolates with diverse daptomycin MICs. Antimicrob Agents Chemother 58:4527–4534. doi: 10.1128/AAC.02686-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller C, Kong J, Tran TT, Arias CA, Saxer G, Shamoo Y. 2013. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob Agents Chemother 57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clinical and Laboratory Standards Institute. 2017. Performance standards for antimicrobial susceptibility testing; 27th ed Supplement M100. CLSI, Wayne, PA. [Google Scholar]
- 34.Guzman Prieto AM, Wijngaarden J, Braat JC, Rogers MR, Majoor E, Brouwer EC, Zhang X, Bayjanov JR, Bonten MJ, Willems RJ, van Schaik W. 2017. The two-component system ChtRS contributes to chlorhexidine tolerance in Enterococcus faecium. Antimicrob Agents Chemother 61:e02122-16. doi: 10.1128/AAC.02122-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hurlimann LM, Corradi V, Hohl M, Bloemberg GV, Tieleman DP, Seeger MA. 2016. The heterodimeric ABC transporter EfrCD mediates multidrug efflux in Enterococcus faecalis. Antimicrob Agents Chemother 60:5400–5411. doi: 10.1128/AAC.00661-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 37.Davis DR, McAlpine JB, Pazoles CJ, Talbot MK, Alder EA, White C, Jonas BM, Murray BE, Weinstock GM, Rogers BL. 2001. Enterococcus faecalis multi-drug resistance transporters: application for antibiotic discovery. J Mol Microbiol Biotechnol 3:179–184. [PubMed] [Google Scholar]
- 38.Davis DV, Rogers BL, White AC. October 2001. Multidrug resistance (mdr) efflux pump polypeptides. Google Patent WO 2001079257 A2.
- 39.Mechold U, Murphy H, Brown L, Cashel M. 2002. Intramolecular regulation of the opposing (p)ppGpp catalytic activities of Rel(Seq), the Rel/Spo enzyme from Streptococcus equisimilis. J Bacteriol 184:2878–2888. doi: 10.1128/JB.184.11.2878-2888.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. 2004. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response. Cell 117:57–68. doi: 10.1016/S0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- 41.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 42.Theilacker C, Sava I, Sanchez-Carballo P, Bao Y, Kropec A, Grohmann E, Holst O, Huebner J. 2011. Deletion of the glycosyltransferase bgsB of Enterococcus faecalis leads to a complete loss of glycolipids from the cell membrane and to impaired biofilm formation. BMC Microbiol 11:67. doi: 10.1186/1471-2180-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theilacker C, Sanchez-Carballo P, Toma I, Fabretti F, Sava I, Kropec A, Holst O, Huebner J. 2009. Glycolipids are involved in biofilm accumulation and prolonged bacteraemia in Enterococcus faecalis. Mol Microbiol 71:1055–1069. doi: 10.1111/j.1365-2958.2008.06587.x. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez-Rodriguez MV, Dopico-Garcia MS, Noguerol-Cal R, Carballeira-Amarelo T, Lopez-Vilarino JM, Fernandez-Martinez G. 2010. Application of liquid chromatography in polymer nonionic antistatic additives analysis. J Sep Sci 33:3595–3603. doi: 10.1002/jssc.201000460. [DOI] [PubMed] [Google Scholar]
- 45.Russell AD. 2002. Introduction of biocides into clinical practice and the impact on antibiotic-resistant bacteria. Symp Ser Soc Appl Microbiol 2002:121S–135S. doi: 10.1046/j.1365-2672.92.5s1.12.x. [DOI] [PubMed] [Google Scholar]
- 46.Costa SS, Viveiros M, Amaral L, Couto I. 2013. Multidrug efflux pumps in Staphylococcus aureus: an update. Open Microbiol J 7:59–71. doi: 10.2174/1874285801307010059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajamohan G, Srinivasan VB, Gebreyes WA. 2010. Novel role of Acinetobacter baumannii RND efflux transporters in mediating decreased susceptibility to biocides. J Antimicrob Chemother 65:228–232. doi: 10.1093/jac/dkp427. [DOI] [PubMed] [Google Scholar]
- 48.Hassan KA, Jackson SM, Penesyan A, Patching SG, Tetu SG, Eijkelkamp BA, Brown MH, Henderson PJ, Paulsen IT. 2013. Transcriptomic and biochemical analyses identify a family of chlorhexidine efflux proteins. Proc Natl Acad Sci U S A 110:20254–20259. doi: 10.1073/pnas.1317052110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tattawasart U, Maillard JY, Furr JR, Russell AD. 2000. Outer membrane changes in Pseudomonas stutzeri resistant to chlorhexidine diacetate and cetylpyridinium chloride. Int J Antimicrob Agents 16:233–238. doi: 10.1016/S0924-8579(00)00206-5. [DOI] [PubMed] [Google Scholar]
- 50.Westergren G, Emilson CG. 1980. In vitro development of chlorhexidine resistance in Streptococcus sanguis and its transmissibility by genetic transformation. Scand J Dent Res 88:236–243. [DOI] [PubMed] [Google Scholar]
- 51.Tally FP, Zeckel M, Wasilewski MM, Carini C, Berman CL, Drusano GL, Oleson FB Jr. 1999. Daptomycin: a novel agent for Gram-positive infections. Expert Opin Invest Drugs 8:1223–1238. doi: 10.1517/13543784.8.8.1223. [DOI] [PubMed] [Google Scholar]
- 52.Akins RL, Rybak MJ. 2001. Bactericidal activities of two daptomycin regimens against clinical strains of glycopeptide intermediate-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus faecium, and methicillin-resistant Staphylococcus aureus isolates in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob Agents Chemother 45:454–459. doi: 10.1128/AAC.45.2.454-459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alder JD. 2005. Daptomycin: a new drug class for the treatment of Gram-positive infections. Drugs Today (Barc) 41:81–90. doi: 10.1358/dot.2005.41.2.882660. [DOI] [PubMed] [Google Scholar]
- 54.Munita JM, Murray BE, Arias CA. 2014. Daptomycin for the treatment of bacteraemia due to vancomycin-resistant enterococci. Int J Antimicrob Agents 44:387–395. doi: 10.1016/j.ijantimicag.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hachmann AB, Sevim E, Gaballa A, Popham DL, Antelmann H, Helmann JD. 2011. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob Agents Chemother 55:4326–4337. doi: 10.1128/AAC.01819-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muraih JK, Pearson A, Silverman J, Palmer M. 2011. Oligomerization of daptomycin on membranes. Biochim Biophys Acta 1808:1154–1160. doi: 10.1016/j.bbamem.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 57.Jung D, Rozek A, Okon M, Hancock RE. 2004. Structural transitions as determinants of the action of the calcium-dependent antibiotic daptomycin. Chem Biol 11:949–957. doi: 10.1016/j.chembiol.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 58.Ho SW, Jung D, Calhoun JR, Lear JD, Okon M, Scott WR, Hancock RE, Straus SK. 2008. Effect of divalent cations on the structure of the antibiotic daptomycin. Eur Biophys J 37:421–433. doi: 10.1007/s00249-007-0227-2. [DOI] [PubMed] [Google Scholar]
- 59.Straus SK, Hancock RE. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta 1758:1215–1223. doi: 10.1016/j.bbamem.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 60.Muller A, Wenzel M, Strahl H, Grein F, Saaki TN, Kohl B, Siersma T, Bandow JE, Sahl HG, Schneider T, Hamoen LW. 2016. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci U S A 113:E7077–E7086. doi: 10.1073/pnas.1611173113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277:139–158. doi: 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tran TT, Munita JM, Arias CA. 2015. Mechanisms of drug resistance: daptomycin resistance. Ann N Y Acad Sci 1354:32–53. doi: 10.1111/nyas.12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abranches J, Martinez AR, Kajfasz JK, Chavez V, Garsin DA, Lemos JA. 2009. The molecular alarmone (p)ppGpp mediates stress responses, vancomycin tolerance, and virulence in Enterococcus faecalis. J Bacteriol 191:2248–2256. doi: 10.1128/JB.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gaca AO, Kajfasz JK, Miller JH, Liu K, Wang JD, Abranches J, Lemos JA. 2013. Basal levels of (p)ppGpp in Enterococcus faecalis: the magic beyond the stringent response. mBio 4:e00646-13. doi: 10.1128/mBio.00646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Honsa ES, Cooper VS, Mhaissen MN, Frank M, Shaker J, Iverson A, Rubnitz J, Hayden RT, Lee RE, Rock CO, Tuomanen EI, Wolf J, Rosch JW. 2017. RelA mutant Enterococcus faecium with multiantibiotic tolerance arising in an immunocompromised host. mBio 8:e00066-17. doi: 10.1128/mBio.02124-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adams HM, Li X, Mascio C, Chesnel L, Palmer KL. 2015. Mutations associated with reduced surotomycin susceptibility in Clostridium difficile and Enterococcus species. Antimicrob Agents Chemother 59:4139–4147. doi: 10.1128/AAC.00526-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mechler L, Herbig A, Paprotka K, Fraunholz M, Nieselt K, Bertram R. 2015. A novel point mutation promotes growth phase-dependent daptomycin tolerance in Staphylococcus aureus. Antimicrob Agents Chemother 59:5366–5376. doi: 10.1128/AAC.00643-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thurlow LR, Thomas VC, Hancock LE. 2009. Capsular polysaccharide production in Enterococcus faecalis and contribution of CpsF to capsule serospecificity. J Bacteriol 191:6203–6210. doi: 10.1128/JB.00592-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Perez-Casal J, Caparon MG, Scott JR. 1991. Mry, a trans-acting positive regulator of the M protein gene of Streptococcus pyogenes with similarity to the receptor proteins of two-component regulatory systems. J Bacteriol 173:2617–2624. doi: 10.1128/jb.173.8.2617-2624.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/y59-099. [DOI] [PubMed] [Google Scholar]
- 71.Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CR, Guan Z. 2012. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc Natl Acad Sci U S A 109:16504–16509. doi: 10.1073/pnas.1212797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li C, Tan BK, Zhao J, Guan Z. 2016. In vivo and in vitro synthesis of phosphatidylglycerol by an Escherichia coli cardiolipin synthase. J Biol Chem 291:25144–25153. doi: 10.1074/jbc.M116.762070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adams HM, Joyce LR, Guan Z, Akins RL, Palmer KL. 2017. Streptococcus mitis and S. oralis lack a requirement for CdsA, the enzyme required for synthesis of major membrane phospholipids in bacteria. Antimicrob Agents Chemother 61:e02552-16. doi: 10.1128/AAC.02552-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leenhouts K, Buist G, Bolhuis A, ten Berge A, Kiel J, Mierau I, Dabrowska M, Venema G, Kok J. 1996. A general system for generating unlabelled gene replacements in bacterial chromosomes. Mol Gen Genet 253:217–224. doi: 10.1007/s004380050315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.