

Abstract
Metformin is the most commonly prescribed treatment for Type II diabetes and related disorders, however molecular insights into its mode(s) of action have been limited by an absence of structural data. Structural considerations along with an increasing body of literature demonstrating its effects on one-carbon metabolism suggest the possibility of folate mimicry and anti-folate activity. Motivated by increasing recognition that anti-diabetic biguanides may act directly upon the gut microbiome, we have determined structures of the complexes formed between the anti-diabetic biguanides: phenformin, buformin, and metformin and E. coli dihydrofolate reductase (ecDHFR) based on NMR, crystallographic and molecular modeling studies. Inter-ligand Overhauser effects indicate that metformin can form ternary complexes with p-aminobenzoyl-L-glutamate (pABG) as well as other ligands that occupy the region of the folate binding site that interacts with pABG, however DHFR inhibition is not cooperative. The biguanides inhibit the activity of ecDHFR competitively, with the phenformin inhibition constant 100-fold lower than that of metformin. This inhibition may be significant at concentrations present in the gut of treated individuals, and inhibition of DHFR in intestinal mucosal cells may also occur if accumulated levels are sufficient. Perturbation of folate homeostasis can alter the pyridine nucleotide redox ratios that are important regulators of cellular metabolism.
Graphical Abstract
Introduction
Metformin (MFM) is the most widely prescribed antihyperglycemic drug for treatment of type 2 diabetes (T2D) and related conditions including polycystic ovary syndrome and metabolic syndrome 1–3. It is currently undergoing extensive evaluation for treatment of some cancers and other illnesses 4, 5. Extensive physiological and biochemical studies have identified a large number of indirect targets, including AMP-activated kinase 6, the LKB1 master kinase7, and mTOR 8 that are influenced by a relatively limited number of direct targets. The primary direct targets of metformin that have been identified are mitochondrial: complex I of the respiratory chain 9, 10 as well as more recently identified targets including hexokinase II 11 and glycerophosphate dehydrogenase 12. Directly-targeted non-mitochondrial enzymes include AMP deaminase13, and other structures such as the cell membrane14 have also been proposed. Increasing evidence identifies the gut microbiome, and more generally the gastrointestinal tract, as important mediators of biguanide effects 15–17. Not only do these targets experience the highest concentrations of metformin 18, 19, but oral/intestinal treatment of metformin results in a greater drop of blood glucose than IV or even portal treatment 20. Further, growing bacteria are characterized by large, negative membrane potentials21, somewhat analogous to mitochondria for which membrane potential-dependent MFM accumulation appears essential to reach inhibitory concentrations9, 22.
Although the possibility that metformin and other biguanides could target folate metabolizing enzymes is supported by structural relationships 23–26, most reviews covering metformin activity have given little consideration to the impact that direct interference with folate metabolism would have on the cell 27, 28. Summarizing some of the evidence for folate pathway-mediated effects: 1) Elevated homocysteine levels have been observed in patients undergoing biguanide treatment 29, 30 that have been reversed with supplemental folate treatment 31. 2) Parallels between the responses to biguanides and anti-folate drugs such as methotrexate have often been noted 32, 33. 3) Conversely, metabolomic analyses of patients receiving anti-folate drugs such as methotrexate (MTX) exhibit a response consistent with activation of AMP-activated kinase (AMPK), considered a major indirect target of metformin 34. Inhibition of nucleotide synthesis by methotrexate can lead to the accumulation of biosynthetic intermediates that activate AMPK 35, 36. 4) Recent studies of E. coli-fed C. elegans demonstrated that both metformin and the bacterially-targeted DHFR inhibitor trimethoprim (TMP) can extend the lifespan of C. elegans, and it was concluded that MFM and TMP exhibit a shared mechanism of action 37. 5) Like methotrexate, metformin treatment is associated with reduced incidence and improved prognosis of certain cancers 5, as well as anti-inflamatory effects resulting in efficacy against rheumatoid arthritis and other illnesses related to chronic inflammation 38–40. 6) Teratogenic effects similar to those resulting from folate deficiency have been observed in mice treated with biguanides 41.
Based on these considerations and the complete absence of structural data for a metformin-target complex, we have investigated the interaction of metformin and related biguanides phenformin and buformin with E. coli dihydrofolate reductase (DHFR). Although the biguanides could target multiple folate-dependent enzymes, bacterial DHFR was selected for initial studies based on the extensive structural and functional database available, and the recent evidence for a microbiome-based pathway of activity.
MATERIALS AND METHODS
Expression and purification of unlabeled and labeled ecDHFR
Expression
The E. coli DHFR sequence used in these studies corresponds to UniProt P0ABQ4 that contains Asn37. The gene was synthesized by Genscript and has an additional C-terminal His tag (GGGGHHHHHH). The gene was cloned into a pET-21a vector. BL21-DE3-RIL cells were transformed for protein expression. Unlabeled protein was expressed in E. coli grown on 2XYT media to an OD ~0.7, induced with 1 mM IPTG, then allowed to express overnight at 20°C. Cells expressing 15N-labeled DHFR were grown on M9 media supplemented with 15NH4Cl (CIL) M9 salts and 2.5 ml/L of (15N)-Celtone base powder (CIL). Cells expressing 13C/15N-labeled DHFR were grown on M9 media supplemented with 2g/L of U-[13C6]-D-glucose (CIL), 15NH4Cl M9 salts and 2.5 ml/L of (13C/15N) Celtone base powder (CIL). Cells expressing δ-13C isoleucine-labeled [13CH3-Ile]ecDHFR were grown on M9 media, to which 60 mg/L 2-ketobutyric acid-methyl-13C-3,3-d2 (CIL) was added 30 m prior to induction. Cells were grown to OD~0.7, induced with 1 mM IPTG, then allowed to express overnight at 20°C.
A second, C-terminal His-tagged construct contained an additional N-terminal methionine residue (M0). A third, untagged construct was used in one of the crystal structures. An N-terminal His tag with a TEV cleavage site was added to the ecDHFR by PCR. Removal of the tag resulted in an M1G residue substitution. Additionally, a point mutation C152S was introduced using QuickChange II kit (Agilent), eliminating a residue that is highly prone to oxidation.
Purification
Cells were lysed by sonication in the presence of protease inhibitor cocktail (Roche), and the supernatant collected following centrifugation. Protein was purified by immobilized metal affinity chromatography (IMAC), followed by a Superdex S75 GL sizing column. Samples were then dialyzed into the appropriate buffer. To obtain the untagged construct, the His-tagged protein was first dialyzed into buffer containing 50mM Tris (pH 7.8), 50 mM maltose, 0.4 mM DTT, 0.25 mM EDTA, 2% ethanol. TEV protease (1mg) was added, then gently rocked at room temperature for 3 days. Approximately half the resulting protein had the tag removed by this procedure. Cleaved/uncleaved protein was then separated by IMAC chromatography, and the cleaved portion was then further purified on a Superdex S75 column and dialyzed.
Ligand Kd determinations
Kd values were determined based on NMR measurements of [13CH3-Ile]ecDHFR or isothermal titration calorimetry, as described below.
Isothermal Titration Calorimetry (ITC)
ITC binding studies were carried out on a MicroCal VP-ITC calorimeter. Samples were prepared in MTA polybuffer (100 mM Tris, 50 mM MES, 50 mM Acetic acid) containing 1 mM EDTA and 5 mM β–mercaptoethanol, pH 7.0. Titrations consisted of 75 injections of 3 μL at 25 °C with 240 seconds between each injection. Two types of experiments were performed to examine MFM binding to ecDHFR. In the first experiment, 30–40 mM MFM (syringe concentration) was titrated into 110–120 μM ecDHFR in the apo form, or with 120 μM NADPH bound. Similar titrations were performed for phenformin binding to ecDHFR. A second experiment indirectly determined MFM binding by using competitive displacement 42 of MFM from ecDHFR by DHF. 150–400 μM DHF was titrated into 10–12 μM ecDHFR in buffer containing 200 μM NADP+ and 20 mM MFM. Control experiments of 450–500 μM DHF titrated into 10–12 μM EcDHFR with 200 μM NADP+ were also performed. Similar experiments were performed for R67 DHFR, a non-homologous homotetramer with a single active site pore. R67 DHFR was purified as previously described 43.
Data were analyzed in Origin 7.0 supplied by MicroCal and were globally fit in SEDPHAT 44. For competitive displacement experiments, the data for DHF binding to ecDHFR in the presence and absence of MFM were globally fit to the “Competing B and C for A” model to obtain a Kd for MFM 45.
NMR Kd determinations
Studies to determine the position and binding affinity of MFM utilized [13CH3-Ile]ecDHFR. The 2D 1H-13C HMQC experiments were performed on an Agilent 800 MHz DD2 spectrometer equipped with a cryogenic triple resonance probe, using the BioPack (Agilent Inc., Santa Clara, CA) gChmqc pulse sequence. The 2D experiments were acquired with 14 ppm and 10 ppm sweep widths in the direct 1H and indirect 13C dimensions, respectively, with acquisition times of 92 and 63 ms and a 1 s relaxation delay. The residual water and metformin resonances were suppressed using a WET sequence applied during the relaxation period. Various ligands were titrated into 0.1 – 0. 2 mM labeled ecDHFR in a buffer containing 20 mM Tris-HCl, pH = 7.2 (uncorrected), 120 mM NaCl, 1 mM TCEP, 0.5 mM EDTA, 0.25 mM NaN3, in D2O. The shifts of the labeled Ile resonances were monitored as a function of ligand concentration. Shift data were fit assuming a single binding site using a Mathematica program (version 9, Wolfram Research).
Enzyme Inhibition
A Ki value as well as the mode of inhibition were determined based on initial rate measurements of ecDHFR activity. Kinetics were determined at 30 °C using pH 7.0 MTA buffer containing 1 mM EDTA and 5 mM β-mercaptoethanol. Enzyme and NADPH (70 μM) were preincubated for 2 minutes to avoid any hysteresis associated with the presence of two apo enzyme conformers 46, 47. The reaction was initiated by addition of enzyme and NADPH to a mixture of DHF and the inhibitor. Final concentrations ranged from 2 to 90 μM DHF, 20 to 200 mM MFM, or 0.25 to 5 mM PFM or 1 to 20 mM pABG. As the enzyme is quite active, low concentrations were used in the assay (0.5 to 1 nM ecDHFR). To maintain enzyme stability over the course of the assays, bovine serum albumin was added to the enzyme stock (37 μM) for a final BSA concentration in the assay of 125 to 375 nM. Data were initially analyzed by Lineweaver-Burk (LWB) plots 48. Ki values were determined by plotting the slope from the LWB plot vs. the inhibitor concentration and/or by a non-linear fit of all the data 49.
To determine the cooperativity between MFM and pABG, constant concentrations of DHF (7.5 μM) and NADPH (70 μM) were used, while the inhibitor concentrations were varied. Data were analyzed using the following equation from Segel:
| (1) |
where v is the velocity, kcat is the catalytic rate constant, [S], [I] and [X] are the substrate, inhibitor I and inhibitor X concentrations respectively, Km is the Michaelis constant, Ki and Kx are the inhibition constants for I and X respectively, and α describes the cooperativity between inhibitors I and X. Data were plotted as Dixon plots as per 48, with α > 1 indicating negative cooperativity.
NMR Spectroscopy
ILOE experiments
Formation of ternary complexes formed by pairs of ligands that interact with the pteridine- and pABG-binding subsites of the folate binding site of ecDHFR was evaluated using inter-ligand Overhauser effect (ILOE) studies 50–52. Samples contained 0.1 mM ecDHFR, and 5 mM of each ligand, unless otherwise indicated, in the screening buffer: 25 mM phosphate, pH 6.8 (uncorrected), 100 mM NaCl, 1 mM TCEP, 0.25 mM NaN3 in D2O. Samples were run at 25 °C. Due to the poor solubility of Fenbufen, samples were limited to 2 mM and also contained 2 % dimethylformamide-d7. ILOE screening utilized either 1D or 2D NOE experiments as described below.
Initial 2D ILOE NOESY experiments were carried out either on a Varian INOVA 500 MHz NMR spectrometer equipped with a gradient triple resonance probe or an Agilent DD2 800 MHz spectrometer equipped with a cryogenic triple resonance probe, using the BioPack (Agilent Inc., Santa Clara CA) tnnoesy pulse sequence. The 2D NOESY experiments were acquired with a 700 ms mixing time, 14 ppm sweep widths in both dimensions, acquisition times of 256 and 36 ms in the directly and indirectly detected dimensions, and a 1 s relaxation delay. The residual water peak was suppressed using presaturation during the relaxation delay and the mixing time. The 2D spectra were processed using NMRPipe 53 and analyzed using NMRViewJ 54. The 2D spectra often produced weak ILOE cross-peaks with interference from F1 apodization noise due to intense methyl resonance from metformin and some of the other ligands at the high concentrations (~5 mM) used in these studies. Improved signal-to-noise and artifact free spectra were obtained using 1D ILOE experiments 55 obtained using an Agilent 800 MHz NMR spectrometer equipped with a cryogenic triple resonance probe. The 1D ILOE experiments were carried out using the ChemPack (Agilent Inc, Santa Clara CA) NOESY1D experiment. The 1D NOESY experiments were usually acquired with a 700 ms mixing time, 14 ppm sweep width, acquisition time of 1 s with 2 s relaxation delay. In some cases, buildup curves of the ILOE peaks were obtained by acquiring a series of experiments with 100, 300, 500 and 700 ms. The spectra were usually acquired with 1024 scans or 4096 scans to improve signal-to-noise. The metformin methyl resonance was inverted using 0.02 ppm Gaussian pulses. Zero quantum filtering was employed using a 50 ms/10204.8 Hz WURST2 inversion pulse 56 during the mixing time. The 1D spectra were processed and analyzed using Agilent VnmrJ 4.2 software. In many cases, ILOE cross peaks were verified by complementary studies in which the resonances of the ligand occupying the pABG subsite were inverted. Additional control studies were performed in the absence of DHFR to verify that no ILOE resonances that might involve aggregated species were present.
ecDHFR isoleucine methyl assignments
Isoleucine methyl resonances of ecDHFR were assigned for a ternary complex containing 750 μM U-[13C,15N]ecDHFR in the presence of 50 mM pABG, 5 mM NADP+, and 100 mM MFM in a buffer containing 25mM Tris (pH 7.0), 0.5mM EDTA, 1mm TCEP, 0.25mM azide, 0.1% Triton X-100 in 10% D2O. The ternary complex produced very heterogeneous 1H-15N HSQC and HNCO spectra that were not suitable for backbone resonance assignments. The heterogeneity of the amide-detected spectra may result from intermediate exchange broadening due to fluctuations of the Met20 loop. Due to this heterogeneity, the 3D methyl-methyl NOESY experiment was used to assign the methyl groups based on a 3D 13C,13C methyl NOESY experiment 57, that was acquired using a pulse sequence obtained from the Lewis Kay group (University of Toronto, Department of Chemistry). The single constant time version of the experiment was acquired on an Agilent DD2 600 MHz spectrometer equipped with a cryogenic triple resonance probe with a 100 ms mixing time. The experiment was obtained with sweep width of 14 ppm in the directly detected 1H dimension (F3) and sweep widths of 21.22 ppm in each of the indirectly detected 13C dimensions (F1 and F2). The acquisition times in each of the three dimensions were 24.3 ms, 9.4 ms and 84 ms in the F1, F2, and F3 dimensions respectively. The isoleucine methyl resonances were assigned by mapping their methyl-methyl NOEs onto a crystal structure (1RX2, 58) of ecDHFR in complex with folate and NADP+. The 3D spectrum was processed using NMRPipe 53 and analyzed using NMRViewJ 54.
Summary of Screened compounds
Hippurate analogs of two types were studied: the first included various substitutions on the benzoyl ring, and the second included benzoyl propionate analogs in which the amide NH group of glycine is replaced with a methylene group. The latter were used to extend the measurements to hippurate analogs that were not commercially available. Analogs in group 1 included 4-methylhippurate, 3,4-dimethoxyhippurate, 3,4,5-trimethoxyhippurate, 2-bromohippurate, 4-bromohippurate, and 4-nitrohippurate. Analogs in the second group included 3-(2,4-dimethylbenozyl)propionate (DMBP), 3-(2,3,4,5-tetramethylbenozyl)propionate (TMBP), 3-(4-isopropylbenozyl)propionate (IPBP), and 3-(2,4,5-trimethoxybenozyl)propionate. We also looked more broadly at several NSAIDs, conjugated fatty acids, glutathione derivatives, and peptides. The examples discussed in the manuscript and the supplementary materials generally gave the most significant ILOE results.
Protein crystallization/data collection and refinement
Ternary crystals of His-tagged ecDHFR construct in complex with phenformin and NADP+ were obtained at room temperature in space group p21 using the sitting drop vapor diffusion technique with 250 nl of protein solution at 20 mg/ml concentration in 10mM Tris pH 7.4, 40 mM NaCl, 0.25 mM sodium azide, 1.15 mM NADP+ and 5 mM phenformin mixed with 250nl of the reservoir solution consisting of 10mM trisodium citrate and 33% PEG 6K. For data collection, a cryosolution consisting of 500nl of a 10% ethylene glycol, 9 mM trisodium citrate and 29.7% PEG 6K solution was added directly to the crystal drop. The crystal was harvested and flash frozen in liquid nitrogen. Original diffraction of the crystal was poor, but a 5 second anneal of the crystal improved diffraction to 1.65 Å resolution.
Crystals of the quaternary complex of His-tagged ecDHFR construct in the presence of NADP+, 2,4-diaminopyrimidine (DAP), and N-(4aminobenzoyl)-L-glutamate (pABG) were obtained at room temperature in space group p212121 using sitting drop vapor diffusion by mixing 250 nl of protein solution consisting of 40mg/ml protein in 10mM Tris pH 7.4, 5mM BME, 40mM NaCl, 0.25mM sodium azide, 2mM NADP+, 10mM DAP and 50mM pABG with 250 nl of a reservoir solution consisting of 200mM magnesium formate and 20 % PEG 3350. For data collection 1μl of a cryo solution consisting of 17% ethylene glycol, 166mM magnesium formate and 16.6 % PEG 3350 was added to the drop. The crystal was harvested and flash frozen in liquid nitrogen. Crystals diffracted to a resolution of 1.93 Å resolution.
A second quaternary complex using an untagged ecDHFR(C152S) construct was obtained at room temperature in space group P1 using the sitting drop vapor diffusion technique by mixing 1μl of ecDHFR(C152S) at 30mg/ml in 10mM Tris pH 7.4, 40mM NaCl, 0.25mM sodium azide, 2mM NADP+, 10mM DAP and 50mM pABG with 1ul of the reservoir solution consisting of 47.5mM MES pH 6.5, 76mM magnesium acetate and 28.5 % PEG 4K. For data collection 1μl of a cryo solution consisting of 10% ethylene glycol, 43mM MES pH 6.5, 68mM magnesium acetate, and 25.6% PEG 4K was added directly to the drop. The crystal was harvested and flash frozen in liquid nitrogen. Crystals diffracted to a resolution of 1.90 Å.
A ternary complex of DHFR with buformin and NADP+ was obtained using the sitting drop vapor diffusion technique by mixing 250 nl of 40 mg/ml of the untagged ecDHFR(C152S) construct in 10mM Tris pH 7.4, 40mM NaCl, 0.25 mM sodium azide, 2mM NADP+ and 10mM buformin with 250nl of the reservoir solution consisting of 100mM HEPES pH 7.5, 200mM CaCl2, and 30% PEG400. For data collection, 1ml of the cryo solution consisting of 85% reservoir and 15% ethylene glycol was added to the drop followed by direct transfer to 100% cryo-solution and subsequent flash freezing in liquid nitrogen. Data were collected to 1.35 Å resolution.
Data for all four crystals were collected on a Rigaku MicroMax007HF generator equipped with VariMaxHF mirrors and a Saturn92 (PFM complex + P1 quaternary complex) or a Saturn944 detector. Data were processed using either HKL2000 (Saturn92) or HKL3000 (Saturn944) 59. Molecular replacement using Phaser 60 in Phenix 61 using modified coordinates from pdbcode:1RC4 was used as the starting model for the ternary phenformin complex. Molecular replacement was carried out in a similar fashion for the P212121 and P1 quaternary complexes using modified coordinates from pdbcode:1RX2 and 1RA3, respectively. The PFM model provided the starting coordinates for the BFM data set molecular replacement. All structures were refined with iterative cycles of refinement in Phenix and manual model building in Coot 62. The phenformin ternary complex structure contains one molecule of ecDHFR in the asymmetric unit with bound phenformin and NADP+ in an inactive conformation. The P212121 quaternary data set contains five ecDHFR molecules in the asymmetric unit with three molecules (A, C, E) containing DAP, pABG and NADP+ in an inactive conformation (nicotinamide ring outside the active site), one molecule (D) containing DAP, pABG and NADP+ in an active conformation and the fifth (B) containing only bound DAP. The P1 quaternary structure contains two molecules of ecDHFR in the asymmetric unit with both molecules containing DAP and pABG with the NADP+ found in the inactive conformation. The C2 BFM crystal structure contains one molecule in the asymmetric unit, and the NADP+ nicotinamide out of the active site. All data collection and refinement statistics are found in Supplementary Table S1.
Metformin modeling
Metformin was modeled into the active site of ecDHFR starting from the complex containing DAP, pABG, and NADP+. Due to the complexity of biguanide conformations and potential environmental influences on the geometry, the model of metformin with pABG was optimized in the binding pocket using the hybrid QM/MM potential with the ONIOM(MO:MM) framework 63 in Gaussian-09-E1 64. The QM region was defined by metformin, pABG, the carboxyl group of Asp27, and the guanidino groups of Arg51 and Arg56, and was treated using B3LYP exchange-correlation function and 6–31g* basis set. The MM environment consisted of residues 2–57 and 91–115, with coordinates constrained except for Leu28 and Phe31. The glutamate carboxyl of pABG was protonated and constrained in refinement to reduce bias from calculations in a vacuum.
Results
Crystal structures of phenformin and buformin complexes with ecDHFR
Initial studies targeted the phenformin (PFM) and buformin (BFM) ecDHFR complexes in the expectation that the aryl and alkyl groups would enhance binding affinity by interacting with hydrophobic residues in the active site that normally recognize the p-aminobenzoylglutamate (pABG) moiety. Isothermal titration calorimetry and NMR studies indicated that phenformin (PFM) has modest, sub-millimolar affinity for this enzyme, and further revealed significant binding cooperativity in the presence of NADP+ and NADPH (Table 1) which proved sufficient to obtain a crystal structure of the complex. The crystal structures of ternary complexes of ecDHFR-NADP+ with both phenformin and buformin show that the biguanide forms a salt bridge with the Asp27 “folate hook” residue, as well as additional H-bonds with the carbonyl groups of Ile5 and Ile94, and, more weakly, with Tyr100 (Figure 1A,B).
Table 1.
Summary of Kd and Ki values.
| Ligand | Kd (mM) | Ligand2a (pABGsubsite) | Ligand3 (cofactor) | Method/Info |
|---|---|---|---|---|
| I: Dissociation constants | ||||
| Phenformin | 0.716±.045 | - | ITCb | |
| 0.188±0.018 | NADP+ | ITCb | ||
| 0.098±0.007 | NADPH | ITCb | ||
| pABG | 7.64±0.53 | NMRc; Ile50 | ||
| 8.09±0.52 | NMRc; Ile61 | |||
| 8.00±0.37 | NMRc; Ile94 | |||
| (Mean 7.91) | ||||
| Metformin | 14.8±6.2 | NMRc; Ile5 | ||
| 15.9±2.0 | NMRc; Ile5 | |||
| 17.9±8.9d | - | NADP+ | ITCb; ΔH = 0 kcal/mol) | |
| Metformin | 6.89±0.56 | 50 mM pABG | NMRe; Ile50′ | |
| 5.36±1.15 | 50 mM pABG | NMRe; Ile61′ | ||
| 5.27±0.71 | 50 mM pABG | 2 mM NADP+ | NMRe; Ile50′ | |
| 7.0±0.16 | 75 mM DMBP | NMR; Ile50 | ||
| II: Inhibition constants (Ki) in mM | ||||
| Metformin | 24±7f | NADPH | Steady state kinetics | |
| Phenformin | 0.17±0.02 | NADPH | Steady state kinetics | |
| pABG | 1.8 ± 0.3 | NADPH | Steady state kinetics | |
Additional ligand bound to the pABG subsite.
Buffer: 100 mM TrisHCl, 50 mM MES, 50 mM Acetic Acid, 1 mM EDTA, 5 mM BME, pH 7.0; 25° C
Buffer: 20 mM Tris-HCl, pH = 7.2 (uncorrected), 120 mM NaCl, 1 mM TCEP, 0.5 mM EDTA, 0.25 mM NaN3, in D2O.
An initial fit with variable ΔH gave a Kd = 26.7±11.0 mM, ΔH = −1.1±3.2 kcal/mol; the large error in ΔH prompted an alternate fit with ΔH set = 0.
Ile50 and Ile61 each exhibit two resonances, only one of which titrates upon MFM addition.
A non-linear, global fit of all the data gives a Ki of 17.9 ±3.5 mM.
Figure 1. Structures of ecDHFR complexed with phenformin and buformin.

A) Ribbon diagram showing the active site of the ecDHFR-phenformin complex. Major sidechain interactions with PFM are indicated. B) Ribbon diagram showing a similar complex with buformin. C) Ribbon diagram illustrating the analogous interactions with bound methotrexate (pdb: 1RA3). Hydrogen bonds are indicated with black dashed lines.
Surprisingly, the alkyl substitution patterns in the two inhibitors influence binding differently. In the buformin complex, the N1-substituted nitrogen does not interact directly with Asp27, allowing the N1-butyl group to stack against the Phe31 sidechain. In the phenformin complex, the N1-subsituted nitrogen interacts directly with Asp27, positioning the phenethyl sidechain for interactions with Leu28 and, more weakly, with Phe31 and Ile50. The binding site orientation in the buformin complex is more similar to related structures previously reported (pdb: 2ANQ65 ; pdb: 3DGA25). The PFM complex also exhibits a uniquely positioned Met20 loop, in which Met16 is recruited into the active site where it interacts with the phenyl ring of the inhibitor (Figure S1). Overall, the H-bonding pattern of both inhibitors is similar to that seen with various amino-substituted folate analogs such as methotrexate (MTX) (Figure 1C). Similar to MTX, both PFM and BFM form H-bonds with Ile94 - an interaction that cannot form with either oxidized or reduced folate. In contrast with MTX, both biguanide structures are non-planar, exhibiting a relative tilt between the two guanidino groups of ~ 42° for PFM, and ~ 25° for BFM that reduces intramolecular steric conflict (Figure S1).
Interaction of metformin with ecDHFR
The affinity of metformin (MFM) for ecDHFR was expected to be significantly weaker than the affinity of phenformin due to its more limited ability to interact with hydrophobic residues of the active site. NMR generally provides a useful approach for the identification of ligand binding sites and determination of the dissociation constants for weak to moderate affinity ligands. For ecDHFR, the 1H-15N HSQC spectrum is rather poor, characterized by uneven peak intensities and limited resolution, presumably as a consequence of the dynamic characteristics of the enzyme (e.g. 66). Additionally, metformin represents a challenging ligand for study, lacking aromatic groups that can produce significant shift perturbations, as well as H-bond accepting groups that more directly perturb amide resonances. Following unsuccessful attempts to evaluate the interaction using the 15N labeled enzyme, we used [13CH3-Ile]ecDHFR in order to detect the effect of binding on the isoleucine residues, several of which are located near the presumed metformin binding site. This strategy proved viable, with small shifts observed for several Ile residues. Of these, the Ile5 resonance was most readily analyzed due to its spectral isolation. NMR titration data are shown in Figure 2, and the results of various Kd determinations are in Table 1. Analysis of the MFM Kd using ITC also proved challenging; a Kd value was ultimately estimated based on a displacement assay 67 in which bound MFM was replaced by higher affinity dihydrofolate (DHF). These studies gave similar values, with a mean Kd of 17.9 mM (Figure 2; Table 1).
Figure 2. NMR and ITC Metformin binding data.

A) NMR titration of the Ile5 1H resonance as a function of [MFM] in a sample containing 0.2 mM [13CH3-Ile]ecDHFR. B) 1H NMR shift for Ile50 as a function of [MFM] in a sample of labeled enzyme also containing 50 mM pABG; C) 1H NMR shift of Ile50 as a function of [MFM] in a sample of labeled enzyme also containing 75 mM hippurate analog DMBP. Data in the center and lower panels correspond to the MFM-sensitive Ile50 resonance. D) As direct titration of MFM into ecDHFR did not provide an enthalpic signal, a displacement assay was used (see Methods). The raw ITC thermogram showing the change in power (ΔP) versus time for 153 μM DHF titrated into 9.4 μM ecDHFR complexed with NADP+ (200 μM in the solution) in MTA buffer including 1 mM EDTA, 5 mM β - mercaptoethanol, pH 7.0 buffer at 25 °C is shown. E) The fit of the integrated ITC thermogram, along with the fit residuals. F) The ITC thermogram for the titration of 254 μM DHF into 11 μM ecDHFR complexed with NADP+ and MFM (200 μM and 20 mM present in solution, respectively). G) The fit of the thermogram and the residuals for the individual fit. A global fit of duplicate titrations of DHF into the ecDHFR-NADP+ binary complex and DHF into MFM bound to ecDHFR-NADP+ were analyzed in SEDPHAT (see Methods) to obtain the binding parameters for MFM. Allowing the enthalpy of binding to float in the fitting procedure resulted in an error exceeded 100%. Due to this problem as well as the absence of an enthalpic signal in the MFM titration study, ΔH was set = 0, resulting in a Kd of 18 ± 9 mM and a stoichiometry of 0.65±0.01 from the global fit.
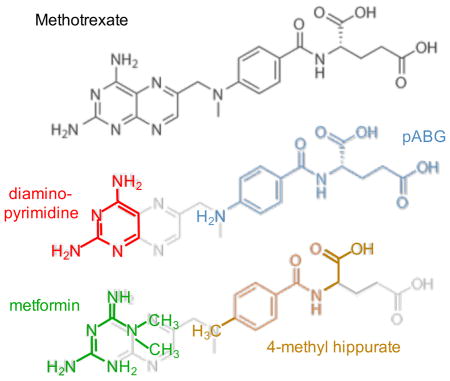
As in the case of PFM (Table 1), it was anticipated that MFM would also be subject to cooperative binding effects with the pyridine nucleotide cofactor. However, an additional cooperative binding contribution is potentially available for metformin involving another ligand that occupies the remainder of the folate binding site. Birdsall et al. 68 have observed very strong cooperativity between a pair of ligands, 2,4-diaminopyrimidine (DAP) and p-aminobenzoyl-L-glutamate (pABG) corresponding to methotrexate fragments, that occupy the pteridine and pABG binding subsites of L. casei DHFR (Figure 3A; Figure S2).
Figure 3. Schematic illustrating the pteridine- and pABG-binding subsites of the DHFR folate binding site.

The panels illustrate how the subsites are occupied by (A) folate; (B) 2,4-diaminopyrimidine (DAP) and pABG; (C) metformin (MFM) and pABG; (D) MFM and a 4-methyl-substituted hippurate; (E) MFM and the NSAID Fenbufen. Double-headed arrows indicate ILOE interactions.
In contrast with the results for the L. casei enzyme, the E. coli DHFR exhibits substantially lower affinity for pABG (Kd = 7.9 mM; Figure S3), and the fraction of bound pABG is insufficient to produce transferred NOE signals. However, when both ligands are present, the pABG aromatic protons do exhibit intra-ligand transferred NOE signals, as well as inter-ligand Overhauser effect (ILOE) signals, with the strongest connecting the DAP H-5 and pABG H-3,5 protons. In this case, the higher affinity DAP is able to function as a recruiter ligand for pABG due to the large cooperative binding effect, so that the fraction of bound pABG is increased and the bound NOE exchange-transferred to the free pABG.
Determination of inhibition constants
Inhibition of the DHFR-dependent reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) was determined using steady state kinetics based on the change in absorption at 340 nm (see Methods). The DHF concentration was varied for a series of MFM concentrations up to 200 mM, and a double reciprocal plot demonstrated competitive binding with a Ki = 24 ± 7 mM (Figure 4). A global fit of the data using SAS software gave a somewhat lower value: Ki = 17.9 ± 3.5 mM (Figure S4) (Table 1). These values are in good agreement with the Kd values summarized in Table 1. Similarly, inhibition constants were obtained for pABG and PFM (Figure 4, Table 1). The Ki for pABG was 1.8 ± 0.3 mM. The Kd/Ki ratio for pABG of 4.4 reflects the presence of NADPH in the assay, and is very similar to the Kd ratio of 4.5 reported for binding of pABG to L. casei DHFR in the absence and presence of NADPH 68. For PFM, the Ki (0.17 ± 0.02 mM), was intermediate between the Kd values measured in the presence of NADP+ and NADPH (Table 1). The global fitting procedure noted above yielded Ki values of 1.3 ± 0.2 mM and 0.21 ± 0.02 mM for pABG and PFM, respectively.
Figure 4. Determination of metformin inhibition constant for ecDHFR.

A) Lineweaver-Burk plot for the inhibition of ecDHFR with Metformin (MFM). All of the lines intersect at the same point on the y-axis indicating competitive inhibition of MFM with DHF. The inset shows the slopes plotted versus [MFM]. Error bars from fits of the slopes are shown. A fit of the data (R2 = 0.970) yields a Ki of 24 ± 7 mM for MFM. A non-linear fit of all of the data yielded a Ki = 17.9 ± 3.5 mM. B) Lineweaver-Burk plot of PFM inhibition of ecDHFR. The pattern of intersecting lines indicates competitive inhibition. The slopes are plotted versus [PFM] in the inset plot. Error bars for the slopes are shown. A fit of the data (R2 = 0.992) yields a Ki of 0.17 ± 0.02 mM.
ILOE screening for ternary complexes containing Metformin
Measurements of inter-ligand Overhauser effects (ILOEs) 50–52 provide an extremely useful basis for identifying and characterizing the ternary complexes formed by metformin and additional ligands that interact with the pABG binding subsite of DHFR. Remarkably, although neither metformin nor pABG exhibits transferred NOE signals under our standard experimental conditions (see Methods), ILOE signals connecting the MFM methyl resonances with the pABG aromatic resonances were present. The affinity of the ecDHFR for MFM is ~ 20-fold weaker than for DAP, so that metformin is not as effective as DAP for recruitment of pABG. The dependence of the ILOE signals on the mixing time was obtained using a 1D NOE experiment (Figure 5A). The observation of significant ILOE peaks in the absence of transferred NOE peaks can occur for two reasons 51: 1) immobilization of the ligands is much greater in the ternary compared with the binary complex, so that they more effectively sense the slow rotational correlation time of the macromolecule, and/or 2) averaged NOE contributions from the free (positive) and bound (negative) ligand become dominated by the positive contributions from the uncomplexed ligand due to low affinity binding, while the uncomplexed ligands do not contribute to the ILOE. Loss of transferred NOE information due to the second effect, which we believe provides the main explanation for the data obtained in these studies, is a general characteristic of studies involving low affinity ligands. The ILOE spectra in Figure 5 confirm the cobinding of the two ligands, as well as providing information on the relative orientation. Thus, the metformin methyls are positioned closest to the 3,5-protons on the p-aminophenyl ring, and a bit further from the 2,6-protons. Due to averaging of the contributions from the two methyl groups and the equivalent p-aminophenyl protons, no further quantitation was attempted.
Figure 5. ILOE build-up data for MFM and ligands bound to the pABG subsite.

A) A series of 1D NOE spectra obtained at the indicated mixing times by irradiating the MFM methyl resonance at 3.04 ppm and observing the pABG aromatic resonances. The corresponding interacting nuclei are shown on the right hand side. B) 1D ILOE build up data for a sample containing 5 mM MFM + 5 mM hippurate analog DMBP. C) 1D ILOE build up data for a sample containing 5 mM MFM + 2 mM Fenbufen. Samples for A and B contained the 0.1 mM ecDHFR, 5 mM of the ligands under evaluation, in the standard screening buffer: 25 mM phosphate, pH 6.8 (uncorrected electrode reading), 100 mM NaCl, 1 mM TCEP, 0.25 mM NaN3 in D2O. Sample for C also contained 2% DMF to improve the solubility of Fenbufen. Spectra were run at 25 °C. Double-headed arrows indicate ILOE interactions.
Due to its low cellular concentration, pABG is unlikely to be a significant ligand for the pABG subsite under typical physiological conditions 69, although polyglutamylated analogs might have sufficient affinity for the human enzyme to become significant 70. A more common metabolite, hippurate (N-benzoylglycine), is a structurally similar metabolic end product often observed in metabolomic studies, and substituted hippurate molecules can be formed from substituted benzoic acid 71. We therefore screened various hippurate analogs as well as other structurally related compounds in the presence of metformin. Although neither transferred NOE nor ILOE MFM-hippurate cross peaks were observed, ILOE signals were obtained for metformin and 4-methylhippurate, and stronger ILOE signals were obtained with analogs containing additional hydrophobic substituents, including 2,4-dimethyl, 2,3,4,5-tetramethyl, and 4-isopropyl analogs. As shown in Figure 5B, ILOE signals are readily observed connecting the MFM methyl protons with the 2-CH3, H-3, and 4-CH3 groups of the 2,4-dimethyl analog, while weaker signals are observed for H-5 and H-6. This pattern is consistent with the expected orientation of the substituted benzoyl group, resulting from a hydrophobic interaction of the 2-CH3 protons with the DHFR Leu28 and Phe31 sidechains. 3,4-Dimethoxyhippurate exhibited weak ILOE cross peaks with metformin, while 3,4,5-trimethoxhippurate did not, consistent with the conclusion that this more highly substituted analog defines the transition from cooperative to competitive binding. In this case, one of the methoxy groups probably intrudes into the space occupied by the MFM methyl group(s). Illustrative ILOE build-up curves are shown in Figure 5, and additional examples are included as Figure S5. Consistent data were obtained in studies involving irradiation of the hippurate resonances instead of the metformin methyls.
Efforts to develop DHFR inhibitors targeting infective microorganisms have revealed that the pABG subsites of the corresponding enzymes can accommodate a wide range of structural variation. These structural results further suggest that the low affinity of pABG itself results from its inability to effectively fill this subsite. Based on DHFR complexes that contain biphenyl groups in the pABG subsite 72, we identified several NSAIDs as binding candidates. Both Diflunisal and Fenbufen exhibited transferred NOE signals consistent with binding, and strong metformin-Fenbufen ILOE signals were observed (Figure 5C). As a result of its limited solubility, an approximate Kd value of 460 μM was obtained for Fenbufen, ~20-fold below the pABG value.
pABG analogs that inhibit dihydropteroate synthase target a different step in folate biosynthesis, but can also occupy the pABG subsite of DHFR. NMR studies of Bowden et al.73 showed that benzenesulfonamides can bind cooperatively with DAP to L. casei DHFR. ILOE data for the ecDHFR – MFM – sulfamethoxazole system yielded cross peaks with both aromatic rings, suggesting that the sulfamethoxazole can bind in either of two orientations (Figure S5). Modeling of the alternate conformation is consistent with the conclusion that binding affinity is mainly dependent on hydrophobic interactions and the main specific interaction between the sulfonyl oxygens and Arg52 can be present in either orientation.
The studies summarized above demonstrate co-binding of MFM and pABG analogs, and provide insight into the relative orientations of these molecules in ternary complexes with ecDHFR.
Structure of related quaternary complexes and the modeled metformin position
In an effort to better understand the basis of the cooperativity between ligands that bind to the two folate subsites and to have a structure more directly useful for modeling the metformin position, crystal structures were determined for ecDHFR in the presence of DAP, pABG, and NADP+ (Figure 6A). Two crystal forms were obtained, one containing two equivalent molecules in the unit cell with the nicotinamide ring of NADP+ occupying a non-productive position out of the active site, and a second containing five molecules in the unit cell in various ligation states and conformations. The relative orientation of the DAP and pABG molecules is similar in all of the structures containing both ligands, and the distances between the DAP C-5 and the closer pABG meta carbon are 3.8–4.0 Å. The corresponding separation in bound methotrexate (pdb: 1RA3) is 4.7 Å. The quaternary complexes in which the nicotinamide ring is in (molecule D) or out of the active site (molecule A) both correspond to closed conformations of the Met20 loop, although the loop conformations are not identical (Figure S6).
Figure 6. Structures of DHFR quaternary complexes.

A) Active site of crystal structure corresponding to the ecDHFR-NADP+-DAP-pABG complex. B) Gaussian-generated model of the active site containing MFM and pABG. Hydrogen bonds are indicated with black dashed lines and the red arrows indicate ILOE interactions. Lower panels show overlaid structures for modeled MFM with bound PFM (C) or bound BFM (D). Intramolecular steric conflict is reduced by relative rotation of the planes containing the two guanidine groups. The PFM and MFM rotation is in the opposite sense, while the angles for MFM and BFM are more similar.
To better understand how MFM might bind to DHFR, modeling of MFM to DHFR was carried out using a Gaussian analysis with the starting position based on three observations: 1) the structural analogies with the buformin and DAP-pABG complexes; 2) the selective perturbation of Ile δ-CH3 resonances positioned near the binding site, and 3) the extensive ILOE observations that connect the metformin methyl groups with ligands occupying the pABG subsite. The challenge in positioning metformin in the pteridine subsite of DHFR involves optimizing both the metformin conformation and its active site interactions. The result of a Gaussian analysis of metformin in the DHFR active site is shown in Figure 6B. Our model adopts a torsion between the guanidino planes that is larger than that observed in the BFM structure, and in the opposite sense characterizing the PFM structure (Figures 6C, 6D). In order to accommodate the MFM methyl groups, the p-aminophenyl group of the pABG moves away from Ile50 and toward Leu28 on the opposite site of the binding cavity. The MFM shows the same set of favorable H-bond interactions that is observed for the BFM and PFM complexes, and the two methyl carbon atoms are positioned 4.2 Å and 4.8 Å from the closer pABG meta carbon. Thus, the distance of the closer methyl group is similar to the closest distance in the DAP – pABG structure. Based on this structure, the two methyl groups of the 2,4-dimethyl hippurate analog bracket the closer MFM methyl group, consistent with the structural relationships indicated by the ILOE data.
Binding cooperativity
Attempts to quantify the cooperative nature of MFM and pABG binding revealed additional conformational complexities of the ecDHFR-ligand system. The NMR spectra of [13CH3-Ile]ecDHFR exhibit additional minor resonances, that are most readily resolved for Ile50 and Ile61 (Figure S3). Titration of the sample with pABG indicates that the major and minor resonances exhibit similar pABG-dependent shift behavior. However, subsequent titration of the ecDHFR-pABG complex with MFM indicates that only one set of resonances shifts, while the second set is not affected by the addition of MFM (Figure S7). The Kd values obtained from measurements of the MFM-sensitive resonances are lower than those obtained in the absence of pABG, consistent with cooperative binding (Table 1), however the cooperativity is much smaller than reported for DAP and pABG 68. In the fully ligated sample used for NMR assignments (see Methods), only one set of methyl resonances was observed, and the Ile50 shift corresponded to the titration-sensitive Ile50 resonance (Figure S7). Thus, this form is apparently favored in the presence of high ligand concentrations.
The basis for the conformational heterogeneity is unclear, however one possibility is that pABG recruits the Met20 loop to a position that blocks the pteridine-binding subsite (Figure 3). Some support for this interpretation was obtained from a preliminary low resolution crystallographic study of the ecDHFR-pABG complex. Although density for many of the loop sidechains is not observed, it appears that Glu17 on the Met20 loop extends into the pteridine-binding subsite (Figure S6). Since there are substantial sequence differences among the loops of DHFR from different species, these results are likely to be specific for ecDHFR. In summary, in the E. coli enzyme, pABG exerts both positive cooperativity as well as an indirect, enzyme-mediated negative cooperativity on metformin binding. Consistent with contributions from both effects, positive cooperativity of MFM and pABG in DHFR inhibition studies was not observed.
Cooperative inhibition for pABG and MFM was also evaluated using steady state kinetics (data not shown). Dixon plots showing the effect of pABG and MFM on the other’s inhibition indicated that the two ligands bind to separate sites on ecDHFR. The nature of the interaction between the two inhibitors can be determined from the x-value at which all the lines intersect 74. The resulting α value, which describes the cooperative inhibition by MFM and pABG, was 3.3 ± 1.3. An α value greater than 1 indicates a net antagonistic interaction between pABG and MFM, although binding is not mutually exclusive – as also demonstrated by the ILOE studies. This is qualitatively consistent with the NMR data summarized above and in Figures S6, showing both pABG-enhanced binding and pABG-insensitive binding for the resolved Ile50 and Ile61 resonances in response to MFM.
Similar co-binding behavior was observed for MFM and 3-(2,4-dimethylbenzoyl)propionate (DMBP), and in this case, the spectra were simpler (Figure S7). The Kd values from serial titration studies with DMBP and then MFM in the presence of 75 mM DMBP were 5.4 mM (DMBP) and 7.0 mM (MFM). In this example, the cooperative effect is very clear, but the reduction in Kd is only about 2.3.
Discussion
Despite years of intense study, the mechanisms underlying the complex physiological effects of biguanides remain poorly understood. Recent evidence has accumulated implicating the gut microbiome as a significant target of these drugs 16, 18. The microbiome represents an attractive target for a weakly interactive drug from two points of view: 1) Metformin dosages are typically large, 0.5–2.5 g/day, and gut concentrations are correspondingly elevated, and 2) similar to evolutionarily-related mitochondria, gut microbes typically exhibit high, negative membrane potentials, supporting accumulation of cationic compounds. According to Bot and Prodan 21, E. coli cells in suspension exhibit a membrane potential of −220 mV in early exponential phase, falling to −140 in late exponential phase. These membrane potentials correspond to intra/extracellular ratios of ~ 5,000 and 225, for monocationic species that are distributed according to a Nernst equilibrium. It consequently becomes much more reasonable to achieve millimolar intracellular concentrations in these cells. Membrane potential-dependent biguanide accumulation in both cells and mitochondria has been demonstrated in many studies9, 22, 75, although the ratios generally do not approach the values that would correspond to a true Nernst equilibrium. In the present study, we have investigated the obvious and apparently neglected target of folate-dependent enzymes, selecting E. coli DHFR as a representative target of a gut enzyme. The structural information, binding affinities and inhibition constants for the E. coli DHFR are by far the most extensive data available for interaction of biguanides with any proposed biguanide target. The likely variation of biguanide-DHFR Ki values for different species of gut bacteria provides one possible basis for species selection in metformin-treated patients 16, 17. Indeed, the first, A–B loop in DHFR derived from different species shows extreme sequence variability, and as discussed above, the binding affinity for pABG differs substantially between the E. coli and L. casei enzymes. Another effect mediated by gut bacteria is based on observations that these microorganisms provide a portion of the vitamins utilized by their host 76, 77. Perturbation of folate metabolism in these organisms by biguanides may directly impact the composition of the available folate pool, and indirectly influence the availability of other nutrients.
The study of E. coli-fed C. elegans by Cabreiro et al. 37 demonstrating that both metformin and the bacterially-targeted DHFR inhibitor trimethoprim (TMP) extend the lifespan of C. elegans, utilized MFM concentrations in the 50–100 mM range, 2.5–5-fold above the MFM Ki value for ecDHFR. Although other effects may also contribute to the effect on lifespan, the Ki values determined here indicate that significant DHFR inhibition will occur at these concentrations.
Binding promiscuity is a characteristic of both folate substrates and anti-folate drugs. Folate itself is both a poor substrate and an inhibitor of DHFR 78, dihydrofolate is a potent inhibitor of folylpoly-γ-glutamate synthetase69, and dihydrobiopterin is an alternate substrate for DHFR 79. Methotrexate has been reported to target 17 different proteins 80–82. As shown here, biguanides behave as pteridine fragments, and as such may interact with a broad range of pterin-dependent enzymes with low binding affinity. This behavior also introduces the possibility of cumulative metabolic perturbations resulting from weak, multi-enzyme inhibition. In addition, the small size of these drugs allows interactions with co-binding ligands that occupy the remainder of the folate-binding site, as demonstrated for ecDHFR in the present study. Although it is not possible to draw further inferences at this time, the present results support further investigation of interactions with other pterin-binding proteins, particularly those that bind reduced pterins that are mimicked by the diaminopyrimidine and biguanide structures.
In addition to effects mediated by the microbiome, the gut mucosal cells of the host organism are exposed to the high gut levels of these drugs, and multiple studies indicate significant accumulations of metformin 18, 19, 83, 84 and phenformin 85, 86. Further, these cells are reported to have limited ability to reduce folate 87. Although it is unclear whether inhibitory concentrations can be reached, biguanide accumulation in these cells will thus target a weak step in folate reduction, possibly increasing the blood levels of oxidized/reduced folates if there is significant DHFR inhibition. An increased ratio of oxidized/reduced folates may then impact hepatic metabolism, since human liver DHFR has been reported to exhibit low and variable activity and to be inhibited by folate 78. As noted above, accumulated DHF can then inhibit folylpoly-γ-glutamate synthetase, resulting in further dysregulation of the folate co-enzyme pool, which plays a central role in regulating pyridine nucleotide homeostasis. Perturbation of the ratios of oxidized/reduced pyridine nucleotide pools have been frequently noted in the metformin literature and attributed to effects on other targets 12, 88, 89.
The high MFM concentration required to inhibit DHFR raises the possibility that additional, weaker-binding targets may come into play that do not contribute to the response at physiological doses. Although an imperfect solution, comparative evaluations using different biguanides provide one approach to address this question, based on the proposition that lower concentrations of high affinity phenformin provide a reasonable mimic of high metformin concentration. Although there are differences in metabolism, uptake and export pathways, there appear to be few if any qualitative differences in the responses to these two biguanides 89. Thus, concentrations of phenformin that more closely approach physiological values appear to be having similar effects, supporting the validity of metformin studies at higher concentration. These results support a common target(s) for MFM and PFM with differing affinity such as ecDHFR. Alternate explanations of differential metformin/phenformin sensitivity have been advanced for other targets, but data supporting these analyses are limited and generally rely on unsupported assumptions.
The pervasive effects of biguanides on cellular function and the complex network of metabolite interactions make identification of cause and effect relationships difficult to unravel. MFM and the other anti-diabetic biguanides can act directly upon DHFR, and probably other pterin-dependent enzymes. The studies presented here on the interaction of biguanides with ecDHFR provide a point of reference for further evaluation of the anti-folate effects of these drugs.
Supplementary Material
Acknowledgments
The work presented utilized suggestions and other input provided by the protein expression, crystallography, and mass spectrometry core facilities of the NIEHS. Valuable discussions with Jun Zhang, U. of Alabama-Birmingham, are greatly appreciated.
Funding. Research in this publication was supported in part by Research Project Number ZIA-ES050111 to REL, and ZIA-ES102645 to LCP in the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health, and by GM 110669 to EEH at the University of Tennessee.
ABBREVIATIONS
- BFM
buformin
- DMBP
3-(2,4-dimethylbenozyl)propionate
- ecDHFR
E. coli dihydrofolate reductase
- IPBP
3-(4-isopropylbenozyl)propionate
- MFM
metformin
- pABG
p-aminobenzoyl-L-gltuamate
- PFM
phenformin
- TMBP
3-(2,3,4,5-tetramethylbenozyl)propionate
Footnotes
Deposition of Structural Data. Four related pdb structures, corresponding to the ecDHFR-NADP+- PFM complex (pdbidcode: 5UIH), ecDHFR-NADP+-BFM complex (pdbidcode: 5UII), and ecDHFR-NADP+-DAP-pABG complex in either of two different space groups (pdbidcodes: 5UIO and 5UIP), have been deposited in the protein data bank, and will be made available upon publication of this manuscript.
NOTES. The authors declare no competing financial interests.
Supporting Information. Supporting information includes 7 Figures and one table summarizing crystallographic data statistics and is available free of charge on the ACS Publications Web site.
References
- 1.Bailey CJ, Turner RC. Drug therapy - Metformin. New Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 2.Lord JM, Flight IHK, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. Brit Med J. 2003;327:951–955. doi: 10.1136/bmj.327.7421.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orchard TJ, Temprosa M, Goldberg R, Haffner S, Ratner R, Marcovina S, Fowler S, Grp DPPR. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: The diabetes prevention program randomized trial. Ann Intern Med. 2005;142:611–619. doi: 10.7326/0003-4819-142-8-200504190-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cicero AFG, Tartagni E, Ertek S. Metformin and its clinical use: new insights for an old drug in clinical practice. Arch Med Sci. 2012;8:907–917. doi: 10.5114/aoms.2012.31622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dowling RJO, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. Bmc Med. 2011;9 doi: 10.1186/1741-7015-9-33. article 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou GC, Myers R, Li Y, Chen YL, Shen XL, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, DePinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dowling RJO, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 9.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 10.Bridges HR, Jones AJY, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462:475–487. doi: 10.1042/BJ20140620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salani B, Marini C, Rio AD, Ravera S, Massollo M, Orengo AM, Amaro A, Passalacqua M, Maffioli S, Pfeffer U, Cordera R, Maggi D, Sambuceti G. Metformin impairs glucose consumption and survival in Calu-1 cells by direct inhibition of hexokinase-II. Sci Rep. 2013;3:2070. doi: 10.1038/srep02070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA, Prigaro BJ, Wood JL, Bhanot S, MacDonald MJ, Jurczak MJ, Camporez JP, Lee HY, Cline GW, Samuel VT, Kibbey RG, Shulman GI. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. doi: 10.1038/nature13270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ouyang JY, Parakhia RA, Ochs RS. Metformin Activates AMP Kinase through Inhibition of AMP Deaminase. Journal of Biological Chemistry. 2011;286:1–11. doi: 10.1074/jbc.M110.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samart N, Beuning CN, Haller KJ, Rithner CD, Crans DC. Interaction of a Biguanide Compound with Membrane Model Interface Systems: Probing the Properties of Antimalaria and Antidiabetic Compounds. Langmuir. 2014;30:8697–8706. doi: 10.1021/la501600s. [DOI] [PubMed] [Google Scholar]
- 15.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Krogh Pedersen H, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Igor Costea P, Kultima JR, Li J, Jorgensen T, Levenez F, Dore J, Meta HITc, Nielsen HB, Brunak S, Raes J, Hansen T, Wang J, Ehrlich SD, Bork P, Pedersen O. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528:262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer PV, Duca FA. Targeting the gastrointestinal tract to treat type 2 diabetes. J Endocrinol. 2016;230:R95–R113. doi: 10.1530/JOE-16-0056. [DOI] [PubMed] [Google Scholar]
- 17.Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut. 2014;63:1513–1521. doi: 10.1136/gutjnl-2014-306928. [DOI] [PubMed] [Google Scholar]
- 18.McCreight LJ, Bailey CJ, Pearson ER. Metformin and the gastrointestinal tract. Diabetologia. 2016;59:426–435. doi: 10.1007/s00125-015-3844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Proctor WR, Bourdet DL, Thakker DR. Mechanisms underlying saturable intestinal absorption of metformin. Drug Metab Dispos. 2008;36:1650–1658. doi: 10.1124/dmd.107.020180. [DOI] [PubMed] [Google Scholar]
- 20.Stepensky D, Friedman M, Raz I, Hoffman A. Pharmacokinetic-pharmacodynamic analysis of the glucose-lowering effect of metformin in diabetic rats reveals first-pass pharmacodynamic effect. Drug Metab Dispos. 2002;30:861–868. doi: 10.1124/dmd.30.8.861. [DOI] [PubMed] [Google Scholar]
- 21.Bot CT, Prodan C. Quantifying the membrane potential during E. coli growth stages. Biophys Chem. 2010;146:133–137. doi: 10.1016/j.bpc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 22.Davidoff F. Effects of Guanidine Derivatives on Mitochondrial Function .3. Mechanism of Phenethylbiguanide Accumulation and Its Relationship to in-Vitro Respiratory Inhibition. Journal of Biological Chemistry. 1971;246:4017–4027. [PubMed] [Google Scholar]
- 23.Bag S, Tawari NR, Queener SF, Degani MS. Synthesis and biological evaluation of biguanide and dihydrotriazine derivatives as potential inhibitors of dihydrofolate reductase of opportunistic microorganisms. J Enzym Inhib Med Ch. 2010;25:331–339. doi: 10.3109/14756360903179443. [DOI] [PubMed] [Google Scholar]
- 24.Vanichtanankul J, Taweechai S, Uttamapinant C, Chitnumsub P, Vilaivan T, Yuthavong Y, Kamchonwongpaisan S. Combined Spatial Limitation around Residues 16 and 108 of Plasmodium falciparum Dihydrofolate Reductase Explains Resistance to Cycloguanil. Antimicrob Agents Ch. 2012;56:3928–3935. doi: 10.1128/AAC.00301-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dasgupta T, Chitnumsub P, Kamchonwongpaisan S, Maneeruttanarungroj C, Nichols SE, Lyons TM, Tirado-Rives J, Jorgensen WL, Yuthavong Y, Anderson KS. Exploiting Structural Analysis, in Silico Screening, and Serendipity To Identify Novel Inhibitors of Drug-Resistant Falciparum Malaria. Acs Chem Biol. 2009;4:29–40. doi: 10.1021/cb8002804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayer S, Daigle DA, Brown ED, Khatri J, Organ MG. An expedient and facile one-step synthesis of a biguanide library by microwave irradiation coupled with simple product filtration. Inhibitors of dihydrofolate reductase. J Comb Chem. 2004;6:776–782. doi: 10.1021/cc049953+. [DOI] [PubMed] [Google Scholar]
- 27.Rosilio C, Ben-Sahra I, Bost F, Peyron JF. Metformin: A metabolic disruptor and anti-diabetic drug to target human leukemia. Cancer Lett. 2014;346:188–196. doi: 10.1016/j.canlet.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Pernicova I, Korbonits M. Metformin-mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- 29.Carlsen SM, Folling I, Grill V, Bjerve KS, Schneede J, Refsum H. Metformin increases total serum homocysteine Levels in non-diabetic male patients with coronary heart disease. Scand J Clin Lab Inv. 1997;57:521–527. doi: 10.3109/00365519709084603. [DOI] [PubMed] [Google Scholar]
- 30.Wulffele MG, Kooy A, Lehert P, Bets D, Ogterop JC, Van der Burg BB, Donker AJM, Stehouwer CDA. Effects of short-term treatment with metformin on serum concentrations of homocysteine, folate and vitamin B12 in type 2 diabetes mellitus: a randomized, placebo-controlled trial. J Intern Med. 2003;254:455–463. doi: 10.1046/j.1365-2796.2003.01213.x. [DOI] [PubMed] [Google Scholar]
- 31.Aarsand AK, Carlsen SM. Folate administration reduces circulating homocysteine levels in NIDDM patients on long-term metformin treatment. J Intern Med. 1998;244:169–174. doi: 10.1046/j.1365-2796.1998.00361.x. [DOI] [PubMed] [Google Scholar]
- 32.Corominas-Faja B, Quirantes-Pine R, Oliveras-Ferraros C, Vazquez-Martin A, Cufi S, Martin-Castillo B, Micol V, Joven J, Segura-Carretero A, Menendez JA. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging-Us. 2012;4:480–498. doi: 10.18632/aging.100472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menendez JA, Joven J. One-carbon metabolism: An aging-cancer crossroad for the gerosuppressant metformin. Aging-Us. 2012;4:894–898. doi: 10.18632/aging.100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tedeschi PM, Johnson-Farley N, Lin H, Shelton LM, Ooga T, Mackay G, Van Den Broek N, Bertino JR, Vazquez A. Quantification of folate metabolism using transient metabolic flux analysis. Cancer Metab. 2015;3:6. doi: 10.1186/s40170-015-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pirkmajer S, Kulkarni SS, Tom RZ, Ross FA, Hawley SA, Hardie DG, Zierath JR, Chibalin AV. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes. 2015;64:360–369. doi: 10.2337/db14-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beckers A, Organe S, Timmermans L, Vanderhoydonc F, Deboel L, Derua R, Waelkens E, Brusselmans K, Verhoeven G, Swinnen JV. Methotrexate enhances the antianabolic and antiproliferative effects of 5-aminoimidazole-4-carboxamide riboside. Mol Cancer Ther. 2006;5:2211–2217. doi: 10.1158/1535-7163.MCT-06-0001. [DOI] [PubMed] [Google Scholar]
- 37.Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cocheme HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D. Metformin Retards Aging in C. elegans by Altering Microbial Folate and Methionine Metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang KY, Kim YK, Yi H, Kim J, Jung HR, Kim IJ, Cho JH, Park SH, Kim HY, Ju JH. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol. 2013;16:85–92. doi: 10.1016/j.intimp.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Ma W, Zhong H, Liu W, Sun Q. Metformin inhibits proliferation of human keratinocytes through a mechanism associated with activation of the MAPK signaling pathway. Exp Ther Med. 2014;7:389–392. doi: 10.3892/etm.2013.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin YM, Choi SC, Xu ZW, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Normalization of CD4(+) T cell metabolism reverses lupus. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denno KM, Sadler TW. Effects of the Biguanide Class of Oral Hypoglycemic Agents on Mouse Embryogenesis. Teratology. 1994;49:260–266. doi: 10.1002/tera.1420490405. [DOI] [PubMed] [Google Scholar]
- 42.Zhang YL, Zhang ZY. Low-affinity binding determined by titration calorimetry using a high-affinity coupling ligand: a thermodynamic study of ligand binding to protein tyrosine phosphatase 1B. Anal Biochem. 1998;261:139–148. doi: 10.1006/abio.1998.2738. [DOI] [PubMed] [Google Scholar]
- 43.Chopra S, Dooling RM, Horner CG, Howell EE. A balancing act between net uptake of water during dihydrofolate binding and net release of water upon NADPH binding in R67 dihydrofolate reductase. Journal of Biological Chemistry. 2008;283:4690–4698. doi: 10.1074/jbc.M709443200. [DOI] [PubMed] [Google Scholar]
- 44.Houtman JC, Brown PH, Bowden B, Yamaguchi H, Appella E, Samelson LE, Schuck P. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 2007;16:30–42. doi: 10.1110/ps.062558507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao H, Piszczek G, Schuck P. SEDPHAT--a platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods. 2015;76:137–148. doi: 10.1016/j.ymeth.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cayley PJ, Dunn SMJ, King RW. Kinetics of Substrate, Coenzyme, and Inhibitor Binding to Escherichia-Coli Dihydrofolate-Reductase. Biochemistry. 1981;20:874–879. doi: 10.1021/bi00507a034. [DOI] [PubMed] [Google Scholar]
- 47.Penner MH, Frieden C. Substrate-Induced Hysteresis in the Activity of Escherichia-Coli Dihydrofolate-Reductase. Journal of Biological Chemistry. 1985;260:5366–5369. [PubMed] [Google Scholar]
- 48.Segel IH. Enzyme Kinetics. Wiley & Sons, Inc; New York: 1975. [Google Scholar]
- 49.Smiley RD, Saxton AM, Jackson MJ, Hicks SN, Stinnett LG, Howell EE. Nonlinear fitting of bisubstrate enzyme kinetic models using SAS computer software: application to R67 dihydrofolate reductase. Anal Biochem. 2004;334:204–206. doi: 10.1016/j.ab.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 50.Barsukov IL, Lian LY, Ellis J, Sze KH, Shaw WV, Roberts GCK. The conformation of coenzyme A bound to chloramphenicol acetyltransferase determined by transferred NOE experiments. Journal of molecular biology. 1996;262:543–558. doi: 10.1006/jmbi.1996.0534. [DOI] [PubMed] [Google Scholar]
- 51.London RE. Theoretical analysis of the inter-ligand Overhauser effect: A new approach for mapping structural relationships of macromolecular ligands. J Magn Reson. 1999;141:301–311. doi: 10.1006/jmre.1999.1897. [DOI] [PubMed] [Google Scholar]
- 52.Li DW, DeRose EF, London RE. The inter-ligand Overhauser effect: A powerful new NMR approach for mapping structural relationships of macromolecular ligands. J Biomol Nmr. 1999;15:71–76. doi: 10.1023/a:1008360208627. [DOI] [PubMed] [Google Scholar]
- 53.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - a Multidimensional Spectral Processing System Based on Unix Pipes. J Biomol Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 54.Johnson BA, Blevins RA. Nmr View - a Computer-Program for the Visualization and Analysis of Nmr Data. J Biomol Nmr. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 55.Kapilashrami K, Bommineni GR, Machutta CA, Kim P, Lai CT, Simmerling C, Picart F, Tonge PJ. Thiolactomycin-based beta-ketoacyl-AcpM synthase A (KasA) inhibitors: fragment-based inhibitor discovery using transient one-dimensional nuclear overhauser effect NMR spectroscopy. The Journal of biological chemistry. 2013;288:6045–6052. doi: 10.1074/jbc.M112.414516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kupce E, Freeman R. Optimized adiabatic pulses for wideband spin inversion. J Magn Reson Ser A. 1996;118:299–303. [Google Scholar]
- 57.Zwahlen C, Gardner KH, Sarma SP, Horita DA, Byrd RA, Kay LE. An NMR experiment for measuring methyl-methyl NOEs in C-13-labeled proteins with high resolution. J Am Chem Soc. 1998;120:7617–7625. [Google Scholar]
- 58.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 59.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 60.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vreven T, Byun KS, Komaromi I, Dapprich S, Montgomery JA, Morokuma K, Frisch MJ. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J Chem Theory Comput. 2006;2:815–826. doi: 10.1021/ct050289g. [DOI] [PubMed] [Google Scholar]
- 64.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian, Inc; 2009. [Google Scholar]
- 65.Summerfield RL, Daigle DM, Mayer S, Mallik D, Hughes DW, Jackson SG, Sulek M, Organ MG, Brown ED, Junop MS. A 2.13 angstrom structure of E-coli dihydrofolate reductase bound to a novel competitive inhibitor reveals a new binding surface involving the M20 loop region. Journal of Medicinal Chemistry. 2006;49:6977–6986. doi: 10.1021/jm060570v. [DOI] [PubMed] [Google Scholar]
- 66.Beierlein JM, Deshmukh L, Frey KM, Vinogradova O, Anderson AC. The Solution Structure of Bacillus anthracis Dihydrofolate Reductase Yields Insight into the Analysis of Structure-Activity Relationships for Novel Inhibitors. Biochemistry. 2009;48:4100–4108. doi: 10.1021/bi802319w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang YL, Zhang ZY. Low-affinity binding determined by titration calorimetry using a high-affinity coupling ligand: A thermodynamic study of ligand binding to protein tyrosine phosphatase 1B. Anal Biochem. 1998;261:139–148. doi: 10.1006/abio.1998.2738. [DOI] [PubMed] [Google Scholar]
- 68.Birdsall B, Burgen ASV, Miranda JRD, Roberts GCK. Cooperativity in Ligand-Binding to Dihydrofolate-Reductase. Biochemistry. 1978;17:2102–2110. doi: 10.1021/bi00604a013. [DOI] [PubMed] [Google Scholar]
- 69.Kwon YK, Lu WY, Melamud E, Khanam N, Bognar A, Rabinowitz JD. A domino effect in antifolate drug action in Escherichia coli. Nat Chem Biol. 2008;4:602–608. doi: 10.1038/nchembio.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schirch V, Strong WB. Interaction of Folylpolyglutamates with Enzymes in One-Carbon Metabolism. Arch Biochem Biophys. 1989;269:371–380. doi: 10.1016/0003-9861(89)90120-3. [DOI] [PubMed] [Google Scholar]
- 71.Pero RW. Health consequences of catabolic synthesis of hippuric acid in humans. Curr Clin Pharmacol. 2010;5:67–73. doi: 10.2174/157488410790410588. [DOI] [PubMed] [Google Scholar]
- 72.Lamb KM, Lombardo MN, Alverson J, Priestley ND, Wright DL, Anderson AC. Crystal Structures of Klebsiella pneumoniae Dihydrofolate Reductase Bound to Propargyl-Linked Antifolates Reveal Features for Potency and Selectivity. Antimicrob Agents Ch. 2014;58:7484–7491. doi: 10.1128/AAC.03555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bowden K, Hall AD, Birdsall B, Feeney J, Roberts GC. Interactions between inhibitors of dihydrofolate reductase. Biochem J. 1989;258:335–342. doi: 10.1042/bj2580335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yonetani T, Theorell H. Studies on Liver Alcohol Dehydrogenase Complexes .3. Multiple Inhibition Kinetics in Presence of 2 Competitive Inhibitors. Arch Biochem Biophys. 1964;106:243–251. doi: 10.1016/0003-9861(64)90184-5. [DOI] [PubMed] [Google Scholar]
- 75.Chien HC, Zur AA, Maurer TS, Yee SW, Tolsma J, Jasper P, Scott DO, Giacomini KM. Rapid Method To Determine Intracellular Drug Concentrations in Cellular Uptake Assays: Application to Metformin in Organic Cation Transporter 1-Transfected Human Embryonic Kidney 293 Cells. Drug Metab Dispos. 2016;44:356–364. doi: 10.1124/dmd.115.066647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24:160–168. doi: 10.1016/j.copbio.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 77.Kopp M, Durr K, Steigleder M, Clavel T, Rychlik M. Measurements of Intra- and Extra-Cellular 5-Methyltetrahydrofolate Indicate that Bifidobacterium Adolescentis DSM 20083T and Bifidobacterium Pseudocatenulatum DSM 20438T Do Not Actively Excrete 5-Methyltetrahydrofolate In vitro. Front Microbiol. 2017;8:445. doi: 10.3389/fmicb.2017.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bailey SW, Ayling JE. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:15424–15429. doi: 10.1073/pnas.0902072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sawabe K, Wakasugi KO, Hasegawa H. Tetrahydrobiopterin uptake in supplemental administration: Elevation of tissue tetrahydrobiopterin in mice following uptake of the exogenously oxidized product 7,8-dihydrobiopterin and subsequent reduction by an anti-folate-sensitive process. J Pharmacol Sci. 2004;96:124–133. doi: 10.1254/jphs.fp0040280. [DOI] [PubMed] [Google Scholar]
- 80.Haupt VJ, Daminelli S, Schroeder M. Drug Promiscuity in PDB: Protein Binding Site Similarity Is Key. PloS one. 2013;8:e65894. doi: 10.1371/journal.pone.0065894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matherly LH, Hou ZJ, Deng YJ. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer Metast Rev. 2007;26:111–128. doi: 10.1007/s10555-007-9046-2. [DOI] [PubMed] [Google Scholar]
- 82.Allegra CJ, Chabner BA, Drake JC, Lutz R, Rodbard D, Jolivet J. Enhanced Inhibition of Thymidylate Synthase by Methotrexate Polyglutamates. Journal of Biological Chemistry. 1985;260:9720–9726. [PubMed] [Google Scholar]
- 83.Bailey CJ, Wilcock C, Scarpello JHB. Metformin and the intestine. Diabetologia. 2008;51:1552–1553. doi: 10.1007/s00125-008-1053-5. [DOI] [PubMed] [Google Scholar]
- 84.Han TX, Proctor WR, Costales CL, Cai H, Everett RS, Thakker DR. Four Cation-Selective Transporters Contribute to Apical Uptake and Accumulation of Metformin in Caco-2 Cell Monolayers. J Pharmacol Exp Ther. 2015;352:519–528. doi: 10.1124/jpet.114.220350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gardner ML, Langlow DR. The absorption of phenformin and its effects on glucose and water absorption in isolated perfused rat small intestine. Q J Exp Physiol Cogn Med Sci. 1977;62:247–255. doi: 10.1113/expphysiol.1977.sp002396. [DOI] [PubMed] [Google Scholar]
- 86.Kruger FA, Altschuld RA, Hollobaugh SL, Jewett B. Studies on the site and mechanism of action of phenformin. II. Phenformin inhibition of glucose transport by rat intestine. Diabetes. 1970;19:50–52. doi: 10.2337/diab.19.1.50. [DOI] [PubMed] [Google Scholar]
- 87.Patanwala I, King MJ, Barrett DA, Rose J, Jackson R, Hudson M, Philo M, Dainty JR, Wright AJA, Finglas PM, Jones DE. Folic acid handling by the human gut: implications for food fortification and supplementation. Am J Clin Nutr. 2014;100:593–599. doi: 10.3945/ajcn.113.080507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD(+) metabolism and SIRT1 activity. Nature. 2009;458:1056–U1140. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Janzer A, German NJ, Gonzalez-Herrera KN, Asara JM, Haigis MC, Struhl K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:10574–10579. doi: 10.1073/pnas.1409844111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.