

Abstract
Multiple organ dysfunction syndrome (MODS) is a detrimental clinical complication in critically ill patients with high mortality. Emerging evidence suggests that oxidative stress and endothelial activation (induced expression of adhesion molecules) of vital organ vasculatures are key, early steps in the pathogenesis. We aimed to ascertain the role and mechanism(s) of enhanced extracellular superoxide dismutase (EcSOD) expression in skeletal muscle in protection against MODS induced by endotoxemia. We showed that EcSOD overexpressed in skeletal muscle-specific transgenic mice (TG) redistributes to other peripheral organs through the circulation and enriches at the endothelium of the vasculatures. TG mice are resistant to endotoxemia (induced by lipopolysaccharide [LPS] injection) in developing MODS with significantly reduced mortality and organ damages compared with the wild type littermates (WT). Heterogenic parabiosis between TG and WT mice conferred a significant protection to WT mice, whereas mice with R213G knock-in mutation, a human single nucleotide polymorphism leading to reduced binding EcSOD in peripheral organs, exacerbated the organ damages. Mechanistically, EcSOD inhibits vascular cell adhesion molecule 1 expression and inflammatory leukocyte adhesion to the vascular wall of vital organs, blocking an early step of the pathology in organ damage under endotoxemia. Therefore, enhanced expression of EcSOD in skeletal muscle profoundly protects against MODS by inhibiting endothelial activation and inflammatory cell adhesion, which could be a promising therapy for MODS.
Keywords: Endotoxemia, MODS, EcSOD, Skeletal muscle, Free radicals, Oxidative stress, Endothelial activation, Parabiosis, VCAM-1
Graphical Abstract
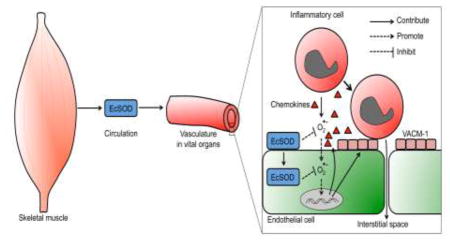
Introduction
Multiple organ dysfunction syndrome (MODS) is a serious clinical syndrome as a result of trauma, hemorrhagic shock and sepsis (see Review[1]). Starting as an inflammatory response in the originally affected tissues/organs, it perpetuates to damage all organs if left unchecked. Despite extensive research, MODS remain the main cause of death in the intensive care units with an extremely high rate of mortality (30–80%)[2]. To date, there is no effective therapy for MODS[2], probably due to an incomplete understanding of the pathogenic process.
The pathogenesis of MODS involves cascades of events including systemic inflammatory responses, local cell activation, inflammatory cell infiltration, coagulation, ischemia and eventually failure of the vital organs[3]. Emerging evidence suggests that oxidative stress and activation of the endothelium are key, early steps in the etiology [3]. Free radicals produced by leukocytes and/or endothelial cells [4] activate endothelial cells to expression cell surface adhesion molecules and promote inflammatory cell adhesion and transmigration into the interstitial space. These will eventually cause organ damage, as well as platelet adhesion and the consequent coagulation in the vasculature, which further impairs the circulation and metabolism of the vital organs in a vicious cycle. At the center of the pathology is the induction of oxidant and free radicals by leukocytes and/or endothelial cells that trigger endothelial activation[4]. Although general antioxidants have been shown to have promising effects against MODS in preclinical studies, they failed in clinical trials[5] probably due to lack of target specificity, which leads to ineffectiveness and sometimes fatal side effects [5].
Extracellular superoxide dismutase (EcSOD) functions in scavenging biologically toxic superoxide in the extracellular space, and is widely expressed in all tissues with the highest expression in the lung and kiney[6, 7]. EcSOD expression decreases in affected tissues in a variety of chronic diseases[8–10], and genetic approaches in animal models have provided strong evidence for functional roles of ectopic expression of EcSOD in cardiovascular, pulmonary and neuronal disorders[11–19]. We have previously shown that EcSOD is expressed by myofibers in adult skeletal muscle, which can be enhanced by exercise training, leading to elevated EcSOD levels in the blood and peripheral organs[20, 21]. Using molecular genetics model in mice, we showed that EcSOD expressed in muscle-specific EcSOD transgenic mice (TG) also redistributes to other peripheral organs through the circulation[20, 21]. These TG mice are resistant to dexamethasone- and heart failure-induced muscle wasting [21] as well as diabetic cardiomyopathy[20], supporting a role of EcSOD in protecting peripheral organs against oxidative stress. Therefore, induced EcSOD expression in skeletal muscle may mediate the health benefits of exercise in protection against chronic disease. We speculated that enhanced EcSOD expression in skeletal muscle might provide protection against severe disease conditions, such as MODS. The premise is that enhanced expression of EcSOD in skeletal muscle enriches at and enters endothelium of the peripheral organs [21, 22], which can effectively mitigate oxidative stress and endothelial activation, the two nodal steps in MODS development.
Herein, we took advantage of gain- and loss of function molecular genetics approaches in endotoxemia-induced MODS model in mice in combination with powerful parabiosis and intraperitoneal serum transfusion to determine if enhanced expression of muscle-derived EcSOD protects against MODS and to elucidate the underlying mechanism(s) with a focus on endothelial activation and organ damage. Since skeletal muscle is the largest organ (~40% body mass), positive findings would justify using exercise training and/or muscle-mediated gene therapy to prevent and/or treat MODS.
Materials and methods
Animals
Mice were housed in temperature-controlled (22 °C) quarters with a 12:12-h light-dark cycle and free access to water and normal chow (Harlan). Muscle-specific EcSOD transgenic mouse line was generated at the University of Virginia Gene Targeting and Transgenic Facility as described[21] and backcrossed into pure C57BL/6J background (≥10 generations). CX3CR1-GFP mice were purchased from the Jackson Laboratory. To induce endotoxemia, mice received a single injection of LPS (20 mg/kg i.p.)(Sigma, St. Louis) with normal saline or PBS injection (same volume) as control. All animal protocols were approved by the University of Virginia Institutional Animal Care and Use Committee.
Parabiosis
Parabiosis surgery was performed as previously described[23]. Briefly, after anesthetizing and aseptically preparing the mouse, a 1-cm flap of skin was removed running the length from the forelimb to the hindlimb on opposing dorsal sides of animals to be surgically united. Elbows and knees were united using sterile chromic gut suture (5-0) and sterile braided silk suture was used to close the skin opening. Baytril (100 mg/ml) in drinking water was given to help protect against infection for the first 10 days post-surgery. Ketoprofen was administered immediately after surgery and for 3 days post-surgery to help minimize pain. Parabiotic unions were euthanized if any adverse effects were observed, including loss of body mass (>10%), lethargy, mucus in the eyes, or tearing of the suture. Blood sharing was confirmed at 2 weeks post-surgery by analyzing serum EcSOD levels from a tail vein bleed. Endotoxemia was induced at 4 weeks post-surgery.
Intravital microscopy imaging of monocyte adhesion and rolling
To measure monocyte rolling and adhesion the vasculature using a dorsal skinfold window chamber. Briefly, mice were anesthetized using isoflurane (2 – 3% in oxygen) for the duration of the surgery and imaging. The entire back was shaved and depilated. The dorsal skin was tented and pinned to a corkboard placed under the sterile surgical area. Three small holes were punched into the double layer of skin using a 1.5 mm biopsy punch. The dorsal skinfold window chamber frame was placed on the skin, lining up with the three skin punctures. Sterile steel nuts and bolts fixed the frame to the dorsal skin. The skin directly under the window portion of the chamber was carefully removed to expose the cutaneous microcirculation. Saline was applied to keep the exposed vasculature from drying out during imaging. Intravital microscopy was performed on a Nikon Eclipse 80i microscope equipped with an EXFO XCite 120 xenon light source for epifluorescence imaging. Video was taken using a high-speed Photometrics HQ2 CCD camera controlled by Nikon NIS Elements Advanced Research (Laboratory Imaging) with a 20x objective. All mice were imaged immediately after their cutaneous vessels were exposed. Fields of view were selected so that vessels were clearly visible and had blood flow. Videos of GFP+ cells were taken for 60 seconds. Each animal had a minimum of 3 vessels imaged. Each video was analyzed for adhered and rolling GFP+ cells. To be considered “adhered”, a cell had to be at rest at the start of the video, and remain arrested for the length of the video. To count rolling cells, a line was made perpendicular to the blood vessel using the NIS Elements software. Only rolling cells which crossed this line during the video recording were counted. Rolling cells were identified by their variable velocity (stop-and-go behavior). The vessel diameter was measured where the perpendicular line was made, and the vessel area was determined by outlining the vessel in NIS Elements.
Pulmonary function measurement
The functional properties of the lung i.e. the airway and vascular mechanics are characterized by different lung function parameters, including the pulmonary compliance, the airway resistance and pulmonary artery pressure. The airway resistance is an index for the resistive forces against the airflow in the airways and depends on the diameter and length of the airways. The airway resistance can be calculated from the relation between transpulmonary pressure and airflow velocity. The airway resistance increases as a consequence of narrowing of the airways due to bronchoconstriction or obstructive processes, e.g. bronchial edema or enhanced mucus deposition. The pulmonary compliance is a marker for the functional stiffness of the lung and can be calculated from the relation between tidal volume and transpulmonary pressure. The pulmonary compliance decreases during restrictive pathological processes e.g. atelectasis, fibrosis, pulmonary edema or disturbed surfactant secretion. The increase in pulmonary artery pressure is an index of vasoconstriction of lung and represents an underlying pathophysiology resulting in pulmonary edema or changes in pulmonary vascular resistance. We measured the lung function using a buffer-perfused mouse lung system (Hugo Sachs Elektronik). Briefly, mice were anesthetized with ketamine and xylazine and ventilated with room air at 100 strokes/min with a tidal volume of 7 μl/g body weight with a positive end expiratory pressure of 2 cm H2O using the MINIVENT mouse ventilator (Hugo Sachs Elektronik). The animals were exsanguinated by transecting the inferior vena cava. The pulmonary artery was cannulated via the right ventricle, and the left ventricle was immediately tube-vented through a small incision at the apex of the heart. The lungs were then perfused at a constant flow of 60 μl/g body weight/min with Krebs-Henseleit buffer containing 2% albumin, 0.1% glucose, and 0.3% HEPES. The perfusate buffer and isolated lungs were maintained at 37°C throughout the experiment. Isolated lungs were allowed to equilibrate on the apparatus during a 5-min stabilization period. After equilibration, data were recorded for an additional 10 minutes. Hemodynamic and pulmonary parameters were continuously recorded during this period by the PULMODYN data acquisition system (Hugo Sachs Elektronik).
Serum markers analysis
Blood was collected at the orbital vein prior to animal sacrifice to minimize any influence from organ harvest of serum markers. Serum was isolated from the blood and the following kits were used to assess the effect of endotoxemia and organ failure: Creatinine Serum Detection Kit (Arbor Assays, Ann Arbor, MI, USA), Urea Nitrogen (BUN) Colorimetric Detection Kit (Arbor Assays), Lactate Dehydrogenase Activity Assay Kit (Sigma-Aldrich), and Alanine Transaminase Colorimetric Activity Assay Kit (Cayman Chemical, Ann Arbor, MI, USA). All samples were run in triplicate and according to the manufacturer’s instructions.
Lung open airspace analysis
The entire lung cross-section images were analyzed for lung open airspace. First, the image was opened in ImageJ and converted to 8-bit. Then, the image was inverted and threshold applied so that the tissue was a grey color and the open space was colored red. Next, area fraction was selected in the Set Measurements-Analyze tool. For a selected area, ImageJ reported the %area that was red (or open airspace) using the measure tool under Analyze. The entire lung was quantified and average open airspace calculated for each sample.
Serum EcSOD activity analysis
EcSOD activity was measured in serum samples from TG and WT littermates using Superoxide Dismutase Colorimetric Activity Kit from a commercial source (EIASODC, Thermo Fisher Scientific, Massachusetts) following the manufacturer’s instructions.
Immunoblot analysis
Immunoblot analysis was performed as described[24] with the following primary antibodies: EcSOD (R&D, Minneapolis), VCAM-1. Immunoblots were analyzed by Odyssey Infrared Imaging System (LI-COR Biosciences, NE) and quantified by Scion Image software.
Immunohistochemistry
Tissue was harvested and fixed in 10% neutral buffered formalin, dehydrated and infiltrated with paraffin and sectioned at 4 μm thickness. General structure was demonstrated by staining with the Hematoxylin-Eosin procedure (HHS-128, Sigma, St. Louis MO). Picrosirius red staining was used to identify collagen deposition (Polysciences #09400, Warrington PA). For immunohistochemistry, sections were pretreated to quench endogenous peroxidase (H2O2 in methanol) and endogenous biotin (Avidin-Biotin blocking kit, Vector Laboratories, Burlingame, CA). Oxidative damage was assessed with antibodies against 4-hydroxynonenal (ab485606, Abcam, Cambridge MA, 1:2000 dilution) and 8 Hydroxyguanosine (ab48508, Abcam, Cambridge MA, 1:100 dilution)
Intraperitoneal serum transfusion
WT mice received intraperitoneal transfusion of serum (1 ml) from either WT or TG mice through intraperiotoneal injection followed by LPS injection 12 hours after transfusion.
Microbubble imaging of VCAM-1 expression in vivo
Microbubbles were manufactured as described[25]. Ultrasound imaging was performed as reported with minor modifications[26]. Briefly, at 6 hours post LPS injection, each mouse under anesthesia (1% isoflurane in oxygen) was injected via the retro-orbital vein (i.v.) with a bolus of VCAM-1 targeted microbubbles (1.7 × 106). All images were acquired with an Acuson Sequoia 512 ultrasound system (Siemens, Issaquah, WA) with a 15L8 high-frequency linear assay transducer. The acoustic focus was set proximal to the left kidney to acquire images before microbubble injection and for 5 minutes, at which point all unbound microbubbles had been cleared. To verify the binding specificity of targeted MBs, a high power destructive pulse was applied (8 MHz; MI=1.9). All images were digitally stored on magneto-optical (MO) disk and transferred onto a personal computer for subsequent off-line analysis using ImageJ. The relative intensity was calculated as ratio of stable signal to peak signal normalized by baseline signal.
Statistics
The results are presented as mean ± SE. Survival curves were analyzed using the Kaplan-Meier analysis. Comparisons between two paired groups were analyzed by a paired student’s t-test. Comparisons among groups were analyzed by one-way ANOVA, and when appropriate a Tukey’s HSD post-hoc test was used for detection of differences across specific means. All data passed ANOVA assumptions for normality and equal variance. A p-value <0.05 was required to report significance.
Results
Enhanced EcSOD expression in skeletal muscle profoundly protects against MODS with reduced mortality under endotoxemia
TG mice had significantly increased EcSOD in all peripheral organs except for the brain (Fig. 1A and S4) probably due to the blood-brain barrier, and a 6-fold increase of serum EcSOD assessed by western blot (p < 0.001; Fig. S1A)[21] as well as colorimetric measurement of the enzyme activity (4.27 ± 1.96 U/ml in WT mice vs 25.2 ± 1.61 U/ml in TG mice; p < 0.001) (Fig. S1A). This finding confirms that the transgene-encoded EcSOD is biologically active with antioxidant enzyme activity. TG mice had completely normal morphology in peripheral organs by H&E staining (Fig. S1B) and showed increased EcSOD at endothelium of the blood vessels in the lung compared with WT mice by immunohistochemistry (Fig. 1B). Since the transgene is under the control of the creatine kinase promoter, which is skeletal muscle-specific with weak leaky activity in the heart[27], the great majority of increased EcSOD protein in the circulation and peripheral tissues is of skeletal muscle origin. To determine if enhanced expression of EcSOD in skeletal muscle reduces mortality caused by endotoxemia, we injected a lethal dose of lipopolysaccharide (LPS, 20 mg/kg, i.p.) to WT and TG mice (referred to WT-LPS and TG-LPS, respectively). All WT-LPS mice died by 91 hours post-injection with a 50% survival at 37.5 hours (Fig. 1C). The survival was significantly improved in TG-LPS mice (p < 0.001) with more than half the mice (54.5%) viable 10 days post injection. These findings demonstrate a profound protection against endotoxemic death in TG mice.
Fig. 1. Skeletal muscle-specific EcSOD transgenic mice are protected from LPS-induced MODS.

A) Immunoblot images showing that EcSOD levels are increased in various peripheral tissues and serum in muscle-specific EcSOD transgenic mice under the control of muscle creatine kinase promoter, but not in the brain, compared with WT littermate mice. The vertical lines separates images from different parts of the same membrane or different membranes with the exact same experimental condition (probed together) and exposure; B) EcSOD was detected at smooth muscles of blood vessel in WT lung, not so much at vascular endothelial cells (v) (arrows) bronchial (br) and alveoli (al). Significantly elevated EcSOD was detected at smooth muscles, vascular endothelial cells (arrows) and alveoli, but not at bronchial in TG mice. Experiments were repeated three times for each conditions; C) Mann-Whitney survival curves for TG and WT following LPS injection (20 mg/kg) (n = 10–11). *** denotes p < 0.001); D) Quantification of BUN, SCR, LDH and ALT (n = 7–12). ** and *** denote p < 0.01 and p < 0.001, respectively; and E) Quantitative measurements of lung physiological function in airway resistance, pulmonary compliance and pulmonary artery pressure (n = 6). ** and *** denote p < 0.01 and p < 0.001, respectively.
Since this lethal dose of LPS injection causes MODS in mice[28], we elected to determine if enhanced EcSOD expression reduces the severity of MODS by comparing biomarkers of organ damages of kidney, liver, heart and skeletal muscle between WT-LPS and TG-LPS mice using WT mice with saline injection (WT-Saline) as control. Kidney injury was evident in WT-LPS mice as shown by significantly elevated levels of blood urea nitrogen (BUN, 80.9 ± 1.7 mg/dl; p < 0.001) and serum creatinine (SCR, 1.67 ± 0.19 mg/dl; p < 0.001) compared with WT-Saline mice (25.6 ± 0.44 mg/dl for BUN and 0.64 ± 0.02 mg/dl for SCR) (Fig. 1D). TG-LPS mice had significantly reduced BUN (50.5 ± 1.7 mg/dl; p < 0.001) and SCR (0.76 ± 0.13 mg/dl; p < 0.01) compared with WT-LPS mice. Serum lactate dehydrogenase (LDH), a marker of muscle damage and other types of cell death, was significantly elevated in WT-LPS (175.4 ± 6.3 mU/ml; p < 0.001) compared to WT-Saline mice (45.3 ± 12.0 mU/ml), and was attenuated in TG-LPS mice (118.3 ± 12.3 mU/ml; p < 0.01 vs. WT-LPS) (Fig. 1D). LPS also induced liver injury in WT-LPS mice, indicated by significantly increased serum alanine aminotransferase (ALT) activity (26.98 ± 2.23 mU/ml) compared to WT-Saline mice (5.66 ± 1.85 mU/ml; p < 0.001) (Fig. 1D). TG-LPS mice had significantly reduced serum ALT activity (10.78 ± 0.91 mU/ml) compared to WT-LPS mice (p < 0.001). These findings clearly demonstrate that TG mice are resistant to endotoxemia-induced MODS.
To further examine the organ damages by endotoxemia and the protection by enhanced EcSOD expression in skeletal muscle, we evaluated the morphological changes by H&E staining of the lungs, kidney, liver, and heart at 6 hours after LPS injection. WT-LPS mice showed profound pathology of vascular coagulation and signs of inflammatory cell infiltration in the lung, kidney and liver with unappreciable changes in the heart (Fig. S1C). In particular, WT-LPS mice demonstrated a significant loss of open space of the alveoli in the lungs likely due to serum exudate, neutrophils and inflammatory monocytes (Fig. S1C). When WT-LPS lungs were placed in 10% formalin, the increase density made them sank (Fig. S1D), whereas, TG-LPS lungs had greater open airspace (Fig. S1C) and remained floating in fixative, similar to WT-Saline lung (Fig. S1D). Therefore, morphological data also strongly support that TG mice had significantly less severe MODS, particularly in the lung, compared with WT mice under the condition of endotoxemia.
We then assessed oxidative stress in the lung and kidney, the two most affected organs by endotoxemia. Using immunohistochemistry with antibodies against 4-hydroxynonenal (4-HNE) for lipid peroxidation, we found that WT-LPS mice had significantly increased 4-HNE staining in the lung, which was significantly reduced in TG-LPS mice (Fig. S1E). These findings were associated with significantly less collagen deposition. Picrosirius red staining showed restricted staining to the blood vessels in WT-Saline mice, but it became widespread in WT-LPS mice. Importantly, TG-LPS mice showed significantly less collagen deposition (Fig. S1F). To investigate oxidative stress in the kidney, 8-hydroxydeoxyguanosine (8-OHdG, a marker of DNA oxidative stress) was measured immunohistochemically. WT-LPS mice had profoundly positive nuclear staining, but not in WT-Saline or TG-LPS mice (Fig. S1G). Taken together, these findings provide direct evidence of increased oxidative stress in vital organs under the condition of endotoxemia, which are mitigated in transgenic mice with muscle-specific overexpression of EcSOD.
Finally, we assessed pulmonary function using an isolated perfused lung system for physiologically relevant respiration and perfusion ex vivo[29]. We observed a significant increase in airway resistance from 1.39 ± 0.06 cm H2O/μl/sec in WT-saline mice to 2.18 ± 0.21 in WT-LPS mice (p < 0.01), which was completely abolished in TG-LPS mice (1.40 ± 0.10; p < 0.01) (Fig. 1E). Similarly, LPS injection resulted in reduced pulmonary compliance (2.79 ± 0.29 μl/cm H2O) and increased pulmonary artery pressure (11.53 ± 0.15 cm H2O) compared with WT-Saline mice (7.50 ± 0.52, p < 0.001 and 5.45 ± 0.09, p < 0.001, respectively), both of which were completely abolished in TG mice (6.61 ± 0.71, p < 0.001 and 6.50 ± 0.25, p < 0.001, respectively) (Fig. 1E). These functional data provide direct evidence for profound protection of pulmonary function in TG mice.
Parabiosis reveals a protection from TG to WT mice through the circulation against MODS
To determine if a blood-borne factor(s) in TG mice is responsible for the protection against MODS, we performed surgery to induce heterogenic parabiosis between WT and TG mice (WT-TG) using isogenic parabiosis between WT mice (WT-WT) as control (Fig. 2A, top). Four weeks after the surgery, WT-TG and WT-WT pairs were administered LPS (20 mg/kg to both mice in the pair), and tissues and serum samples were collected at 6 hours post-injection. Blood sharing after parabiosis was confirmed by detection of increased EcSOD in the sera of heterogenetic WT mice compared to isogenic WT-WT pairs (Fig. 2A, bottom, and S5). Lungs from WT-WT pairs sank when placed in fixative at 6 hours post LPS injection, while TG-WT lungs remained floating (Fig. 2B). These findings were further confirmed by histology as WT-WT pairs had reduced lung open airspace to ~40–50% (similar to WT-LPS mice) while TG-WT mice had ~70% open space similar to WT-Saline control mice (p < 0.001 vs. WT-LPS) (Fig. 2C and S2A). To further investigate EcSOD-mediated protection in the lung, we performed immunofluorescence to assess macrophage infiltration and activation (iNOS-positive) in lung sections from TG-WT and WT-WT pairs (Fig. 2D). TG-WT heterogenic pairs had significantly reduced total macrophages (−44%, p < 0.01) and activated macrophages (−50%, p < 0.001) compared to WT-WT isogenic pairs (Fig. S2B).
Fig. 2. Heterogenic parabiosis confers protection of WT mice against MODS from TG mice and R213G KI mice are more vulnerable to organ damage.

A) Graphical representation of WT, TG and isogenic (WT-WT) and heterogenic (WT-TG) parabiotic mice (top) and representative immunoblot of serum EcSOD protein demonstrating elevated serum EcSOD in WT mice of the WT-TG parabiotic pairs, but not in WT-WT parabiotic pairs (bottom); B) Representative image of lung in fixative (float or sink) demonstrating greater lung density in WT-WT compared with WT-TG pairs following LPS injection; C) Representative H&E histological images of the lungs; D) Representative pan-macrophage and iNOS immunohistochemistry images; E) Quantification of BUN, SCR, LDH, and ALT (n = 4). * and *** denote p < 0.05 and p < 0.001, respectively. F) PCR-based genotyping for KI and WT mice showing the detection of the KI and WT alleles (top) and immuoblot analysis of EcSOD proteins in the kidney and serum using GAPDH and Ponseau staining as controls for loading. The vertical lines separates images from different parts of the same membrane; and G) Quantification of BUN in homozygous KI and WT mice following LPS injection (6 hrs) (n = 4–5). * denotes p < 0.05.
Importantly, TG-WT pairs had significantly reduced BUN (52.9 ± 1.8 mg/dl, p < 0.001) and SCR (0.58 ± 0.07 mg/dl, p < 0.05) in the serum compared to those in WT-WT mice (67.6 ± 1.6 and 1.29 ± 0.15, respectively) (Fig. 2E). In addition, TG-WT pairs had significantly reduced serum LDH (124.2 ± 3.4, p < 0.001) and ALT (9.13 ± 2.07, p < 0.001) activities compared with WT-WT pairs (187.1 ± 6.2 and 24.6 ± 3.27, respectively) (Fig. 2E). Therefore, blood sharing via parabiosis bestowed a clear protection from TG mice to WT mice against MODS. These are the first findings using parabiosis to confirm that a blood-borne factor(s), presumably EcSOD, is sufficient to protect MODS.
R213G knock-in mice are more vulnerable to organ damage under endotoxemia
A common single nucleotide polymorphism (SNP) in the EcSOD gene in humans with R213G codon change and loss of binding of EcSOD protein to sulfated polysaccharides has been shown to predispose patients to poor prognosis in oxidative stress-related diseases[30–32]. This adverse phenotypic change has been recapitulated in R213G knock-in mice (KI)[33, 34]. We compared EcSOD protein levels in the serum and different organs between KI mice and their wild type littermates (Fig. 2F, S2C, S2D, S5 and S8). We found that homozygous KI mice have significantly elevated EcSOD protein levels in the serum, but reduced levels in the lung and kidney with reduced trend in the skeletal muscle, heart and liver (Fig. 2F, S2C, S2D, S5 and S8), confirming that R213G mutation leads to loss of EcSOD binding to sulfated polysaccharides in peripheral organs. Under endotoxemia, KI mice demonstrated significantly more severe kidney injury as measured by BUN (51.4 ± 6.8% in KI vs 78.6 ± 4.7 in WT, p < 0.05) (Fig. 2G). These findings with a loss-of-function molecular genetic approach that mimics a clinically relevant human SNP support the notion that binding of increased EcSOD to peripheral organs is critical for protection against organ damage under endotoxemia. This information could potentially be of great clinical value for prevention and treatment of MODS.
Enhanced EcSOD expression in skeletal muscle inhibits endothelial activation
Since endothelial activation is a key, early step in MODS, we explored if increased EcSOD reduces vascular cell adhesion protein 1 (VCAM-1) expression in vital organs under the condition of endotoxemia. Immunohistochemistry confirmed a low degree of VCAM-1 expression at the endothelium in the control WT-Saline lung (Fig. 3A). LPS injection resulted in a significant increase of VCAM-1 in WT-LPS mice, which was significantly reduced in TG-LPS mice (Fig. 3A). This finding was further confirmed by immunoblot showing a marked induction (~4.5-fold, p < 0.001) of VCAM-1 protein in WT-LPS mice compared WT-Saline injection, which was significantly mitigated in TG-LPS mice (p < 0.05, vs. WT-LPS) (Fig. 3B and S6). Since kidney is also sensitive of endotoxemia with evident pathology (Fig. S1C) and oxidative stress (Fig. S1G), we determined if endotoxemia resulted in endothelial activation in the kidney. Immunoblot showed ~4-fold greater VCAM-1 protein expression in WT-LPS (p < 0.001) compared with WT-Saline mice, which was reduced to 2.5-fold in TG-LPS (p < 0.01 vs. WT-Saline) (Fig. 3C and S6). Taken together, these findings demonstrate that endotoxemia induces profound endothelial activation in vital organs, such as the lung and kidney, in WT mice. This endothelial activation is significantly attenuated in mice with muscle-specific overexpression of EcSOD.
Fig. 3. TG mice are resistant to endotoxemia-induced endothelial activation.

A) Representative VCAM-1 immunohistochemistry image of the lung demonstrating less endothelial cell activation in TG-LPS compared to WT-LPS. The experiment was done in 4–5 mice for each condition with similar findings; B) Representative immunoblot and quantitative representation of VCAM-1 protein in the lung (n = 5–11). The vertical lines separates images from different parts of the same membrane. * and *** denote p < 0.05 and p < 0.001, respectively; and C) Representative immunoblot and quantitative representation of VCAM-1 protein in the kidney (n = 5–9). *, ** and *** denote p < 0.05, p < 0.01 and p < 0.001, respectively.
EcSOD is sufficient to inhibit endothelial activation in vitro and in vivo
To test whether it is elevated EcSOD that directly inhibits endothelial activation, we pre-treated human umbilical vein endothelial cell (HUVEC) monolayer with WT or TG serum (containing significantly different levels of EcSOD) (Fig. S1A) followed by tumor necrosis factor α (TNF-α) treatment, which is known to induce mitochondrial superoxide production and VCAM-1 expression [35] in a free radical-dependent manner[36]. This cell culture model recapitulates endothelial activation in vivo as endothelial cells are stimulated by cytokines released during inflammatory cell-endothelial cell interaction. We first pre-incubated the HUVEC monolayer with serum from WT or TG mice followed by TNF-α treatment and assessed VCAM-1 expression by immunoblot and mitochondrial superoxide production by MitoSOXTM Red staining. HUVEC monolayers pre-treated with WT serum had a 2-fold increase of VCAM-1 protein expression (Fig. 4A, S3B and S7) and a significant increase of MitoSOX fluorescence (Fig. S3A) following TNF-α stimulation, both of which were abolished in HUVECs pre-incubated with TG serum (Fig. 4A, S3A, S3B and S7). These findings suggest that endothelial cells targeted by EcSOD with binding in the extracellular space and/or endocytosis-mediated entry are resistant to cytokine-induced endothelial activation with reduced mitochondrial ROS production.
Fig. 4. Serum EcSOD is sufficient to reduce endothelial cell activation in vitro and in vivo.

A) Immunoblot of VCAM-1 protein in HUVEC cells pre-incubated with WT or TG serum followed by TNF-α treatment; B) Representative immunoblot for serum EcSOD protein in recipient WT mice before and after receiving WT or TG serum (Pre vs. Post serum, respectively), and following LPS injection (Post LPS) (n = 6–10); C) Representative ultrasound-based images of VCAM-1 target microbubbles in WT recipient mice that received WT or TG serum followed by LPS injection. Kidneys were imaged prior to microbubble injection (Baseline), immediately (Peak signal) and 5 minutes after injection (Stable signal), and after application of a high power destructive pulse (After burst); D) Quantification of stable signal (n = 6–10). * denotes p < 0.05; and E) Quantification of BUN (n = 4–7). * and *** denotes p < 0.05 and p < 0.001, respectively.
To further test if EcSOD in TG serum per se is sufficient to reduce endothelial activation in vivo, we performed intraperitoneal serum transfusion, which is free of blood cells. We first confirmed that a bolus injection of TG serum (1 ml) resulted in elevated EcSOD in the blood in WT mice as early as 30 min with a peak at 12 hours (Fig. S3C and S9). LPS injection in the receipient mice (12 hours post serum transfusion) did not lead to significant reduction of EcSOD at 6 hours (Fig. 4B and S7), indicating no aberrant degradation of EcSOD by LPS injection. We performed Ultrasound-based imaging with intravenous bolus injection of VCAM-1 targeted microbubbles to assess endothelial activation in the kidney. WT mice receiving WT serum transfusion showed a 2-fold increase (p < 0.05) in ultrasound retention signal after LPS injection, which was blunted in mice receiving TG serum (Fig. 4C and 4D). These mice also showed a moderate, but significant reduction of blood urea nitrogen compared with mice receiving WT serum transfusion (Fig. 4E). Altogether, these in vitro and in vivo findings support that it is the elevated EcSOD, rather than cells in the blood, that prevents endothelial activation and protects against MODS under endotoxemia. In addition, the findings may support the potential practicality of using serum transfusion in MODS treatment.
EcSOD inhibits inflammatory cell adhesion to endothelial cells under endotoxemia
To ascertain if reduced endothelial activation in TG mice leads to reduced inflammatory cell adhesion in the vasculature in vivo, we took advantage of intravital imaging with fluorescent labeled inflammatory cells in transgenic mice. We first crossbred transgenic reporter mice of CX3C chemokine receptor 1 (CX3CR1, also known as fractalkine receptor)-green-fluorescent protein (GFP) with TG mice to generate CX3CR1-GFP;TG transgenic mice (hereafter referred to as GFP-TG) and littermate GFP-CX3CR1-GFP;WT controls (GFP-WT), both of which have GFP-positive circulating monocytes. A vascular window chamber was surgically installed on the back of GFP-WT and GFP-TG mice 6 hours after LPS injection, and GFP monocytes rolling and adhesion were visualized and recorded using a fluorescent microscopy (Fig. 5A). GFP-WT mice after LPS injection showed nearly a 2-fold increase in GFP monocytes adhesion per vessel surface area compared with GFP-WT-PBS, which was blunted in GFP-TG-LPS (Fig. 5B). The total number of rolling cells decreased dramatically in WT mice following LPS injection (p < 0.001 vs. GFP-WT-PBS), which was completely intact in GFP-TG-LPS mice. These findings provide in vivo evidence that elevated EcSOD mitigates monocyte adhesion through inhibition of endothelial activation, which is a mechanism by which enhanced EcSOD expression in skeletal muscle protects against MODS under endotoxemia.
Fig. 5. Muscle-specific EcSOD transgenic mice are resistant the endotoxemia-induced monocyte adhesion.

A) CX3CR1-GFP (Monocyte-GPF reporter mice)-TG double transgenic mice or CX3CR1-GFP-WT control mice receiving LPS injection were fitted with a vessel-chamber window (left panel) at 6 hours post LPS injection to observe and record vascular monocyte adhesion and rolling (middle and right panels). Red arrows indicate GFP-monocytes adhered to the vessel walls; B) Quantification of monocyte adhesion per vessel wall surface area for CX3CR1-GFP-WT controls (WT) and CX3CR1-GFP-TG (TG) mice after LPS injection (n = 4–9); C) Quantification of rolling monocytes per vessel wall surface area (n = 5–9). * and *** denote p < 0.05 and p < 0.001, respectively; and D) A working model of EcSOD-mediated protection against endothelial cell activation and monocyte adhesion under endotoxemia. Endotoxemia induces expression of adhesion and chemokines in endothelial cells via NFκB mediated transcription to promote inflammatory cell interaction and adhesion. This process will in turn promote free radical generation from both inflammatory and endothelial cells, exacerbating the inflammatory responses. EcSOD could scavenge the free radicals at and inside the endothelial cells and attenuate the inflammatory cell-endothelial cell interaction.
Discussion
MODS is a major cause of mortality in intensive care units, and accumulating evidence suggests that oxidative stress and endothelial activation are major contributing factors to the pathology[5]. Although general antioxidants have been shown to be effective in various MODS models in animals, clinical trials have failed, probably due to lack of targeting specificity of the antioxidants[5]. We therefore need antioxidants that target the vital organs to break the vicious cycle of endothelial activation and inflammatory cell-mediated damage. Fortunately, our body produces EcSOD, which is secreted by producing cells and gets to redistribute to other tissues/organs through the circulation to execute its physiological function. Herein, we tested the hypothesis that enhanced EcSOD expression in skeletal muscle protects against MODS. Our premise is that enhanced EcSOD expression gets redistributed to vital organs and targets endothelial cells with great specificity. We showed that TG mice were profoundly protected from LPS-induced mortality and MODS with reduced endothelial activation in the lung and kidney, whereas a genetic mutation of EcSOD with loss of extracellular binding leads to exacerbation of MODS. To definitely prove that EcSOD mediates the protection, we performed heterogenic parabiosis between TG and WT mice. We showed that parabiosis led to elevated circulating EcSOD in WT mice and that the WT mice became resistant to endotoxemia, but not in isogenic WT-WT pairs. Furthermore, we have used culture human endothelial cells and serum transfusion and obtained direct evidence to support that EcSOD rather than the blood cells was sufficient to inhibit endotoxemia-induced endothelial activation in vitro and in vivo. These findings for the first time show comprehensive evidence of the protective role of muscle-derived EcSOD in protection against MODS and provide strong rationale for further exploration of EcSOD as a therapy for MODS.
The pathogenesis of endotoxemia-induced MODS is a complex process with cascades of events in a vicious cycle[3]. Many of these events occur rapidly making it difficult to elucidate the causal relationship. Nevertheless, emerging evidence suggests that endothelial injury with oxidative stress and endothelial activation in vital organ vasculatures are key, early steps in the etiology[3]. These events promote adhesion and transmigration of inflammatory cells into the interstitial space, further exacerbating the circulation to and metabolism of the vital organ. Endothelial injury may also lead to vascular dysfunctions, such as leaky vessels and impaired vasoreactivity, with clinical symptoms of failure in maintaining normal blood pressure.
EcSOD is a glycoprotein that is secreted by various cells and binds to extracellular matrix[7] due to its high affinity for sulfated polysaccharides. EcSOD can be redistributed to other cells, tissues and organs in a paracrine or endocrine manner and taken up by targeted cells[22]. EcSOD expression decreases in tissues affected by oxidative stress in many diseases[8–10], and loss-of-function and gain-of-function genetic approaches in animal models have confirmed the functional role of ectopic expression of EcSOD[11–19]. The most intriguing and clinically relevant finding of EcSOD function is that a common SNP in humans (R213G) leads to loss of binding of EcSOD to sulfated polysaccharides and predisposes the patients to cardiovascular and other oxidative stress-related diseases with poor prognosis[30–32]. Genetic studies in animals showed loss of protection due to loss of extracellular binding, but not loss of enzyme activity[37, 38], strongly supporting the importance of extracellular binding. This may also explain why delivery of recombinant SOD from bacteria without correct posttranslational modification failed to protect myocardial infarction[39]. To date, there is no evidence that EcSOD protects against MODS, particularly by redistributing from skeletal muscle to and protect other tissues/organs. Here, we provide both gain- and loss-of-function evidence that EcSOD distribution and binding to the peripheral organs from skeletal muscle protect against oxidative stress-induced organ damage under endotoxemia.
The importance of skeletal muscle in organ-organ crosstalk in health and disease has been exemplified by molecules, called myokines, secreted into circulation that initiate or alter signaling and cellular processes in other peripheral tissues[40]. Myokines have been implicated in regulating biological processes in many peripheral organs as well as the central nervous system[41]. Our previous findings with exercise training and current findings with muscle-specific EcSOD transgenic mice and R213G knock-in mice are consistent with the myokine theory; the only difference here is that EcSOD serves as an antioxidant, not a signaling molecule. Our following findings are particularly supportive: 1) exercise training increases EcSOD expression in skeletal muscle with elevated serum EcSOD; 2) enhanced expression of EcSOD in skeletal muscle redistributes to all peripheral tissues through the circulation; and 3) there is elevated serum EcSOD in WT mice through shared circulation by parabiosis from TG mice. We believe that EcSOD from skeletal muscle provides protects against MODS by inhibiting oxidative stress-induced endothelial activation, a critical step in MODS. It remains to be determined whether enhanced expression of EcSOD in skeletal muscle by exercise training is necessary for the protection.
Regular exercise is the most effective intervention against oxidative stress-related diseases[42–44]. Particularly relevant to this study, exercise training has been shown to attenuate vital organ dysfunction/damages induced by endotoxemia or sepsis in animal models[45–49], triggering great interests in the underlying mechanisms[50–53]. Exercise has been shown to promote EcSOD expression in skeletal muscle and peripheral organs. Following an acute bout of exercise in mice, EcSOD mRNA is increased in both skeletal muscle and aorta[54], while exercise training increases EcSOD protein level in the blood[55] and peripheral organs[56, 57]. Our current data show that EcSOD overexpressed in skeletal muscle is sufficient to protect against MODS under endotoxemia, and systemic administration of EcSOD has therapeutic potential. Future studies should focus on the causative role of exercise training-enhanced EcSOD expression in skeletal muscle in disease prevention.
Our collective findings with cultured endothelial cells, parabiosis and serum transfusion combined with intravital imaging provide novel insights into the underlying mechanism by which enhanced expression of EcSOD in skeletal muscle could profoundly protect against MODS. Mechanistically, our findings in cultured HUVAC cells show evidence that treatment with endogenously produced wild type EcSOD is sufficient to block cytokine-induced endothelial activation along with blunted mitochondrial ROS production. This finding is consistent with phenomenon of “ROS-induced ROS release” originally observed in cardiomyocytes[58, 59] and recently found in endothelial cells[60]. A straightforward explanation is that scavenging superoxide near the endothelium that is produced by inflammatory cells and/or by the endothelial cells will block endothelial activation in the cascade events that lead to tissue injuries in MODS. It is also conceivable that the protection is in part mediated by the elevated cytosolic EcSOD due to clathrin-mediated endocytosis of EcSOD[22]. Our findings unveil the elegant antioxidant system of our body with precise targeting against insults to the first line of defense of the vital organs, the endothelium.
We have previously demonstrated that enhanced expression of EcSOD in skeletal muscle is sufficient to protect against muscle atrophy[21] and diabetic cardiomyopathy in mice[20]. Here, we for the first time prove the advantage of enhancing EcSOD expression in skeletal muscle in protection against MODS. These findings raise the question how we might promote EcSOD expression in skeletal muscle, which could be of great translational value. Since the EcSOD gene has been shown to be regulated by transcriptional factors and modulators[61, 62], interventions that promote the expression and/or activity of these regulatory factors may promote EcSOD expression with excellent therapeutic values. In addition, NO signaling has been linked to EcSOD expression in skeletal muscle[21, 63]. Therefore, combinatory approaches may optimally enhance EcSOD expression in vivo as a potential therapeutic intervention for MODS in the future.
Conclusions
We have obtained novel findings to show that EcSOD overexpressed in skeletal muscle redistributes through the circulation to peripheral organs and provides potent protection against MODS under endotoxemia. On the contrary, loss of binding of EcSOD binding to the peripheral organs due to mutation mimicking a human SNP leads to exacerbated organ damages. This EcSOD-mediated protection is a result of decreased oxidative stress and reduced endothelial activation. Importantly, systemic delivery of EcSOD can recapitulates the protective effects in vivo. We now propose a model whereby muscle-derived EcSOD in the circulation adheres to and enters endothelial cells in peripheral organs to mitigate oxidative stress, adhesion molecule expression and inflammatory cell-endothelial cell interaction. Future endeavors should promote EcSOD expression and/or systemic administration of correctly modified recombinant EcSOD as therapeutic option to combat MODS and other oxidative stress-related diseases in clinical settings.
Supplementary Material
Highlights.
Increased EcSOD in vital organs from skeletal muscle protects against MODS.
EcSOD mutation mimicking human R213G SNP exacerbates MODS.
Parabiosis and serum transfusion confirm causation of EcSOD in protection.
The mechanism is inhibition of endothelial activation in endotoxemia.
Acknowledgments
We would like to thank Drs. Jonathan Kipnis and Igor Smirnov for helping setting up the surgical procedure for parabiosis. This work was supported by the National Institutes of Health (R01GM114840 to ZY) and American Heart Association (12POST12030231 Postdoctoral Fellowship to JAC). JAC, JD, ZY were involved in designing research studies, conducting experiments, acquiring data, analyzing data and writing the manuscript. KSM, AKS, XC, JZ, JC, CAG, MO, ZD, VAL, MZ were involved in conducting experiments, acquiring data, analyzing data; BM, BHA, ALK, VEL, SMP were involved in designing research studies and writing the manuscript.
Abbreviations
- 4-HNE
4-hydroxynonenal
- 8-OHdG
8-hydroxydeoxyguanosine
- ALT
alanine aminotransferase
- ARE
antioxidant response element
- BUN
blood urea nitrogen
- CX3CR1
CX3C chemokine receptor 1
- EcSOD
extracellular superoxide dismutase
- GFP
green-fluorescent protein
- HUVEC
human umbilical vein endothelial cell
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- MODS
multiple organ dysfunction syndrome
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- SCR
serum creatinine
- SNP
single nucleotide polymorphism
- TG
transgenic
- TNF-α
tumor necrosis factor α
- VCAM-1
vascular cell adhesion molecule 1
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rossaint J, Zarbock A. Pathogenesis of Multiple Organ Failure in Sepsis. Crit Rev Immunol. 2015;35(4):277–91. doi: 10.1615/critrevimmunol.2015015461. [DOI] [PubMed] [Google Scholar]
- 2.Wheeler AP, Bernard GR. Treating patients with severe sepsis. N Engl J Med. 1999;340(3):207–14. doi: 10.1056/NEJM199901213400307. [DOI] [PubMed] [Google Scholar]
- 3.Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003;101(10):3765–77. doi: 10.1182/blood-2002-06-1887. [DOI] [PubMed] [Google Scholar]
- 4.Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: Clinical evidence and therapeutic implications. Trends Cardiovasc Med. 2014;24(4):165–169. doi: 10.1016/j.tcm.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Slutsky AS, Vincent JL. Oxygen free radicals in ARDS, septic shock and organ dysfunction. Intensive care medicine. 2000;26(4):474–6. doi: 10.1007/s001340051185. [DOI] [PubMed] [Google Scholar]
- 6.Marklund SL. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem J. 1984;222(3):649–55. doi: 10.1042/bj2220649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folz RJ, Guan J, Seldin MF, Oury TD, Enghild JJ, Crapo JD. Mouse extracellular superoxide dismutase: primary structure, tissue-specific gene expression, chromosomal localization, and lung in situ hybridization. Am J Respir Cell Mol Biol. 1997;17(4):393–403. doi: 10.1165/ajrcmb.17.4.2826. [DOI] [PubMed] [Google Scholar]
- 8.Dahl M, Bowler RP, Juul K, Crapo JD, Levy S, Nordestgaard BG. Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. American journal of respiratory and critical care medicine. 2008;178(9):906–12. doi: 10.1164/rccm.200804-549OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider MP, Sullivan JC, Wach PF, Boesen EI, Yamamoto T, Fukai T, Harrison DG, Pollock DM, Pollock JS. Protective role of extracellular superoxide dismutase in renal ischemia/reperfusion injury. Kidney Int. 2010;78(4):374–81. doi: 10.1038/ki.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Hou M, Li Y, Traverse JH, Zhang P, Salvemini D, Fukai T, Bache RJ. Increased superoxide production causes coronary endothelial dysfunction and depressed oxygen consumption in the failing heart. Am J Physiol Heart Circ Physiol. 2005;288(1):H133–41. doi: 10.1152/ajpheart.00851.2003. [DOI] [PubMed] [Google Scholar]
- 11.Lu Z, Xu X, Hu X, Zhu G, Zhang P, van Deel ED, French JP, Fassett JT, Oury TD, Bache RJ, Chen Y. Extracellular superoxide dismutase deficiency exacerbates pressure overload-induced left ventricular hypertrophy and dysfunction. Hypertension. 2008;51(1):19–25. doi: 10.1161/HYPERTENSIONAHA.107.098186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Deel ED, Lu Z, Xu X, Zhu G, Hu X, Oury TD, Bache RJ, Duncker DJ, Chen Y. Extracellular superoxide dismutase protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free radical biology & medicine. 2008;44(7):1305–13. doi: 10.1016/j.freeradbiomed.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gongora MC, Qin Z, Laude K, Kim HW, McCann L, Folz JR, Dikalov S, Fukai T, Harrison DG. Role of extracellular superoxide dismutase in hypertension. Hypertension. 2006;48(3):473–81. doi: 10.1161/01.HYP.0000235682.47673.ab. [DOI] [PubMed] [Google Scholar]
- 14.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995;92(14):6264–8. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, Crapo JD, Rahman I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci U S A. 2010;107(35):15571–6. doi: 10.1073/pnas.1007625107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lund DD, Chu Y, Miller JD, Heistad DD. Protective effect of extracellular superoxide dismutase on endothelial function during aging. Am J Physiol Heart Circ Physiol. 2009;296(6):H1920–5. doi: 10.1152/ajpheart.01342.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin ED. Extracellular superoxide dismutase (EC-SOD) quenches free radicals and attenuates age-related cognitive decline: opportunities for novel drug development in aging. Curr Alzheimer Res. 2005;2(2):191–6. doi: 10.2174/1567205053585710. [DOI] [PubMed] [Google Scholar]
- 18.Iida S, Chu Y, Francis J, Weiss RM, Gunnett CA, Faraci FM, Heistad DD. Gene transfer of extracellular superoxide dismutase improves endothelial function in rats with heart failure. Am J Physiol Heart Circ Physiol. 2005;289(2):H525–32. doi: 10.1152/ajpheart.00108.2005. [DOI] [PubMed] [Google Scholar]
- 19.Prasad KM, Smith RS, Xu Y, French BA. A single direct injection into the left ventricular wall of an adeno-associated virus 9 (AAV9) vector expressing extracellular superoxide dismutase from the cardiac troponin-T promoter protects mice against myocardial infarction. J Gene Med. 2011;13(6):333–41. doi: 10.1002/jgm.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Call JA, Chain KH, Martin KS, Lira VA, Okutsu M, Zhang M, Yan Z. Enhanced skeletal muscle expression of extracellular superoxide dismutase mitigates streptozotocin-induced diabetic cardiomyopathy by reducing oxidative stress and aberrant cell signaling. Circ Heart Fail. 2015;8(1):188–97. doi: 10.1161/CIRCHEARTFAILURE.114.001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okutsu M, Call JA, Lira VA, Zhang M, Donet JA, French BA, Martin KS, Peirce-Cottler SM, Rembold CM, Annex BH, Yan Z. Extracellular superoxide dismutase ameliorates skeletal muscle abnormalities, cachexia, and exercise intolerance in mice with congestive heart failure. Circ Heart Fail. 2014;7(3):519–30. doi: 10.1161/CIRCHEARTFAILURE.113.000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu Y, Piper R, Richardson S, Watanabe Y, Patel P, Heistad DD. Endocytosis of extracellular superoxide dismutase into endothelial cells: role of the heparin-binding domain. Arterioscler Thromb Vasc Biol. 2006;26(9):1985–90. doi: 10.1161/01.ATV.0000234921.88489.5c. [DOI] [PubMed] [Google Scholar]
- 23.Wright DE, Wagers AJ, Gulati AP, Johnson FL, Weissman IL. Physiological migration of hematopoietic stem and progenitor cells. Science. 2001;294(5548):1933–6. doi: 10.1126/science.1064081. [DOI] [PubMed] [Google Scholar]
- 24.Akimoto T, Ribar TJ, Williams RS, Yan Z. Skeletal muscle adaptation in response to voluntary running in Ca2+/calmodulin-dependent protein kinase IV-deficient mice. Am J Physiol Cell Physiol. 2004;287(5):C1311–9. doi: 10.1152/ajpcell.00248.2004. [DOI] [PubMed] [Google Scholar]
- 25.Ferrante EA, Pickard JE, Rychak J, Klibanov A, Ley K. Dual targeting improves microbubble contrast agent adhesion to VCAM-1 and P-selectin under flow. J Control Release. 2009;140(2):100–7. doi: 10.1016/j.jconrel.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoyt K, Warram JM, Wang D, Ratnayaka S, Traylor A, Agarwal A. Molecular Ultrasound Imaging of Tissue Inflammation Using an Animal Model of Acute Kidney Injury. Mol Imaging Biol. 2015;17(6):786–92. doi: 10.1007/s11307-015-0860-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sternberg E, Spizz G, Perry W, Vizard D, Weil T, Olson E. Identification of upstream and intragenic regulatory elements that confer cell-type-restricted and differentiation-specific expression on the muscle creatine kinase gene. Mol Cell Biol. 1988;8(7):2896–909. doi: 10.1128/mcb.8.7.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Maio A, Mooney ML, Matesic LE, Paidas CN, Reeves RH. Genetic component in the inflammatory response induced by bacterial lipopolysaccharide. Shock. 1998;10(5):319–23. doi: 10.1097/00024382-199811000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Sharma AK, LaPar DJ, Stone ML, Zhao Y, Mehta CK, Kron IL, Laubach VE. NOX2 Activation of Natural Killer T Cells Is Blocked by the Adenosine A2A Receptor to Inhibit Lung Ischemia-Reperfusion Injury. American journal of respiratory and critical care medicine. 2016;193(9):988–99. doi: 10.1164/rccm.201506-1253OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arcaroli JJ, Hokanson JE, Abraham E, Geraci M, Murphy JR, Bowler RP, Dinarello CA, Silveira L, Sankoff J, Heyland D, Wischmeyer P, Crapo JD. Extracellular superoxide dismutase haplotypes are associated with acute lung injury and mortality. American journal of respiratory and critical care medicine. 2009;179(2):105–12. doi: 10.1164/rccm.200710-1566OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation. 2004;109(1):59–65. doi: 10.1161/01.CIR.0000105720.28086.6C. [DOI] [PubMed] [Google Scholar]
- 32.Yamada H, Yamada Y, Adachi T, Fukatsu A, Sakuma M, Futenma A, Kakumu S. Protective role of extracellular superoxide dismutase in hemodialysis patients. Nephron. 2000;84(3):218–23. doi: 10.1159/000045580. [DOI] [PubMed] [Google Scholar]
- 33.Hartney JM, Stidham T, Goldstrohm DA, Oberley-Deegan RE, Weaver MR, Valnickova-Hansen Z, Scavenius C, Benninger RK, Leahy KF, Johnson R, Gally F, Kosmider B, Zimmermann AK, Enghild JJ, Nozik-Grayck E, Bowler RP. A common polymorphism in extracellular superoxide dismutase affects cardiopulmonary disease risk by altering protein distribution. Circ Cardiovasc Genet. 2014;7(5):659–66. doi: 10.1161/CIRCGENETICS.113.000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pate KM, Sherk VD, Carpenter RD, Weaver M, Crapo S, Gally F, Chatham LS, Goldstrohm DA, Crapo JD, Kohrt WM, Bowler RP, Oberley-Deegan RE, Regan EA. The beneficial effects of exercise on cartilage are lost in mice with reduced levels of ECSOD in tissues. J Appl Physiol. 1985;118(6):760–7. doi: 10.1152/japplphysiol.00112.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chai H, Wang Q, Huang L, Xie T, Fu Y. Ginsenoside Rb1 inhibits tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Biol Pharm Bull. 2008;31(11):2050–6. doi: 10.1248/bpb.31.2050. [DOI] [PubMed] [Google Scholar]
- 36.Weber C, Erl W, Pietsch A, Strobel M, Ziegler-Heitbrock HW, Weber PC. Antioxidants inhibit monocyte adhesion by suppressing nuclear factor-kappa B mobilization and induction of vascular cell adhesion molecule-1 in endothelial cells stimulated to generate radicals. Arterioscler Thromb. 1994;14(10):1665–73. doi: 10.1161/01.atv.14.10.1665. [DOI] [PubMed] [Google Scholar]
- 37.Iida S, Chu Y, Weiss RM, Kang YM, Faraci FM, Heistad DD. Vascular effects of a common gene variant of extracellular superoxide dismutase in heart failure. Am J Physiol Heart Circ Physiol. 2006;291(2):H914–20. doi: 10.1152/ajpheart.00080.2006. [DOI] [PubMed] [Google Scholar]
- 38.Chu Y, Alwahdani A, Iida S, Lund DD, Faraci FM, Heistad DD. Vascular effects of the human extracellular superoxide dismutase R213G variant. Circulation. 2005;112(7):1047–53. doi: 10.1161/CIRCULATIONAHA.104.531251. [DOI] [PubMed] [Google Scholar]
- 39.Flaherty JT, Pitt B, Gruber JW, Heuser RR, Rothbaum DA, Burwell LR, George BS, Kereiakes DJ, Deitchman D, Gustafson N, et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation. 1994;89(5):1982–91. doi: 10.1161/01.cir.89.5.1982. [DOI] [PubMed] [Google Scholar]
- 40.Petersen A, Pedersen B. The anti-inflammatory effect of exercise. J Appl Physiol. 2005;98(4):1154–62. doi: 10.1152/japplphysiol.00164.2004. [DOI] [PubMed] [Google Scholar]
- 41.Schnyder S, Handschin C. Skeletal muscle as an endocrine organ: PGC-1alpha, myokines and exercise. Bone. 2015;80:115–25. doi: 10.1016/j.bone.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woo J, Hui E, Hui D, Lum CM, Or KH, Kwok T. A pilot study to examine the feasibility and acceptability of a community model for exercise prescription for patients with chronic disease. Hong Kong Med J. 2009;15(Suppl 2):12–6. [PubMed] [Google Scholar]
- 43.Gielen S, Adams V, Niebauer J, Schuler G, Hambrecht R. Aging and heart failure--similar syndromes of exercise intolerance? Implications for exercise-based interventions. Heart Fail Monit. 2005;4(4):130–6. [PubMed] [Google Scholar]
- 44.Rodrigues B, Jorge L, Mostarda CT, Rosa KT, Medeiros A, Malfitano C, de Souza AL, Jr, Viegas KA, Lacchini S, Curi R, Brum PC, De Angelis K, Irigoyen MC. Aerobic exercise training delays cardiac dysfunction and improves autonomic control of circulation in diabetic rats undergoing myocardial infarction. J Card Fail. 2012;18(9):734–44. doi: 10.1016/j.cardfail.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 45.DeBlieux PM, McDonough KH, Barbee RW, Shepherd RE. Exercise training attenuates the myocardial dysfunction induced by endotoxin. J Appl Physiol. 1989;66(6):2805–10. doi: 10.1152/jappl.1989.66.6.2805. [DOI] [PubMed] [Google Scholar]
- 46.Ramos DS, Olivo CR, Quirino Santos Lopes FD, Toledo AC, Martins MA, Lazo Osorio RA, Dolhnikoff M, Ribeiro W, Vieira RP. Low-intensity swimming training partially inhibits lipopolysaccharide-induced acute lung injury. Med Sci Sports Exerc. 2010;42(1):113–9. doi: 10.1249/MSS.0b013e3181ad1c72. [DOI] [PubMed] [Google Scholar]
- 47.de Araujo CC, Silva JD, Samary CS, Guimaraes IH, Marques PS, Oliveira GP, do Carmo LG, Goldenberg RC, Bakker-Abreu I, Diaz BL, Rocha NN, Capelozzi VL, Pelosi P, Rocco PR. Regular and moderate exercise before experimental sepsis reduces the risk of lung and distal organ injury. J Appl Physiol. 2012;112(7):1206–14. doi: 10.1152/japplphysiol.01061.2011. [DOI] [PubMed] [Google Scholar]
- 48.Chen HI, Hsieh SY, Yang FL, Hsu YH, Lin CC. Exercise training attenuates septic responses in conscious rats. Med Sci Sports Exerc. 2007;39(3):435–42. doi: 10.1249/mss.0b013e31802d11c8. [DOI] [PubMed] [Google Scholar]
- 49.Reis Goncalves CT, Reis Goncalves CG, de Almeida FM, Dos Santos Lopes FD, Dos Santos Durao AC, Dos Santos FA, da Silva LF, Marcourakis T, Castro-Faria-Neto HC, Vieira RD, Dolhnikoff M. Protective effects of aerobic exercise on acute lung injury induced by LPS in mice. Crit Care. 2012;16(5):R199. doi: 10.1186/cc11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481(7382):463–8. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aoi W, Naito Y, Takagi T, Tanimura Y, Takanami Y, Kawai Y, Sakuma K, Hang LP, Mizushima K, Hirai Y, Koyama R, Wada S, Higashi A, Kokura S, Ichikawa H, Yoshikawa T. A novel myokine, secreted protein acidic and rich in cysteine (SPARC), suppresses colon tumorigenesis via regular exercise. Gut. 2013;62(6):8. doi: 10.1136/gutjnl-2011-300776. [DOI] [PubMed] [Google Scholar]
- 52.Pedersen BK, Fischer CP. Beneficial health effects of exercise--the role of IL-6 as a myokine. Trends in pharmacological sciences. 2007;28(4):152–6. doi: 10.1016/j.tips.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 53.Febbraio MA, Pedersen BK. Contraction-induced myokine production and release: is skeletal muscle an endocrine organ? Exerc Sport Sci Rev. 2005;33(3):114–9. doi: 10.1097/00003677-200507000-00003. [DOI] [PubMed] [Google Scholar]
- 54.Hitomi Y, Watanabe S, Kizaki T, Sakurai T, Takemasa T, Haga S, Ookawara T, Suzuki K, Ohno H. Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox Rep. 2008;13(5):213–6. doi: 10.1179/135100008X308894. [DOI] [PubMed] [Google Scholar]
- 55.Ookawara T, Haga S, Ha S, Oh-Ishi S, Toshinai K, Kizaki T, Ji LL, Suzuki K, Ohno H. Effects of endurance training on three superoxide dismutase isoenzymes in human plasma. Free Radic Res. 2003;37(7):713–9. doi: 10.1080/1071576031000102132. [DOI] [PubMed] [Google Scholar]
- 56.Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest. 2000;105(11):1631–9. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakao C, Ookawara T, Kizaki T, Oh-Ishi S, Miyazaki H, Haga S, Sato Y, Ji LL, Ohno H. Effects of swimming training on three superoxide dismutase isoenzymes in mouse tissues. J Appl Physiol. 2000;88(2):649–54. doi: 10.1152/jappl.2000.88.2.649. [DOI] [PubMed] [Google Scholar]
- 58.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192(7):1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brady NR, Elmore SP, van Beek JJ, Krab K, Courtoy PJ, Hue L, Westerhoff HV. Coordinated behavior of mitochondria in both space and time: a reactive oxygen species-activated wave of mitochondrial depolarization. Biophys J. 2004;87(3):2022–34. doi: 10.1529/biophysj.103.035097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim YM, Kim SJ, Tatsunami R, Yamamura H, Fukai T, Ushio-Fukai M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am J Physiol Cell Physiol. 2017;312(6):C749–C764. doi: 10.1152/ajpcell.00346.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zelko IN, Folz RJ. Sp1 and Sp3 transcription factors mediate trichostatin A-induced and basal expression of extracellular superoxide dismutase. Free radical biology & medicine. 2004;37(8):1256–71. doi: 10.1016/j.freeradbiomed.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 62.Kamiya T, Nakahara R, Mori N, Hara H, Adachi T. Ten-eleven translocation 1 functions as a mediator of SOD3 expression in human lung cancer A549 cells. Free Radic Res. 2017;51(3):329–336. doi: 10.1080/10715762.2017.1313415. [DOI] [PubMed] [Google Scholar]
- 63.Yu Z, Li P, Zhang M, Hannink M, Stamler J, Yan Z. Fiber type-specific nitric oxide protects oxidative myofibers against cachectic stimuli. PLoS ONE. 2008;3(5):e2086. doi: 10.1371/journal.pone.0002086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.