

Abstract
Objective
Deficiency of α-galactosidase A (αGal-A) in Fabry disease leads to the accumulation mainly of globotriaosylceramide (GL3) in multiple renal cell types. Glomerular podocytes are relatively resistant to clearance of GL3 inclusions by enzyme replacement therapy (ERT). Migalastat, an orally bioavailable small molecule capable of chaperoning misfolded αGal-A to lysosomes, is approved in the European Union for the long-term treatment of patients with Fabry disease and amenable GLA (α-galactosidase A enzyme) mutations. We aimed to examine if migalastat reduces GL3 content of podocytes in Fabry disease.
Methods and analysis
We compared paired renal biopsies of eight adult men with amenable Fabry disease mutations at baseline and after 6 months of treatment with 150 mg migalastat every other day using quantitative unbiased electron microscopic morphometric methods.
Results
Migalastat treatment led to a reduction in mean total GL3 inclusion volume per podocyte in renal biopsies from baseline to 6 months. This reduction correlated precisely with reduced mean podocyte volume. There was also a direct relationship between reduction in podocyte foot process width and the reduction in mean total podocyte GL3 content following 6 months of migalastat treatment, suggestive of reduced podocyte injury.
Conclusion
Migalastat treatment of 6 months duration in eight male patients with Fabry disease demonstrated effective GL3 clearance from the podocyte, an important and relatively ERT-resistant glomerular cell.
Keywords: Fabry, migalastat, chaperone, podocyte, globotriaosylceramide, GL3
Introduction
Deficiency of α-galactosidase A (αGal-A) in Fabry disease (OMIM 301500) leads to the accumulation mainly of globotriaosylceramide (GL3) in multiple cell types and organs,1 often culminating in severe complications including end-stage renal disease, strokes, cardiomyopathy, arrhythmias and premature death.2
While enzyme replacement therapy (ERT) results in rapid clearing of GL3 inclusions in kidney endothelial and mesangial cells and fibroblasts,3 other renal cell types including podocytes, distal tubular cells and arteriolar smooth muscle cells are more resistant to clearance of GL3 by ERT.3 4 We have previously shown that glomerular endothelial and mesangial cell GL3 accumulation does not increase with age and does not correlate with proteinuria in children with Fabry disease. Podocyte GL3 in these young patients increases over time and correlates with increases in podocyte foot process width (FPW) and with urinary protein excretion, both consistent with podocyte damage. Although a long-term ERT trial led to a reduced rate of renal, cardiac and cerebrovascular clinical events,5 there are substantial residual risks despite ERT.6 Thus, other treatment options are needed.
Migalastat is an orally bioavailable small molecule capable of chaperoning misfolded αGal-A to lysosomes. In the FACETS AT1001-011 study in patients with amenable mutations who were ERT-naïve (or off-ERT for >6 months), 6 months of treatment with migalastat demonstrated a decrease in GL3 inclusions in renal interstitial capillary endothelial cells as well as in plasma lyso-GL3 relative to placebo.7 Qualitative assessments of kidney biopsies after 12 months of migalastat treatment in 23 patients with migalastat amenable GLA (α-galactosidase A enzyme) mutations showed decreases in GL3 in podocytes in 22%, in glomerular endothelial cells in 26% and in mesangial cells in 48% of patients.7 Additionally, after 24 months, significant decreases from baseline were observed in left-ventricular-mass index and gastrointestinal symptoms.7
We previously demonstrated that the notion of relative insensitivity of podocyte to ERT-mediated GL3 reduction is, in part, methodological. Thus, based on previously reported scoring methods there were no early (11 months) reductions in podocyte GL3 with ERT,3 and after 54 months there was incomplete clearance in some patients and none in others.4 However, these previously reported scoring methods are insensitive to changes in podocyte size (volume). In fact, after 11–12 months of ERT, using quantitative electron microscopic morphometric methods, we were able to demonstrate substantial reductions in total volume of GL3 inclusions per podocyte (V(Inc/PC)) while there was no change in the fraction of podocyte cytoplasm filled with GL3 inclusions (Vv(Inc/PC)). This was because, with ERT, there was a parallel decrease in mean podocyte volume and V(Inc/PC) while Vv(Inc/PC) remained constant.8
The present report details results of the application of these quantitative methods to renal biopsies of eight male patients with Fabry disease with amenable mutations from the FACETS study after 6 months of treatment with migalastat.
Methods
Patients
Eight male subjects with amenable mutations as determined by the Good Laboratory Practice-validated human embryonic kidney cell assay7 9 had renal biopsies performed at baseline and after 6 months of treatment with migalastat. These were all the male patients with amenable mutations enrolled in the FACETS study for whom informed consent was available to conduct additional kidney biopsy assessments and where adequate both the baseline and month 6 biopsies were available. Treatment consisted of 6 months of migalastat hydrochloride (150 mg) every other day. Biopsies from nine healthy live kidney donors, aged 16–52 years, were used as controls.
Globotriaosylsphingosine
Plasma globotriaosylsphingosine (lyso-Gb3) levels were analysed by means of liquid chromatography–mass spectroscopy as previously described.7 10
Renal biopsies
Biopsies were fixed in 2.5% glutaraldehyde and embedded in Poly/Bed; 1 µm sections were stained with toluidine blue for identification of glomeruli and scoring of GL3 inclusions in podocytes.11 Random glomerular sections were prepared for stereological studies as described.12 Overlapping digital low magnification (~10 000×) images of the entire glomerular profiles were obtained using a JEOL CX100 electron microscope for estimation of podocyte volume as described below. High magnification (~30 000×) images were obtained according to a systematic uniform random sampling protocol for estimation of volume fraction of GL3 inclusions in cytoplasm of glomerular endothelial cells (Vv(Inc/Endo)), mesangial cells (Vv(Inc/Mes)), podocytes (Vv(Inc/PC)), average podocyte volume and FPW as previously described.12
Estimation of podocyte volume and absolute volume of GL3 inclusions per podocyte
Average volume of podocyte nuclei was estimated using the point-sampled intercept method13 with slight modification to reduce the volume-weighted property of the method.8 This provides shape and size independent estimates of the volume. Volume fraction of podocyte nuclei per podocyte (Vv(PCN/PC)) was estimated using point counting (see online supplementary information). The average volume of podocytes () was calculated:
The absolute volume of GL3 per podocyte was then calculated:
All stereological estimates were done by masked observers.
Statistical analyses
Statistica V.12.0 (StatSoft) was used. Parametric or non-parametric tests were used as appropriate based on the variable characteristics and distribution. Data are presented as mean ± SD, except where indicated. Comparison of baseline and postmigalastat variables was done using paired Student’s t-test (parametric) or Wilcoxon matched pairs test (non-parametric). Relationships between variables were evaluated using Pearson correlation. p≤0.05 was considered statistically significant.
Results
The patients were male and ranged in age from 25 to 60 (42.6±11.0 (mean ± SD)) years (table 1). Estimated glomerular filtration rate (eGFR), determined with the use of the Chronic Kidney Disease Epidemiology Collaboration equation,14 ranged from 119 to 41 (92.8±25.8) mL/min/1.73 m2 and decreased with increasing age (r=−0.847, p=0.008) (table 1). All had increased urinary protein excretion rates (291(161–918) mg/24 hours; median (range)). Albumin/creatinine ratio was 6.5 (1–34), median (range), g/g.
Table 1.
Demographical and clinical characteristics of eight male patients with Fabry disease with amenable mutations
| Number | Age (years) | GLA mutation* | Plasma lyso-Gb3 (nmol) | eGFR (mL/min/1.73 m2) | UPr-24 (mg) | ACR (g/g) |
| 1 | 25 | c.[164A>T; 170A>T] (D55V/Q57L) | 92 | 114 | 198 | 6 |
| 2 | 34 | c.6474>G (Y216C) | 128 | 119 | 400 | 16 |
| 3 | 35 | c.431G>T (G144V) | 120 | 105 | 240 | 1 |
| 4 | 45 | c.729G>C (L243F) | 109 | 102 | 161 | 2 |
| 5 | 45 | c.776C>G (P259R) | 113 | 105 | 335 | 7 |
| 6 | 45 | c.466G>A (A156T) | 218 | 74 | 247 | 4 |
| 7 | 52 | c.98A>G (D33G) | 52 | 82 | 367 | 9 |
| 8 | 60 | c.996C>G (D322E) | 82 | 41 | 918 | 34 |
*Nucleotide change (protein sequence change).
ACR, urinary albumin/creatinine ratio; eGFR, estimated glomerular filtration rate; GLA, α-galactosidase A enzyme; GL3, globotriaosylceramide; UPr-24, 24-hour urinary protein excretion.
Podocytes were not cleared from the GL3 inclusions. While podocytes overall appeared to be smaller at 6 months compared with the baseline (figure 1A), they still contained frequent GL3 inclusions at 6 months. However, unbiased stereology revealed that V(Inc/PC) was reduced from baseline to 6 months post-treatment (p=0.02, figure 1B). This was associated with a parallel decrease in mean podocyte volume (figure 1C; p=0.004) while there was no change in Vv(Inc/PC) (figure 1D). In fact, the decrease in mean podocyte volume and V(Inc/PC) was highly correlated (r=0.98, p=0.00003).
Figure 1.
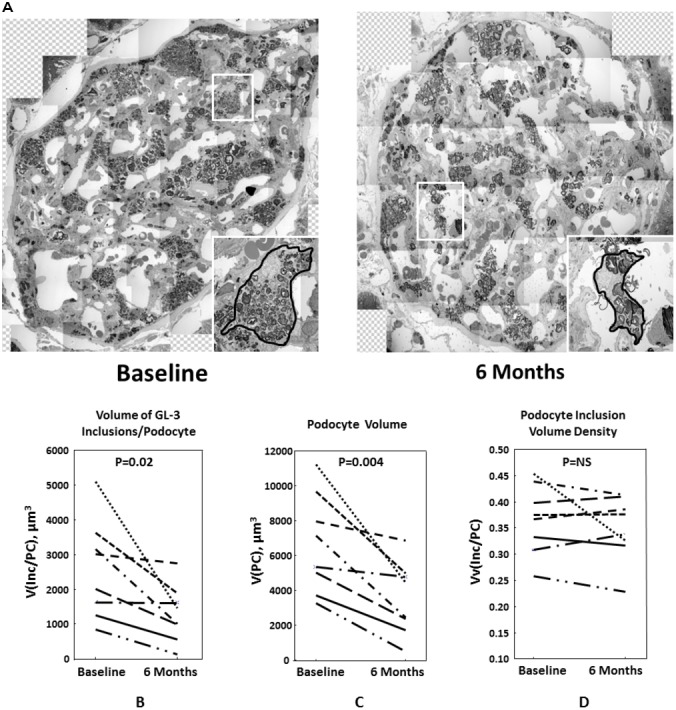
(A) Two representative glomeruli from a male patient with Fabry disease with amenable mutation at baseline (left) and 6 months (right) after migalastat treatment. Inlays show magnified views of two representative podocytes. The podocyte after 6 months migalastat (right) still contains many GL3 (globotriaosylceramide) inclusions, but is smaller than the podocyte at baseline (left). (B–D) Each line represents one case at baseline (left) and 6 months after migalastat treatment (right). (B) Volume of GL3 inclusions/podocyte (V(Inc/PC)) at baseline and after 6 months of migalastat treatment in patients with Fabry disease. (C) Mean podocyte volume (V(PC)) at baseline and after 6 months of migalastat treatment in patients with Fabry disease. (D) Volume density or volume fraction of GL3 inclusions/podocyte (Vv(Inc/PC)) at baseline and after 6 months of migalastat treatment in patients with Fabry disease.
FPW, available in seven of eight subjects, was increased compared with normal controls (n=9) at baseline (p=0.002) and at 6 months (p=0.01, figure 2). FPW was reduced in 5/7 and increased in 2/7 cases after 6 months of migalastat therapy, but this was not statistically significant (figure 2). However, the change from baseline to 6 months in FPW correlated with the changes in mean podocyte volume (r=0.89, p=0.007; figure 3A) and V(Inc/PC) (r=0.82, p=0.02; figure 3B).
Figure 2.

Foot process width in normal controls (circles on the left side) and at baseline and after 6 months of migalastat treatment in patients with Fabry disease. Each line represents one case at baseline (left) and 6 months after migalastat treatment (right).
Figure 3.

(A) Change in the volume of inclusions per podocyte (∆V(Inc/PC)) and change in foot process width at baseline and after 6 months of migalastat treatment in patients with Fabry disease. (B) Change in the mean volume of podocytes (∆V(PC)) and change in foot process width at baseline and after 6 months of migalastat treatment in patients with Fabry disease.
There were no statistically significant changes in eGFR, albuminuria or proteinuria over 6 months treatment (data not shown). There was, however, a trend towards a correlation between the change in 24 hours urine protein excretion and the change in V(Inc/PC) (r=0.69, p=0.06).
Importantly, the decrease in plasma lyso-Gb3 with migalastat treatment correlated with per cent change in Vv(Inc/PC) (r=0.82, p=0.02) and V(Inc/PC) (r=0.79, p=0.02) from baseline to 6 months (figure 4A,B).
Figure 4.

(A) Change in volume fraction of GL3 inclusions/podocyte (∆Vv(Inc/PC)) and change in plasma lyso-Gb3 at baseline and after 6 months of migalastat treatment in patients with Fabry disease. (B) Change in volume of GL3 inclusions/podocyte (∆V(Inc/PC)) and change in plasma lyso-Gb3 at baseline and after 6 months of migalastat treatment in patients with Fabry disease. GL3, globotriaosylceramide.
Discussion
The highlight of our study is observation of partial clearance of Fabry podocytes from GL3 inclusions following 6 months of migalastat treatment. Although qualitative evaluation of podocyte GL3 content showed a reduction in only 22% of cases after 12 months of migalastat,7 our quantitative methods detected a decrease in V(Inc/PC) in all eight male patients with amenable mutations after only 6 months of treatment. This is best explained by the parallel and remarkably highly correlated reduction in mean podocyte volume with no change in Vv(Inc/PC). This lack of change in Vv(Inc/PC) makes the overall reduction in GL3 inclusions per podocyte very difficult to appreciate by scoring. In fact, we have previously shown that a masked renal pathologist could not discern by scoring the marked reduction in GL3 inclusions per podocyte in men with Fabry disease treated with ERT for ~1 year while, as in the present study, there was a regular and parallel decrease in mean podocyte volume and V(Inc/PC).8
In order to determine if this partial clearance of podocytes from GL3 was associated with reduced podocyte injury, we estimated widening or effacement of foot processes, a regular concomitant of podocyte injury.15 Although there was no statistically significant change in FPW following migalastat treatment, the change in FPW and changes in podocyte size and GL3 content were directly correlated, suggesting that partial podocyte GL3 clearance may contribute to a reduction in podocyte injury. This is supported by the statistical trend for a correlation between the change in V(Inc/PC) and the change in urinary protein excretion over the 6 months of the study.
The stabilisation of amenable mutant αGal-A by migalastat given every 2 days has been hypothesised to increase effective intracellular enzyme levels more consistently than ERT given every 2 weeks.16 Given that migalastat is a low-molecular-weight iminosugar that is excreted largely unchanged in the urine, it is possible that it has easier access across the glomerular filtration barrier than the ~100 KD agalsidase-β,17 thus having theoretical advantage for treatment of the relatively ERT resistant podocytes.
Lyso-Gb3 is a deacylated form of GL3 which increases as a result of Fabry disease.18 Elevated plasma lyso-Gb3 has been recognised as a sensitive marker for Fabry disease.19 20
The correlation between the change in plasma lyso-Gb3 and the change in V(Inc/PC) with migalastat treatment in the present study is a potentially important finding. These findings, suggesting that plasma lyso-Gb3 may be a useful minimally invasive biomarker that correlates with podocyte GL3 content, are deserving of further study. Sanchez-Niño et al showed that addition of lyso-Gb3 to conditionally immortalised human podocytes in vitro increases expression of transforming growth factor beta 1, extracellular proteins and CD74, suggesting that lyso-Gb3 may have a role in podocyte injury and glomerulosclerosis.21 In another study, Sanchez-Niño et al showed that lyso-Gb3 activates Notch1, a mediator of podocyte injury, in cultured podocytes.22 Thus, reduction in plasma lyso-Gb3 may reflect reduced podocyte GL3 content, and may indicate reduced podocyte injury through other mechanisms.
Taken together, the present study showing a consistent reduction in podocyte GL3 content after 6 months of migalastat, and the earlier report suggesting migalastat treatment benefits on renal peritubular capillary endothelial cells, left-ventricular-mass index, and gastrointestinal symptoms and the apparent stabilisation in GFR over 24 months7 are consistent with multiorgan benefits of this treatment in patients with Fabry disease with amenable mutations. Longer term studies on the effects of migalastat in the treatment of Fabry disease will be of great interest.
Acknowledgments
We would like to thank Ms Frida Meiers, Karen Zaruba, Ann Palmer and Zour Yang for their electron microscopy work. We also thank the National Institutes of Health Lysosomal Disease Network (a part of the NCATS Rare Diseases Clinical Research Network (RDCRN)) for providing support (U54NS065768) that led to development of the methodology used in this manuscript for quantification of GL3 inclusions in podocytes.
Footnotes
Contributors: BN and MM were involved in conception of the work, data collection, data analysis and interpretation, and drafting the article. AS was involved in data collection. JB, JC, HW and EB were involved in conception of the work, data collection, interpretation and drafting the article. All authors approved the final version of the manuscript for publication.
Funding: This work was funded by Amicus Corporation.
Competing interests: BN is a recipient of investigator initiated Genzyme research grants, a consultant to Genzyme and has received speaker’s honoraria and travel support from Genzyme. He is also a member of the Medical Advisory Board of Amicus and performs kidney biopsy studies for Amicus. These interests have been reviewed and managed by the University of Washington in accordance to its conflict of interest policies. MM is a member of the Genzyme sponsored North American Fabry Registry Advisory Board, a recipient of investigator initiated Genzyme research grants, a consultant to Genzyme for clinical trial design, and a speaker at Genzyme educational meetings. These interests have been reviewed and managed by the University of Minnesota in accordance to its conflict of interest policies. He is also a consultant to and performs kidney biopsy studies for Amicus and has served as grant reviewer for Shire.
Patient consent: Obtained.
Ethics approval: Institutional Review Board and Ethics Committees of participating institutions in the FACETS study.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Desnick RJ, Allen KY, Desnick SJ, Raman MK, Bernlohr RW, Krivit W. Fabry’s disease: enzymatic diagnosis of hemizygotes and heterozygotes. Alpha-galactosidase activities in plasma, serum, urine, and leukocytes. J Lab Clin Med 1973;81:157–71. [PubMed] [Google Scholar]
- 2. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet 2001;38:750–60. 10.1136/jmg.38.11.750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, Desnick RJ, O’Callaghan M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int 2002;62:1933–46. 10.1046/j.1523-1755.2002.00675.x [DOI] [PubMed] [Google Scholar]
- 4. Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, Lee P, Loew T, Vedder AC, Abichandani R, Wilcox WR, Guffon N. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol 2007;18:1547–57. 10.1681/ASN.2006080816 [DOI] [PubMed] [Google Scholar]
- 5. Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ. Fabry Disease Clinical Trial Study Group. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007;146:77–86. [DOI] [PubMed] [Google Scholar]
- 6. Warnock DG, Ortiz A, Mauer M, Linthorst GE, Oliveira JP, Serra AL, Maródi L, Mignani R, Vujkovac B, Beitner-Johnson D, Lemay R, Cole JA, Svarstad E, Waldek S, Germain DP, Wanner C. Fabry Registry. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant 2012;27:1042–9. 10.1093/ndt/gfr420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F, Amartino H, Bratkovic D, Feldt-Rasmussen U, Nedd K, Sharaf El Din U, Lourenco CM, Banikazemi M, Charrow J, Dasouki M, Finegold D, Giraldo P, Goker-Alpan O, Longo N, Scott CR, Torra R, Tuffaha A, Jovanovic A, Waldek S, Packman S, Ludington E, Viereck C, Kirk J, Yu J, Benjamin ER, Johnson F, Lockhart DJ, Skuban N, Castelli J, Barth J, Barlow C, Schiffmann R. Treatment of fabry’s Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med 2016;375:545–55. 10.1056/NEJMoa1510198 [DOI] [PubMed] [Google Scholar]
- 8. Najafian B, Tøndel C, Svarstad E, Sokolovkiy A, Smith K, Mauer M. One Year of Enzyme Replacement Therapy Reduces Globotriaosylceramide Inclusions in Podocytes in Male Adult Patients with Fabry Disease. PLoS One 2016;11:e0152812 10.1371/journal.pone.0152812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, Bronfin B, Williams H, Yu J, Bichet DG, Germain DP, Giugliani R, Hughes D, Schiffmann R, Wilcox WR, Desnick RJ, Kirk J, Barth J, Barlow C, Valenzano KJ, Castelli J, Lockhart DJ. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med 2017;19:430–8. 10.1038/gim.2016.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boutin M, Auray-Blais C. Multiplex tandem mass spectrometry analysis of novel plasma lyso-Gb₃-related analogues in Fabry disease. Anal Chem 2014;86:3476–83. 10.1021/ac404000d [DOI] [PubMed] [Google Scholar]
- 11. Fogo AB, Bostad L, Svarstad E, Cook WJ, Moll S, Barbey F, Geldenhuys L, West M, Ferluga D, Vujkovac B, Howie AJ, Burns A, Reeve R, Waldek S, Noël LH, Grünfeld JP, Valbuena C, Oliveira JP, Müller J, Breunig F, Zhang X, Warnock DG. all members of the International Study Group of Fabry Nephropathy (ISGFN). Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol Dial Transplant 2010;25:2168–77. 10.1093/ndt/gfp528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Najafian B, Svarstad E, Bostad L, Gubler MC, Tøndel C, Whitley C, Mauer M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int 2011;79:663–70. 10.1038/ki.2010.484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gundersen HJ, Jensen EB. Stereological estimation of the volume-weighted mean volume of arbitrary particles observed on random sections. J Microsc 1985;138(Pt 2):127–42. 10.1111/j.1365-2818.1985.tb02607.x [DOI] [PubMed] [Google Scholar]
- 14. Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, Feldman HI, Kusek JW, Eggers P, Van Lente F, Coresh J, Coresh J. CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med 2009;150:604–12. 10.7326/0003-4819-150-9-200905050-00006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV. The podocyte’s response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol 2013;304:F333–F347. 10.1152/ajprenal.00478.2012 [DOI] [PubMed] [Google Scholar]
- 16. Khanna R, Soska R, Lun Y, Feng J, Frascella M, Young B, Brignol N, Pellegrino L, Sitaraman SA, Desnick RJ, Benjamin ER, Lockhart DJ, Valenzano KJ. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol Ther 2010;18:23–33. 10.1038/mt.2009.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bishop DF, Desnick RJ. Affinity purification of alpha-galactosidase A from human spleen, placenta, and plasma with elimination of pyrogen contamination. Properties of the purified splenic enzyme compared to other forms. J Biol Chem 1981;256:1307–16. [PubMed] [Google Scholar]
- 18. Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, Strijland A, Ottenhoff R, van Roomen C, Mirzaian M, Wijburg FA, Linthorst GE, Vedder AC, Rombach SM, Cox-Brinkman J, Somerharju P, Boot RG, Hollak CE, Brady RO, Poorthuis BJ. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008;105:2812–7. 10.1073/pnas.0712309105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rombach SM, Dekker N, Bouwman MG, Linthorst GE, Zwinderman AH, Wijburg FA, Kuiper S, Vd Bergh Weerman MA, Groener JE, Poorthuis BJ, Hollak CE, Aerts JM. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta 2010;1802:741–8. 10.1016/j.bbadis.2010.05.003 [DOI] [PubMed] [Google Scholar]
- 20. Togawa T, Kodama T, Suzuki T, Sugawara K, Tsukimura T, Ohashi T, Ishige N, Suzuki K, Kitagawa T, Sakuraba H. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol Genet Metab 2010;100:257–61. 10.1016/j.ymgme.2010.03.020 [DOI] [PubMed] [Google Scholar]
- 21. Sanchez-Niño MD, Sanz AB, Carrasco S, Saleem MA, Mathieson PW, Valdivielso JM, Ruiz-Ortega M, Egido J, Ortiz A. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol Dial Transplant 2011;26:1797–802. 10.1093/ndt/gfq306 [DOI] [PubMed] [Google Scholar]
- 22. Sanchez-Niño MD, Carpio D, Sanz AB, Ruiz-Ortega M, Mezzano S, Ortiz A. Lyso-Gb3 activates Notch1 in human podocytes. Hum Mol Genet 2015;24:5720–32. 10.1093/hmg/ddv291 [DOI] [PubMed] [Google Scholar]