

Abstract
Previous results have indicated that mitochondrial ATP-sensitive potassium (mitoKATP) channels are associated with the hypoxic proliferation of pulmonary artery smooth muscle cells (PASMCs). However, the mechanism underlying the promotive effects of mitoKATP channels on cell proliferation in response to hypoxia remains unknown. mitoKATP channel opening results in a collapse of mitochondrial membrane potential and generation of mitochondrial reactive oxygen species (ROS). As hypoxia-inducible factor-1α (HIF-1α) is a critical oxygen sensor and major transcriptional regulator of the hypoxic adaptive response, the current study assessed whether mitoKATP opening contributes to the chronic proliferation of human PASMCs (hPASMCs) in collaboration with HIF-1α and its downstream targets under hypoxic conditions. The present study demonstrated that there was crosstalk between mitoKATP channels and HIF-1α signaling in PASMCs under hypoxic conditions. The results suggest that mitoKATP channels are involved in the proliferation of PASMCs during hypoxia through upregulation of the ROS/HIF/microRNA-210/iron-sulfur cluster protein signaling pathway.
Keywords: mitochondrial ATP-sensitive potassium channel, pulmonary artery smooth muscle cells, hypoxia, hypoxia-inducible factor-1α, microRNA-210, iron-sulfur cluster protein, mitochondrial membrane potential
Introduction
Pulmonary arterial hypertension (PAH) is a life-threatening disorder characterized by obstructive remodeling of the pulmonary arteries, which may lead to right-sided heart failure and mortality (1–2). PAH may be defined as a mean pulmonary artery pressure (mPAP) of ≥20 mmHg at rest or >30 mmHg with exercise, along with a pulmonary artery occlusion pressure of ≤15 mmHg (1–2). The estimated incidence of primary pulmonary hypertension is 1–2/million in the global population (1–2). There are currently three treatment pathways, namely phosphodiesterase type 5 inhibitor or soluble guanylate cyclase stimulator, prostacyclin class therapy and endothelium receptor antagonist, which target the imbalances of three substances; nitric oxide, prostacyclin and endothelin, respectively (3). Despite the efficacy of these pharmacological therapies in improving symptoms, they do not prevent the progression of PAH or reduce the mortality rate of patients (3). Therefore, novel approaches that more effectively target PAH are required to control the cellular components associated with pulmonary remodeling. The chronic and continued proliferation of human pulmonary artery smooth muscle cells (hPASMCs), in addition to apoptotic resistance, leads to hypoxic pulmonary arterial remodeling, which is considered to be a key mechanism for the pathological development of PAH (4). ATP-sensitive potassium (KATP) channels are located on the cytoplasmic membrane and on subcellular membranes. Subcellular membrane-associated KATP channels are divided into three types: Sarcolemmal KATP, mitochondrial KATP (mitoKATP) and nuclear KATP. MitoKATP channels, which contribute to the control of mitochondrial volume and energetic status (5–7), exhibit high sensitivity to hypoxia (8). Closed mitoKATP channels under normoxic conditions are opened when cells are subjected to hypoxia, and depolarization of the mitochondrial membrane potential (ΔΨm) is induced accordingly (9). In addition, previous studies indicate that the opening of mitoKATP channels leads to the generation of reactive oxygen species (ROS) (5–7). ROS are considered to be a double-edged sword, and their effect on cells as either a survival or apoptotic signal is controlled by the dosage, duration, type and site of ROS production (5–7). Furthermore, opening of mitoKATP channels followed by ΔΨm depolarization may contribute to increased expression of hypoxia-inducible factor-1α (HIF-1α) and proliferation of PASMCs (10). A previous study demonstrated that mitoKATP channel opening participates in ROS overproduction in hPASMCs through ΔΨm depolarization, which indicates the involvement of mitoKATP in the chronic proliferation and/or enhanced apoptotic resistance of hPASMCs (11). However, the molecular mechanism underlying the role of mitoKATP channels in the abnormal proliferation or apoptotic resistance of hPASMCs remains unknown.
The present study investigated whether mitoKATP channels promote the aberrant prolonged proliferation of hPASMCs by assessing ΔΨm and intracellular ROS. The results indicated that mitoKATP channels contribute to hypoxic pulmonary arterial remodeling via regulation of the ROS/HIF/microRNA (miR)-210/iron-sulfur cluster protein (ISCU) signaling pathway.
Materials and methods
Cell culture and treatment
hPASMCs were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in smooth muscle cell medium (ScienCell Research Laboratories, Inc.) supplemented with 2% fetal bovine serum (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin, 100 µg/ml streptomycin and 1% smooth muscle cell growth supplement (all ScienCell Research Laboratories) in an atmosphere containing 5% CO2 at 37°C. The culture medium was replaced every 2 to 3 days until cell 80% confluence was reached. hPASMCs at passages 4–10 were used in the following experimental assays. Smooth muscle cells were confirmed by immunohistochemistry using anti-α-actin monoclonal antibody (dilution 1:300; sc-130616; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) incubated at 37°C for 2 h. The cells were cultured at 37°C in hypoxic conditions (5% CO2 and 5% O2) or normoxic conditions (1% CO2 and 20% O2) as control for 6, 12 and 24 h respectively, prior to the experiment. Subsequently, after treatment with mitoKATP inhibitor 5-hydroxydecanoate (5-HD; 500 µmol/l), mitoKATP agonist diazoxide (100 µmol/l) or HIF-1α inhibitor 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1; 50 µmol/l; all from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) or smooth muscle cell medium supplemented with 2% fetal bovine serum as control for 1 h, immediately the cells were further maintained at 37°C in a humidified atmosphere of 5% CO2 and 5% O2 for 6,12 and 24 h.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
mRNA and miRNA PCR experiments were performed in triplicate. For mRNA expression assay, total RNA was extracted from treated cells using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). cDNA was synthesized using a ReverTra Ace qPCR RT kit (Toyobo Co., Ltd., Osaka, Japan). RNA concentration and purity were assessed by UV spectrophotometry (1.8<A260/A280<2.0). RNA integrity was assessed using electrophoresis. Reverse transcription reaction was performed using 4 µg of total RNA and the First Strand cDNA Synthesis kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. To generate standard curves, 1 µl of first-strand cDNA was amplified using a Premix Ex Taq Version Kit (Takara Bio, Inc., Otsu, Japan) according to the manufacturer's instructions and quantification of PCR products was assessed to plot standard curves. The qPCR reactions were performed using a SYBR Green PCR Master mix (CWBIO; Beijing, China) in a CFX-96 system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using the following primers: HIF-1α, forward 5′-CTGATCATCTGACCAAAACTC-3′ and reverse 5′-GTTTCAACCCAGACATATCCAC-3′; and β-actin, forward 5′-ACTCTTCCAGCCTTCCTTCC-3′ and reverse 5′-CGTCATACTCCTGCTTGCTG-3′. Real-time PCR was performed using the SYBR Premix Ex TaqTM II (Takara Bio, Inc.) protocol on a Bio-Rad Connect real-time PCR detection system with cycling conditions of 95°C for 3 min, followed by 40 cycles of 95°C for 30 sec and 60°C for 30 sec. β-actin was used as an internal control. The relative mRNA expression levels were determined using the 2−ΔΔCq method and normalized to β-actin mRNA (12,13). For the miR-210 expression assay, total cellular RNA was extracted with TRIzol reagent. cDNA was synthesized by reverse transcription using the miRNART-primer (RibBio Co., Ltd., Guangzhou, China). The qPCR reactions were performed using a SYBR Green PCR Master mix using miR-210 and U6 primer purchased from RibBio Co., Ltd. (sequences not supplied), which served as normalization control. qPCR was performed under the following steps: 95°C for 3 min, followed by 40 cycles of 95°C for 30 sec and 56°C for 30 sec. miR-210 expression relative to U6 control was calculated using the 2−ΔΔCq method (14).
Measurement of ΔΨm and intracellular ROS
The ΔΨm was monitored using flow cytometry according to a modified method (15). Briefly, treated cells were detached by trypsinization and washed twice in PBS. A total of 10 µg/ml rhodamine-123 (R-123, Sigma-Aldrich; Merck KGaA) was then added and incubated for 30 min at 37°C in the dark. The fluorescence intensity of cells was measured using a FACSAria II cytometer (BD643182; BD Biosciences, San Jose, CA, USA).
Measurement of mitochondrial intracellular ROS was performed as described previously (11). The fluorescent probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA) was purchased from Sigma-Aldrich; Merck KGaA.
RNA transfection
A total of 2×105 cells were plated into 6-well plates ins mooth muscle cell medium supplemented with 2% fetal bovine serum in normoxic conditions (1% CO2 and 20% O2) at 37°C for 24 h. Cells were transfected with miR-210 mimic (miR-210) or inhibitor (anti-210) (RibBio Co., Ltd.) at a final concentration of 80 nmol/l, or three small interfering RNAs (siRNA1, siRNA2 and siRNA3) against ISCU (GenePharma, Shanghai, China) at a final concentration of 5 nmol/l using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) with serum-free smooth muscle cell medium, as described previously (11). The follow sequences were used: miR-210 mimic, forward 5′-CUGUGCGUGUGACAGCGGGUGA-3′ and reverse 5′-UUGACACGCACACUGUCGCCGA-3′; and inhibitor, forward 5′-UCAGCCGCUGUCACACGCACAG-3′ and reverse 5′-CAGUACUUUUGUGUAGUACAA-3′. After 6 h, the culture medium was replenished with fresh smooth muscle cell medium supplemented 2% fetal bovine serum and cells were cultured for an additional 24 h at 37°C, then subjected to hypoxia (5% CO2 and 5% O2) and normoxia (1% CO2 and 20% O2) at 37°C for 24 h. Control miR sequences and control siRNA sequences were used (miR-con, anti-con, si-con) as negative control. After miR-210 inhibitor and control transfection for 24 h, cells were treated with 5-HD (500 µmol/l) or diazoxide (100 µmol/l) and exposed to hypoxia for 48 h at 37°C.
Western blot analysis
Cells were lysed with lysis buffer (1% Triton X-100, 50 mmol/l Tris-HCl, pH 7.4, 25 mmol/l glycerophosphate, 150 mmol/l NaCl, 2 mmol/l EDTA, 2 mmol/l ethylene glycol tetraacetic acid, 1 mmol/l phenylmethylsulfonyl fluoride, 10% glycerol and 1% protease and phosphatase inhibitors) for 10 min on ice. The lysates were collected and centrifuged at 15,000 × g for 15 min at 4°C. The supernatant was recovered, and protein concentration was measured using a protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). The proteins loaded (40 mg per lane) were subsequently separated by 12% SDS-PAGE and transferred to a polyvinylidene difluoride membrane (PVDF; Thermo Fisher Scientific, Inc.), blocked at 37°C for 2 h in 5% non-fat milk, and incubated with anti- proliferating cell nuclear antigen (PCNA; sc-25280) or anti-ISCU antibody (sc-271536) or β-actin antibody (sc-47778) or GAPDH (sc-25778) at 1:4,000 or 1:3,000 or 1:10,000 or 1:5,000, respectively (Santa Cruz Biotechnology, Inc.) at 4°C overnight. After washing for 10 min in 1X Tris-buffered saline-Tween-20 solution three times, the target protein was probed with the horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (sc-3841; at 1:6,000 dilution; Santa Cruz Biotechnology, Inc.) at 37°C for 2 h. After washing for 10 min in 1X Tris-buffered saline-Tween-20 solution three times, the bound antibody on the PVDF membrane was detected by chemiluminescence with the ECL Detection Reagent kit (Pierce; Thermo Fisher Scientific, Inc.) and the protein expression was analyzed using Quantity One 4.6.2 (Bio-Rad Laboratories, Inc.) (16).
Statistical analysis
Statistical analysis was performed using one-way analysis of variance and Duncan's test using Graphpad software (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA). The data were expressed as the mean ± standard deviation in three independent experiments. The statistical significance of differences among groups was determined using an analysis of variance or Student's t-test. P<0.05 was considered to represent a statistically significant difference.
Results
MitoKATP increases ROS generation in hPASMCs through the collapse of ΔΨm
Hypoxia-triggered mitoKATP channel opening initiated by ATP depletion leads to the collapse of ΔΨm, which has been implicated in the generation of ROS (17). Consistent with a previous report (11), the present study observed that R-123 fluorescence intensity was significantly increased in hPASMCs subjected to hypoxia for 12 and 24 h (P<0.05 and P<0.01, respectively), relative to normoxic control cells (Fig. 1A), indicating that ΔΨm depolarization had occurred. Furthermore, hPASMCs exhibited significantly increased levels of ROS after 12 and 24 h of hypoxia (P<0.05), as determined by DCFH-DA staining (Fig. 1B). To further investigate whether mitoKATP channels were associated with ROS production in hPASMCs, the levels of intracellular ROS were measured following treatment with diazoxide or 5-HD, to activate or inhibit mitoKATP, respectively. As predicted, opening of mitoKATP significantly increased the level of intracellular ROS, and closure of mitoKATP significantly decreased the level of intracellular ROS under hypoxic conditions (P<0.05; Fig. 1C). Furthermore, under normoxic conditions, the opening of mitoKATP significantly increased the level of intracellular ROS while closure of mitoKATP significantly decreased the level of intracellular ROS (P<0.05; Fig. 1C). These results suggest that mitoKATP channels may be involved in the generation of ROS in hPASMCs under hypoxic conditions, potentially through the collapse of ΔΨm.
Figure 1.

MitoKATP channel opening increases intercellular ROS in hPASMCs. (A) The ΔΨm was assessed with R-123 fluorescence after cells were exposed to hypoxia for 12 or 24 h. (B) Levels of intracellular ROS were assessed by DCFH-DA fluorescence after cells were exposed to hypoxia for 12 or 24 h. (C) The intracellular ROS levels were assessed by DCFH-DA fluorescence after cells were treated with 5-HD or diazoxide upon normoxia or hypoxia. *P<0.05 and **P<0.01 vs. control. Data are expressed as percentage of control (n=3), and were analyzed by a Student's t-test. MitoKATP, mitochondrial ATP-sensitive potassium channel; ROS, reactive oxygen species; hPASMCs, human pulmonary artery smooth muscle cells; ΔΨm, mitochondrial membrane potential; R-123, rhodamine-123; DCFH-DA, 2′,7′-dichlorofluorescin diacetate; 5-HD, 5-hydroxyde-canoate FITC, fluorescein isothiocyanate.
MitoKATP interacts with HIF-1α in a positive feedback manner in hPASMCs
As HIF-1α is a critical oxygen sensor and master transcriptional regulator of the hypoxic adaptive response, it has been suggested that mitoKATP channels function in combination with HIF-1α and its downstream targets, including miR-210 and ISCU (12,18). To investigate whether mitoKATP was associated with the expression of HIF-1α and miR-210 in hPASMCs during hypoxia, the expression of HIF-1α mRNA and miR-210 was measured using RT-qPCR. In hPASMCs subjected to hypoxia, levels of HIF-1α mRNA were significantly increased at 12 and 24 h (P<0.01 and P<0.001, respectively), and miR-210 levels were significantly increased at 6 and 12 h (P<0.05; Fig. 2A). In turn, expression of miR-210 was significantly decreased following treatment with the HIF-1α inhibitor YC-1 (P<0.01; Fig. 2B), and expression of HIF-1α was significantly increased following treatment with the mitoKATP agonist diazoxide (P<0.05; Fig. 2C). Furthermore, the levels of HIF-1α mRNA and miR-210 were significantly decreased following closure of mitoKATP with 5-HD (P<0.001 and P<0.01, respectively; Fig. 2C). To further investigate whether HIF-1α was involved in the regulation of mitoKATP, ΔΨm and ROS levels were measured in hPASMCs treated with YC-1. Significant decreases in the fluorescence intensity of R-123 (P<0.05) and DCFH-DA (P<0.01) were observed in hPASMCs upon inhibition of HIF-1α (Fig. 2D).
Figure 2.

MitoKATP interacts with HIF-1α in a positive feedback manner. (A) Expression of HIF-1α and miR-210 was assessed by qPCR after cells were exposed to normoxia or hypoxia for 6, 12 and 24 h. Data are expressed as the mean ± standard deviation (n=3). (B) hPASMCs were treated with YC-1 upon hypoxia for 12 h, and the expression of miR-210 was measured by qPCR. The value was relative to the control without YC-1, using U6 as a reference. (C) hPASMCs were treated with 5-HD or diazoxide in a hypoxia state for 12 h, and the expression of HIF-1α and miR-210 was measured by qPCR. The value was relative to control (untreated) cells, using β-actin as a reference. (D) hPASMCs were treated with YC-1 upon hypoxia for 12 h. Left: Intracellular ROS content analyzed by DCFH-DA fluorescence. Right: ΔΨm analyzed by R-123 fluorescence. Left and right: Results were expressed as a percentage of control (untreated) cells and were presented as the mean ± standard deviation (n=3). *P<0.05, **P<0.01 and ***P<0.001 vs. control. Data were analyzed by a Student's t-test. MitoKATP, mitochondrial ATP-sensitive potassium channel; HIF-1α, hypoxia-inducible factor-1α; miR, microRNA; qPCR, quantitative polymerase chain reaction; hPASMCs, human pulmonary artery smooth muscle cells; YC-1, 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole; 5-HD, 5-hydroxyde-canoate; DCFH-DA, 2′,7′-dichlorofluorescin diacetate; ΔΨm, mitochondrial membrane potential; R-123, rhodamine-123.
These data suggest that mitoKATP channel opening increases the expression of HIF-1α and its downstream target miR-210, leading to HIF-1α-mediated regulation of mitoKATP through increased ROS levels in a positive feedback manner in hPASMCs under hypoxic conditions.
MitoKATP participates in ROS generation in hPASMCs under hypoxic conditions through miR-210/ISCU
Previous studies have indicated that ISCU is a downstream regulatory target of miR-210 (19,20). ISCU, as an essential component of respiratory electron transfer complexes, is involved in ROS generation in the mitochondria (21). The present study blocked the expression of miR-210 using an inhibitor of miR-210. Under normoxic and hypoxic conditions, significant decreases in the fluorescence intensity of R-123 (P<0.001 and P<0.01, respectively) and DCFH-DA (P<0.01) were observed in hPASMCs upon treatment with miR-210 inhibitor transfection (Fig. 3A and B), indicating an involvement of miR-210 in the alteration of ΔΨm and generation of ROS. The expression of ISCU was subsequently assessed, and was demonstrated to be reduced or increased following treatment with miR-210 mimic or miR-210 inhibitor under normoxic conditions, respectively (Fig. 3C). As shown in Fig. 3C, ISCU expression was reduced under hypoxic conditions compared with normoxic conditions.
Figure 3.

MitoKATP participates in ROS generation in hPASMCs under hypoxic conditions via the miR-210/ISCU pathway. The (A) ΔΨm and (B) intracellular ROS levels were measured in hPASMCs transfected with miR-210 mimic or inhibitor for 24 h, prior to normoxia or hypoxia for an additional 24 h. Data are presented as a percentage of control (n=3), and analyzed by a Student's t-test. (C) Expression of ISCU was assessed using western blot analysis upon hypoxia or normoxia, or cells were transfected with miR-210 mimic or inhibitor upon normoxia for 48 h. The β-actin or GAPDH acted as loading control. (D) hPASMCs were transfected with 5 nM of siRNAs against ISCU or si-Con for 48 h. The silencing efficiencies of ISCU (si-ISCU) were measured by western blot analysis. GAPDH acted as loading control. (E) ΔΨm in hPASMCs were transfected with si-Con or si-ISCU upon normoxia for 24 h. The ΔΨm was analyzed by R-123 fluorescence. Data is expressed as a percentage of the control (n=3), and were analyzed by a Student's t-test. *P<0.05, **P<0.01 and ***P<0.001 vs. control. MitoKATP, mitochondrial ATP-sensitive potassium channel; ROS, reactive oxygen species; hPASMCs, human pulmonary artery smooth muscle cells; miR, microRNA; si-Con, scramble control; si, small interfering; ISCU, iron-sulfur cluster protein; ΔΨm, mitochondrial membrane potential; YC-1, 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole; DCFH-DA, 2′,7′-dichlorofluorescin diacetate; R-123, Rhodamine-123; GAPDH, glyceraldehyde 3-phsphate dehydrogenase; con, control; si, small interfering RNA; FITC, fluorescein isothiocyanate.
Furthermore, in order to determine the role of ISCU in the activity of mitoKATP, specific siRNA against ISCU or scrambled control siRNA was transfected into hPASMCs. Western blot analysis indicated that two of the three siRNAs specifically reduced the expression of ISCU (Fig. 3D). In addition, a significant increase in the fluorescence intensity of R-123 was observed in hPASMCs upon treatment with ISCU siRNA (P<0.01; Fig. 3E), which suggests a role of ISCU in the collapse of ΔΨm, the fluorescence intensity of R-123 was lower in hypoxic environments. Loss of ISCU may impair the function of the electron transfer chain, leading to increased generation of ROS and decreased production of ATP. Decreased levels of ATP may then trigger opening of mitoKATP channels and a collapse of ΔΨm.
These data suggest that mitoKATP channels indirectly participate in ROS generation in hPASMCs under hypoxic conditions via the miR-210/ISCU pathway. This may supplement the potential direct involvement of mitoKATP in ROS generation, mediated by channel opening and ΔΨm collapse.
MitoKATP promotes the proliferation of hPASMCs through miR-210 signaling
As hPASMCs grow relatively slowly, the proliferation of hPASMCs was evaluated by measuring the expression of PCNA, as an established marker of cell proliferation that is synthesized in the early G1 and S phases of the cell cycle (22). Results of western blot analysis showed that PCNA expression was markedly increased in hPASMCs subjected to hypoxia for 48 h, relative to normoxic controls (Fig. 4A). The proliferation of hPASMCs upon activation or inhibition of mitoKATP and inhibition of miR-210 was subsequently assessed. It was observed that activation of mitoKATP increased the expression of PCNA, while inhibition of mitoKATP and miR-210 decreased PCNA expression (Fig. 4B). These results indicate that mitoKATP promotes the proliferation of hPASMCs under hypoxic conditions through the miR-210 signaling pathway.
Figure 4.
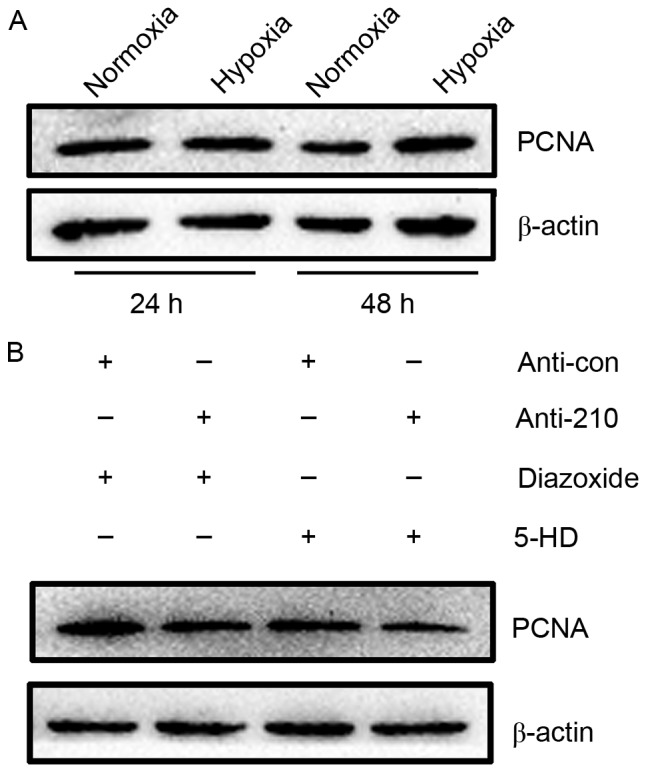
MitoKATP promoted the proliferation of hPASMCs through miR-210 signaling. (A) The expression of PCNA was assessed by western blot analysis in hPASMCs subjected to hypoxia for 24 or 48 h. (B) hPASMCs were transfected with miR-210 inhibitor or control for 24 h, then treated with 5-HD or diazoxide and exposed to hypoxia for 48 h. The expression of PCNA was assessed by western blot analysis. MitoKATP, mitochondrial ATP-sensitive potassium channel; hPASMCs, human pulmonary artery smooth muscle cells; miR, microRNA; PCNA, proliferating cell nuclear antigen; 5-HD, 5-hydroxyde-canoate; con, control.
Discussion
The proliferation and survival of hPASMCs are essential in hypoxic pulmonary arterial remodeling, which typically leads to PAH (4). KATP channels were initially identified in the inner membrane of liver mitochondria (23). Previous results suggest that the opening of mitoKATP channels, which induces the collapse of ΔΨm, contributes to the proliferation and survival of different cell types, including cardiac myocytes (24), renal epithelial cells (25), cerebellar granule neurons (26) and skin cells (27). A previous study also demonstrated that hypoxic proliferation of hPASMCs was associated with mitoKATP channel opening (11). However, the molecular mechanism underlying the promotive effect of mitoKATP channel opening on hPASMC proliferation in PAH remains unclear.
ROS has been implicated as a trigger that causes aberrant proliferation or apoptotic resistance in hPASMCs under hypoxic conditions (28). It has been reported that a collapse of ΔΨm, resulting from mitoKATP channel opening, leads to mitochondrial respiratory chain disorders associated with an increase in complex III-dependent ROS and a decrease in complex I-dependent ROS (21). Thus, complex III is considered to be a major ROS-releasing site during phases of decreased membrane potential mediated by mitoKATP channel opening (28).
However, it is established that HIF-1α is a critical regulator of the hypoxic adaptive response during hypoxia stress (22,23). Under normoxic conditions, HIF-1α is hydroxylated by HIF prolyl-hydroxylases, which enables its recognition and ubiquitination by von Hippel-Lindau E3 ubiquitin ligase, with ubiquitin serving as a signal for proteosomal degradation (24,25). As HIF prolyl-hydroxylase utilizes oxygen as a co-substrate, it is inhibited in response to hypoxia, which allows HIF-1α to function. This negative regulation mediated by HIF prolyl-hydroxylase is an additional mechanism of HIF-1α regulation linked to ROS (11,28). Increases in mitochondrial ROS induced by hypoxia has been demonstrated to contribute to HIF-1α stabilization under hypoxic conditions (29–31). As HIF-1α is a major transcriptional regulator of the cellular and developmental response to hypoxia (32,33), it is possible that crosstalk occurs between HIF-1α and mitoKATP during hPASMC proliferation under hypoxic conditions. In accordance with this assumption, the present data indicated that mitoKATP channel opening participates in the chronic proliferation of hPASMCs in collaboration with HIF-1α under hypoxic conditions.
miR-210, which is upregulated by HIF-1α, is considered to be among the most hypoxia-sensitive miRs and a microregulator of the hypoxia pathway (34). As a downstream target of miR-210 (12,33,35), ISCU is responsible for the assembly of iron sulfur clusters [Fe-S] in mitochondrial respiratory complexes (complexes I, II and III) (21,36). Dysfunction of ISCU leads to an impaired electron transport chain, which results in two consequences: i) ROS generation or ii) a decreased H+ gradient leading to suppressed production of ATP. Subsequently, mitoKATP channels are opened in response to reduced levels of ATP (11,28). Previous data suggest that HIF-1/miR-210/ISCU may act as a critical signaling axis in the regulation of mitochondrial metabolism during hypoxia (19,36–38). Consistent with these findings, results of the present study suggested that mitoKATP promotes the proliferation of hPASMCs via upregulation of the ROS/HIF/miR-210/ISCU signaling pathway.
The present study demonstrated that mitoKATP channels were involved in the proliferation of hPASMCs under hypoxic conditions. An increase in ROS during hypoxia leads to a downregulation in HIF prolyl-hydroxylase, enabling it to function as a regulator of the hypoxic response. Due to dysfunction of respiratory chain complexes in the inner mitochondrial membrane, ROS levels may be generated by two positive feedback loops: ROS/HIF/miR-210/ISCU and ATP/mitoKATP/ ΔΨm. Aberrancies in the levels of ROS ultimately lead to the proliferation of hPASMCs under hypoxic conditions. The potential association between mitoKATP and the chronic proliferation of hPASMCs suggests that targeting of mitoKATP may be a useful strategy for the treatment of hypoxia-associated pulmonary diseases, including PAH.
In conclusion, HIF-1α functions in the hypoxia adaptive response following reduced inhibition by prolyl-hydroxylase, and serves its role due to increased generation of ROS. Elevated levels of ROS, which have been associated with dysfunction of respiratory chain complexes, may be further upregulated by two positive feedback loops: ROS/HIF/miR-210/ISCU and ATP/mitoKATP/ΔΨm.
Figure 5.

Potential mechanism for the involvement of mitoKATP in the proliferation of hPASMCs under hypoxic conditions. The mitochondrial inner membrane is polarized, with a negative matrix side due to a H+ gradient generated by respiratory enzyme complexes. ATP is generated from ADP by ATP synthase and the energy is stored as the ΔΨm. MitoKATP, which may be inhibited by ATP, mediates K+ influx along the ΔΨm, causing a decrease in ΔΨm and regulation of ROS generation. Due to dysfunction of respiratory chain complexes, ROS levels are upregulated by two positive feedback loops: ROS/HIF/miR-210/ISCU and ATP/mitoKATP/ΔΨm. Along with regulation of HIF-1α, the aberrant ROS levels lead to the proliferation of hPASMCs under hypoxic conditions. MitoKATP, mitochondrial ATP-sensitive potassium channel; hPASMCs, human pulmonary artery smooth muscle cells; ATP, adenoside triphosphate; ADP, adenoside diphosphate; ΔΨm, mitochondrial membrane potential; ROS, reactive oxygen species; HIF, hypoxia-inducible factor; miR, microRNA; ISCU, iron-sulfur cluster protein.
Acknowledgements
The present study was supported by the Wuhan Health and Family Planning Commission Project (grant nos. WX14B09 and WX13B07).
Glossary
Abbreviations
- hPASMCs
human pulmonary artery smooth muscle cells
- mitoKATP
mitochondrial ATP-sensitive potassium channel
- HIF
hypoxia-inducible factor
- ROS
reactive oxygen species
- PAH
pulmonary arterial hypertension
- ΔΨm
mitochondrial membrane potential
- miR
microRNA
- 5-HD
5-hydroxydecanoate
- R-123
rhodamine-123
- ISCU
iron-sulfur cluster protein
- DCFH-DA
2′,7′-dichlorofluorescin diacetate
- YC-1
3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole
- siRNA
small interfering RNA
References
- 1.Blok IM, van Riel ACMJ, van Dijk APJ, Mulder BJM, Bouma BJ. From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: Further improvement? Int J Cardiol. 2017;227:51–52. doi: 10.1016/j.ijcard.2016.11.211. [DOI] [PubMed] [Google Scholar]
- 2.Ryan JJ, Archer SL. Emerging concepts in the molecular basis of pulmonary arterial hypertension (PAH): Part I: Metabolic plasticity and mitochondrial dynamics in the pulmonary circulation and right ventricle in pulmonary arterial hypertension. Circulation. 2015;131:1691–1702. doi: 10.1161/CIRCULATIONAHA.114.006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frumkin LR. The pharmacological treatment of pulmonary arterial hypertension. Pharmacol Rev. 2012;64:583–620. doi: 10.1124/pr.111.005587. [DOI] [PubMed] [Google Scholar]
- 4.Ibe JC, Zhou Q, Chen T, Tang H, Yuan JX, Raj JU, Zhou G. Adenosine monophosphate-activated protein kinase is required for pulmonary artery smooth muscle cell survival and the development of hypoxic pulmonary hypertension. Am J Respir Cell Mol Biol. 2013;49:609–618. doi: 10.1165/rcmb.2012-0446OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J Biol Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 6.Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, Heusch G, Cohen MV, Downey JM. Opening of mitochondrial K(ATP) channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.RES.87.6.460. [DOI] [PubMed] [Google Scholar]
- 7.Han J, Kim N, Park J, Seog DH, Joo H, Kim E. Opening of mitochondrial ATP-sensitive potassium channels evokes oxygen radical generation in rabbit heart slices. J Biochem. 2002;131:721–727. doi: 10.1093/oxfordjournals.jbchem.a003157. [DOI] [PubMed] [Google Scholar]
- 8.Teramoto N. Physiological roles of ATP-sensitive K+ channels in smooth muscle. J Physiol. 2006;572:617–624. doi: 10.1113/jphysiol.2006.105973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samavati L, Monick MM, Sanlioglu S, Buettner GR, Oberley LW, Hunninghake GW. Mitochondrial K(ATP) channel openers activate the ERK kinase by an oxidant-dependent mechanism. Am J Physiol Cell Physiol. 2002;283:C273–C281. doi: 10.1152/ajpcell.00514.2001. [DOI] [PubMed] [Google Scholar]
- 10.Zhao JP, Guo Z, Zhou ZG, Chen J, Hu HL, Wang T, Zhang ZX. Effect of opening of mitochondrial ATP-sensitive K(+) channels on the expression of hypoxia inducible factor-1alpha and cell proliferation in pulmonary arterial smooth muscle cells of rats. Sheng Li Xue Bao. 2007;59:157–162. (In Chinese) [PubMed] [Google Scholar]
- 11.Hu HL, Zhang ZX, Chen CS, Cai C, Zhao JP, Wang X. Effects of mitochondrial potassium channel and membrane potential on hypoxic human pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol. 2010;42:661–666. doi: 10.1165/rcmb.2009-0017OC. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Liu Y, Chen J, Tang MJ, Zhang SL, Wei LN, Li CH, Wei DB. Restriction-ligation-free (RLF) cloning: A high-throughput cloning method by in vivo homologous recombination of two PCR products. Genet Mol Res. 2015;14:12306–12315. doi: 10.4238/2015.October.9.19. [DOI] [PubMed] [Google Scholar]
- 13.Jin Z, Sun T, Xia X, Wei Q, Song Y, Han Q, Chen Q, Hu J, Zhang J. Optimized expression, purification of herpes B virus gD protein in Escherichia coli, and production of its monoclonal antibodies. Jundishapur J Microbiol. 2016;9:e32183. doi: 10.5812/jjm.32183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Lu YY, Chen TS, Qu JL, Pan WL, Sun L, Wei XB. Dihydroartemisinin (DHA) induces caspase-3-dependent apoptosis in human lung adenocarcinoma ASTC-a-1 cells. J Biomed Sci. 2009;16:16. doi: 10.1186/1423-0127-16-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Jin Z, Sun T, Jiang Y, Han Q, Song Y, Chen Q, Xia X. Prokaryotic expression, purification and polyclonal antibody production of a truncated recombinant rabies virus L protein. Iranian J Biotechnol. 2015;13:18–24. doi: 10.15171/ijb.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR. Relation between mitochondrial membrane potential and ROS formation. Methods Mol Biol. 2012;810:183–205. doi: 10.1007/978-1-61779-382-0_12. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Wei L, Wei D, Li X, Xu L, Wei L. Testis-specific lactate dehydrogenase (LDH-C4) in skeletal muscle enhances a pika's sprint-running capacity in hypoxic environment. Int J Environ Res Public Health. 2015;12:9218–9236. doi: 10.3390/ijerph120809218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Favaro E, Ramachandran A, McCormick R, Gee H, Blancher C, Crosby M, Devlin C, Blick C, Buffa F, Li JL, et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One. 2010;5:e10345. doi: 10.1371/journal.pone.0010345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee DC, Romero R, Kim JS, Tarca AL, Montenegro D, Pineles BL, Kim E, Lee J, Kim SY, Draghici S, et al. miR-210 targets iron-sulfur cluster scaffold homologue in human trophoblast cell lines: Siderosis of interstitial trophoblasts as a novel pathology of preterm preeclampsia and small-for-gestational-age pregnancies. Am J Pathol. 2011;179:590–602. doi: 10.1016/j.ajpath.2011.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouault TA, Tong WH. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008;24:398–407. doi: 10.1016/j.tig.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Sun K, Ding J, Xu H, Zhu L, Zhang K, Li X, Sun W. Harmine induces apoptosis and inhibits tumor cell proliferation, migration and invasion through down-regulation of cyclooxygenase-2 expression in gastric cancer. Phytomedicine. 2014;21:348–355. doi: 10.1016/j.phymed.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 24.Costa AD, Quinlan CL, Andrukhiv A, West IC, Jabůrek M, Garlid KD. The direct physiological effects of mitoK(ATP) opening on heart mitochondria. Am J Physiol Heart Circ Physiol. 2006;290:H406–H415. doi: 10.1152/ajpheart.00794.2005. [DOI] [PubMed] [Google Scholar]
- 25.Nilakantan V, Liang H, Mortensen J, Taylor E, Johnson CP. Variable effects of the mitoK(ATP) channel modulators diazoxide and 5-HD in ATP-depleted renal epithelial cells. Mol Cell Biochem. 2010;335:211–222. doi: 10.1007/s11010-009-0271-6. [DOI] [PubMed] [Google Scholar]
- 26.Teshima Y, Akao M, Li RA, Chong TH, Baumgartner WA, Johnston MV, Marbán E. Mitochondrial ATP-sensitive potassium channel activation protects cerebellar granule neurons from apoptosis induced by oxidative stress. Stroke. 2003;34:1796–1802. doi: 10.1161/01.STR.0000077017.60947.AE. [DOI] [PubMed] [Google Scholar]
- 27.Cao C, Healey S, Amaral A, Lee-Couture A, Wan S, Kouttab N, Chu W, Wan Y. ATP-sensitive potassium channel: A novel target for protection against UV-induced human skin cell damage. J Cell Physiol. 2007;212:252–263. doi: 10.1002/jcp.21026. [DOI] [PubMed] [Google Scholar]
- 28.Malinska D, Mirandola SR, Kunz WS. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 2010;584:2043–2048. doi: 10.1016/j.febslet.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 30.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 31.Simon MC. Mitochondrial reactive oxygen species are required for hypoxic HIF alpha stabilization. Adv Exp Med Biol. 2006;588:165–170. doi: 10.1007/978-0-387-34817-9_15. [DOI] [PubMed] [Google Scholar]
- 32.Semenza GL. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology (Bethesda) 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 33.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavez A, Miranda LF, Pichiule P, Chavez JC. Mitochondria and hypoxia-induced gene expression mediated by hypoxia-inducible factors. Ann N Y Acad Sci. 2008;1147:312–320. doi: 10.1196/annals.1427.021. [DOI] [PubMed] [Google Scholar]
- 35.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 36.Chan SY, Zhang YY, Hemann C, Mahoney CE, Zweier JL, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang X, Le QT, Giaccia AJ. MiR-210-micromanager of the hypoxia pathway. Trends Mol Med. 2010;16:230–237. doi: 10.1016/j.molmed.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z, Li Y, Zhang H, Huang P, Luthra R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene. 2010;29:4362–4368. doi: 10.1038/onc.2010.193. [DOI] [PubMed] [Google Scholar]