


Abstract
Here we describe the rapid identification and prioritization of novel active marine natural products using an improved dereplication strategy. During the course of our screening of marine natural product libraries, a new cyclic trihydroxamate compound, thalassosamide, was discovered from the α-proteobacterium Thalassospira prof undimaris. Its structure was determined by 2D NMR and MS/MS experiments, and the absolute configuration of the lysine-derived units was established by Marfey’s analysis, whereas that of C-9, 9′, and 9″ was determined via the circular dichroism data of the [Rh2(OCOCF3)4] complex and DFT NMR calculations. Thalassosamide showed moderate in vivo efficacy against Pseudomonas aeruginosa.
Graphical abstract
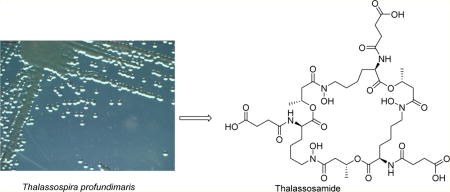
Antimicrobial resistance represents a serious threat to public health and patient safety worldwide.1 Multi-drug-resistant Gram-negative infections are even more worrying and present a major challenge for antibiotic development. Pseudomonas aeruginosa is one of the most common Gram-negative pathogens involved in severe nosocomial infections including in the lungs of cystic fibrosis patients, the bloodstream, the urinary tract, surgical sites, and burn wounds.2 In a recently issued report by the Centers for Disease Control and Prevention, there are 51 000 cases of P. aeruginosa infections reported in the United States each year, representing around 8–10% of all healthcare-associated infections. In more than 6000 of these cases, multi-drug-resistant specimens were identified, of which roughly 400 lead to patient deaths each year.3 As a critical factor in understanding its lethality P. aeruginosa has particularly discriminating outer membrane porins that make its outer membrane impermeable and therefore naturally resistant to many antibiotics. P. aeruginosa readily forms biofilms that can increase resistance to antibiotics by 100–1000-fold.4 Consequently, there is a clear need to identify new agents for the treatment of multi-drug-resistant P. aeruginosa.
Natural products and their derivatives have historically played a dominant role in drug discovery for the treatment of human diseases and are still a viable option as new antibiotics.5 It has been estimated that it takes $50 000 and three months of work to isolate and characterize an active compound found by high-throughput (HTP) screening from natural product sources.6 Therefore, it is critical to identify a known active compound at an early stage, not only for saving time and money but also for allocating resources to pursue more promising candidates.
To streamline the discovery and dereplication process, we developed a discovery platform that includes strain prioritization by metabolomics and an LC/MS fractionation platform to generate high-purity screening libraries. Strain prioritization using metabolomics aims to increase chemical diversity and reduce chemical redundancy. LC/MS-principal component analysis (PCA)7 was used for strain selection followed by an automated, two-step, HTP LC/MS fractionation procedure to generate marine natural product libraries directly into 96-well plates. Drug-resistant bacteria Gram-negative Escherichia coli and P. aeruginosa pathogens and Gram-positive methicillin-resistant Staphylococcus aureus (MRSA) pathogen and the fungal pathogen Candida albicans were chosen for HTP screenings (Figures S14–S17, Supporting Information). HR-LC/MS, nanoscale 1.7 mm CryoProbe NMR spectroscopy, and the AntiBase database were utilized for the dereplication of active wells. In the first stage, 7520 fractions from 94 marine-derived strains were evaluated by HTP screening, and only one hit from Thalassospira profundimaris (WMMC 317), which was isolated from the tunicate Ecteinascidia turbinata, showed activity against P. aeruginosa with no cytotoxicity against fibroblasts. The mass spectra corresponding to the active well indicated a potentially new molecule with an m/z of 991.4 ([M + H]+). Subsequently, LC/MS analysis and isolation of the compound with the target m/z led to a new cyclic trihydroxamate siderophore, thalassosamide (1), which was active against P. aeruginosa both in vitro and in vivo.
The molecular formula of thalassosamide (1) was established as C42H66N6O21 on the basis of HRESIMS data. The 1H and 13C NMR spectra of compound 1 were uncomplicated considering its molecular weight, suggesting that compound 1 had a high degree of symmetry. Analysis of the 1H and 13C NMR and HSQC spectra revealed the presence of 14 carbons, including four carbonyl carbons, one methyl group, seven methylene carbons, and two methines. Therefore, compound 1 should have a C3 symmetry axis in the structure. Interpretation of the 2D NMR data established the amino acid residue lysine, together with succinic acid and β-hydroxybutyric acid units. The exchangeable protons (δH 8.14) were assigned to be 2-NH, 2′-NH, and 2″-NH based on the observed 1H–1H COSY correlations between the exchangeable protons (δH 8.14) and signals at 4.12 ppm (C-2/C-2′/ C-2″). HMBC correlations from H-2 to C-10 and from 2-NH to C-2, C-3, and C-10 indicated connectivity of the succinic acid unit to the lysine residue via an amide bond, whereas the HMBC correlation from H-6 to C-7 revealed a linkage of lysine and β-hydroxybutyric via an amide bond, thus establishing a partial structure of 1. HMBC correlations from H-9/H-9′/H-9″ (δH 5.16) to C-1′, C-1″, and C-1 (δC 171.7) indicated a cyclic triester backbone in compound 1. To satisfy the molecular formula, there were three remaining hydroxy groups that were assigned by default as 6-N-OH, 6′-N-OH, and 6″-N-OH to complete the 2D structure of 1 as depicted. The 2D structure was further confirmed by ESIMS/MS. The [M − H]− ion (m/z 989.4) was chosen as a precursor ion, and the prominent product ions at m/z 889.4, 745.3, 659.3, 559.2, 415.2, and 329.1 confirmed the presence of three structural units: succinic acid, β-hydroxybutyric acid, and N-hydroxylysine (Figure S9, Supporting Information).
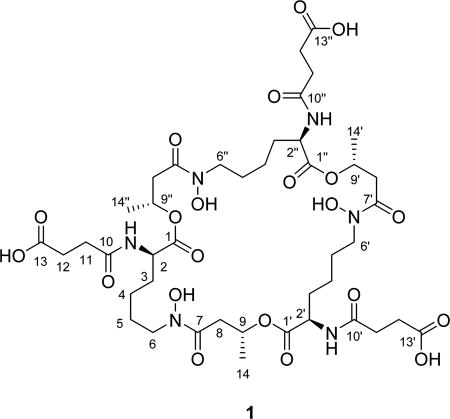
The absolute configuration of C-2, C-2′, and C-2″ was determined by Marfey’s analysis.8 The fluorodinitrophenyl-5-l-leucine amide (FDLA) derivatives of the acid hydrolysate of 1 and corresponding standards containing d- and l-lysines were subjected to LC-MS analysis. On the basis of these studies the lysine in 1 was determined to have the d-configuration. Further efforts to elucidate its absolute configuration involved thalassosamide fragmentation via treatment of 1 with sodium methoxide in methanol and subsequent acidic workup with DOWEX resin to generate diester 2 (Scheme 1). The absolute configuration of the C-9 secondary alcohol in 2 was deduced via the electronic circular dichroism (ECD) data of the in situ formed [Rh2(OCOCF3)4] complex,9 with the inherent contribution subtracted. The Rh complex of 2 showed a negative E band at ca. 350 nm (Figure 1), correlating to the 9R/9′R/9″R absolute configuration by applying the bulkiness rule.9
Scheme 1.

Methanolic Fragmentation of 1
Figure 1.

ECD spectrum of the Rh complex of compound 2 with the inherent ECD spectrum subtracted.
Further efforts to validate the absolute configuration of C-9, C-9′, and C-9″ employed molecular modeling and the density functional theory (DFT) NMR calculations of the two models (2R,2′R,2″R,9R,9′R,9″R- and 2R,2′R,2″R,9S,9′S,9″S-1). Spartan 14 (v.1.1.7 Wavefunction Inc. 2014) was used to identify the lowest energy conformers for each stereoisomer using an equilibrium conformer search with molecular mechanics, followed by further equilibrium geometry (B3LYP/6–31G**) and DFT energy calculation to determine the Boltzmann distribution. Only one conformation was found for each model, which was further used for NMR calculations. The DP4 probability method10 was undertaken to compare the calculated 13C NMR chemical shifts of the two models with the experimental 13C chemical shifts and yielded a 100.0% probability for the R compared to the S configuration (Tables S1 and S2, Supporting Information). Additionally, the model for 9S,9′S,9″S-1 had dihedral angles of 56.10° and 60.78° between H-8a and H-9 and between H-8b and H-9, which both should show small vicinal 3JH coupling constants, while the model for 9R,9′R,9″R-1 had dihedral angles of 57.23° and 176.39°, consistent with the small (4.6 Hz, JH8a,H9) and large coupling constants (8.8 Hz, JH8b,H9) of compound 1.11 Therefore, the 2R, 2′R, 2″R, 9R, 9′R, and 9″R absolute configuration is proposed for compound 1.
The Fe(III)-binding properties of compound 1 were assessed on the basis of the chrome azurol S (CAS) assay;12 deferoxamine mesylate was also analyzed and served as a positive Fe(III)-binding control (Figure 2). Compound 1 was capable of binding Fe(III) based on the color change of the dye from blue to orange, and it appears to form a slightly tighter Fe(III) complex than deferoxamine mesylate; comparable colorimetric changes required lower concentrations of 1 than the deferoxamine species.
Figure 2.

Concentration-dependent chrome azurol S (CAS) assay.
Iron chelators such as deferoxamine and deferasirox have shown antibacterial activity toward Gram (−) organisms, but generally suffer from low potency.13 However, the use of iron chelators in conjunction with tobramycin, an aminocyclitol, was shown to eliminate P. aeruginosa biofilms in models of cystic fibrosis.14 Given the potential of iron chelators as potential adjuvants for treating P. aeruginosa, especially in cystic fibrosis patients, we pursued studies to evaluate the potential of marine-derived 1 as an antibacterial agent, both in vitro and in vivo.
Compound 1 was tested for antibacterial activity against P. aeruginosa and E. coli and showed antibacterial activity with the same MIC value of 64 µg/mL. Compound 1 was also tested in vivo using a murine thigh model for examining infection with P. aeruginosa (ATCC #27853) by an intraperitoneal route (Figure 3). Compared to untreated control animals, modest efficacy was observed at the two highest dosings. Notably, although the potency of 1 was rather low, these studies indicated that, as predicted, toxicity was also low. Therefore, we evaluated the ability of 1 to disrupt P. aeruginosa biofilms as a single agent and in combination with gentamycin; concentrations evaluated ranged from 16 to 64 µg/mL.15 Unfortunately, little effect was observed, and no synergy with gentamycin was observed.
Figure 3.

In vivo activity of single intraperitoneal dose against P. auruginosa (ATCC # 275853) in a neutropenic murine thigh model. Each symbol represents the mean and standard deviation from four thighs of two mice infected with P. auruginosa. The error bars represent the standard deviation. Two single doses (80 and 160 mg/kg) of compound 1 were administered to mice.
In summary, we report the isolation and structure elucidation of thalassosamide (1), a new marine-derived siderophore, with in vivo efficacy in an immunocompromised mouse model of P. aeruginosa infection and no toxicity. The preliminary in vivo data showed modest efficacy. Due to the repeating nature of the units found in thalassosamide, synthesis would be tractable and might allow substitution of the esters for more stable amides; in vivo ester hydrolysis represents a clear metabolic liability. To date, only three natural products have been reported from the marine α-proteobacterium Thalassopira sp., thalassospiramides A, B,16 and G.17 The discovery of thalassosamide adds one more member to this family of natural products and hints at the unexplored and under-rated potential of proteobacteria as producers of new natural products.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were measured on a Perkin–Elmer 241 polarimeter. UV spectra were recorded on an Aminco/OLIS UV–vis spectrophotometer. ECD spectra were recorded on an AVIV model 420 circular dichroism spectrometer. IR spectra were measured with a Bruker Equinox 55/S FT-IR spectrophotometer. 1D and 2D NMR spectra were obtained on a Bruker Avance 600 MHz spectrometer with a 1H{13C/15N} cryoprobe and a 500 MHz spectrometer with a 13C/15N{1H} cryoprobe; chemical shifts were referenced to the residual solvent peaks (CDCl3: δH 7.26, δC 77.23; DMSO-d6: δH 2.50, δC 39.51). HRMS and MSMS data were acquired with a Bruker MaXis 4G QTOF mass spectrometer. RP HPLC was performed using a Shimadzu Prominence HPLC system and a Phenomenex Luna C18 column (250 × 10 mm, 5 µm). The Marfey’s method utilized a Shimadzu Prominence HPLC coupled with a Waters Micromass LCT TOF mass spectrometer.
Biological Material
WMMC 317 was isolated from the tunicate Ecteinascidia turbinata, which was collected on August 13, 2014, in the Florida Keys (24° 39.393, 81° 26.268). A voucher specimen is housed at the University of Wisconsin–Madison. For cultivation, a sample of tunicate (1 cm3) was rinsed with sterile seawater and macerated using a sterile pestle in a microcentrifuge tube, and dilutions were made in sterile seawater, with vortexing between steps to separate bacteria from heavier tissues. Dilutions were separately plated on three media: ISP2 supplemented with artificial seawater, R2A, and M4. Plates were incubated at 28 °C for at least 28 d, and strain WMMC 317 was purified from an R2A isolation plate containing nalidixic acid (25 µg/mL), cycloheximide (50 µg/mL), and nystatin (25 µg/mL).
Sequencing
16S rDNA was sequenced as previously described.18 WMMC 317 was identified as Thalassospira prof undimaris by 850 base BLAST and demonstrated a 100% match with T. prof undimaris fst-13007 (accession number KU578316). The WMMC 317 genome was deposited in GenBank and assigned the accession number KY020405.
Fermentation, Extraction, and Isolation
Three 10 mL seed cultures (25 × 150 mm tubes) in medium ASW-A (20 g soluble starch, 10 g glucose, 5 g peptone, 5 g yeast extract, 5 g CaCO3 per liter of artificial seawater) were inoculated with strain WMMC 317 and shaken (200 rpm, 28 °C) for 7 days. For making artificial seawater, solutions I (415.2 g NaCl, 69.54 g Na2SO4, 11.74 g KCl, 3.40 g NaHCO3, 1.7 g KBr, 0.45 g H3BO3, 0.054 g NaF) and II (187.9 g MgCl2·6H2O, 22.72 g CaCl2·2H2O, 0.428 g SrCl2·6H2O) were made up separately and combined to give a total volume of 20 L. Two-liter flasks (6 × 500 mL) containing Ram2 medium (4 g corn meal, 10 g glucose, 15 g maltose, 7.5 g pharmamedia, 5 g yeast extract per liter of H2O including 500 mL distilled H2O and 500 mL artificial seawater) with Diaion HP20 (7% by weight) were inoculated with 4 mL from the culture tube and shaken at 200 rpm at 28 °C for 7 days. Filtered HP20 was washed with distilled H2O and extracted with acetone. The acetone extract (20 g) was subjected to a liquid–liquid partitioning using 30% aqueous MeOH and CHCl3 (1:1). The 30% aqueous MeOH (1.0 g) was subjected to a Sephadex LH-20 column with MeOH. The fractions containing 1 were further subjected to RP HPLC (10–40% MeCN–H2O with H2O containing 0.1% acetic acid over 25 min, 4.0 mg/mL) using a Phenomenex Luna C18 column (250 × 10 mm, 5 µm), yielding 1 (60 mg, tR 19.4 min).
Thalassosamide (1): colorless oil; [α]25D −11 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 229(1.82) nm; IR (ATR) νmax 3287, 3095, 2939, 2869, 2834, 1726, 1625, 1544, 1447, 1382, 1197, 1130, 1048, 1023, 962, 899, 834, 669 cm−1; 1H and 13C NMR (see Table 1); HRESIMS m/z 1013.4190 [M + Na]+ (calcd for C42H66N6O21Na, 1013.4179).
Table 1.
1H and 13C NMR Data for Thalassosamide (1) (600 MHz for 1H, 125 MHz for 13C, DMSO-d6)
| position | δC, type | δH (J in Hz) | HMBC |
|---|---|---|---|
| 1 | 171.7, C | ||
| 2 | 51.8, CH | 4.12, ddd (7.2) | 1, 3, 10 |
| 2-NH | 8.14, d (7.2) | 2, 3, 10 | |
| 3 | 30.8, CH2 | 1.50, m; 1.61, m | 5 |
| 4 | 22.3, CH2 | 1.23, m | 2, 3, 5, 6 |
| 5 | 26.0, CH2 | 1.45, m | 3, 4, 6 |
| 6 | 46.9, CH2 | 3.43, t (6.7) | 4, 5, 7 |
| 7 | 169.3, C | ||
| 8 | 38.0, CH2 | 2.51, dd (16, 4.6); 2.80, dd (16, 8.8) | 7, 9, 14 |
| 9 | 68.0, CH | 5.16, m | 7, 14, 1′ |
| 10 | 171.4, C | ||
| 11 | 29.9, CH2 | 2.36, m | 10, 12, 13 |
| 12 | 29.3, CH2 | 2.36, m | 10, 11, 13 |
| 13 | 174.0, C | ||
| 14 | 20.0, CH3 | 1.20, d (6.3) | 7, 8, 9 |
| 1′ | 171.7, C | ||
| 2′ | 51.8, CH | 4.12, ddd (7.2) | 1′, 3′, 10′ |
| 2′-NH | 8.14, d (7.2) | 2′, 3′, 10′ | |
| 3′ | 30.8, CH2 | 1.50, m; 1.61, m | 5′ |
| 4′ | 22.3, CH2 | 1.23, m | 2′, 3′, 5′, 6′ |
| 5′ | 26.0, CH2 | 1.45, m | 3′, 4′, 6′ |
| 6′ | 46.9, CH2 | 3.43, t (6.7) | 4′, 5′, 7′ |
| 7′ | 169.3, C | ||
| 8′ | 38.0, CH2 | 2.51, dd (16, 4.6); 2.80, dd (16, 8.8) | 7′, 9′, 14′ |
| 9′ | 68.0, CH | 5.16, m | 7′, 14′, 1″ |
| 10′ | 171.4, C | ||
| 11′ | 29.9, CH2 | 2.36, m | 10′, 12′, 13′ |
| 12′ | 29.3, CH2 | 2.36, m | 10′, 11′, 13′ |
| 13′ | 174.0, C | ||
| 14′ | 20.0, CH3 | 1.20, d (6.3) | 7′, 8′, 9′ |
| 1″ | 171.7, C | ||
| 2″ | 51.8, CH | 4.12, ddd (7.2) | 1″, 3″, 10″ |
| 2″-NH | 8.14, d (7.2) | 2″, 3″, 10″ | |
| 3″ | 30.8, CH2 | 1.50, m; 1.61, m | 5″ |
| 4″ | 22.3, CH2 | 1.23, m | 2″, 3″, 5″, 6″ |
| 5″ | 26.0, CH2 | 1.45, m | 3″, 4″, 6″ |
| 6″ | 46.9, CH2 | 3.43, t (6.7) | 4″, 5″, 7″ |
| 7″ | 169.3, C | ||
| 8″ | 38.0, CH2 | 2.51, dd (16, 4.6); 2.80, dd (16, 8.8) | 7″, 9″, 14″ |
| 9″ | 68.0, CH | 5.16, m | 7″, 14″, 1 |
| 10″ | 171.4, C | ||
| 11″ | 29.9, CH2 | 2.36, m | 10″, 12″, 13″ |
| 12″ | 29.3, CH2 | 2.36, m | 10″, 11″, 13″ |
| 13″ | 174.0, C | ||
| 14″ | 20.0, CH3 | 1.20, d (6.3) | 7″, 8″, 9″ |
Acid Hydrolysis of Thalassosamide (1)
Separate solutions of compound 1 (1.0 mg) in 6 N HI (1 mL) were hydrolyzed at 110 °C for 18 h and dried under vacuum.
Determination of Proteinogenic Amino Acid Configuration
l-FDLA was synthesized as previously reported, and this reagent used to generate hydolysate-derived diastereomers.8 The hydrolysate was mixed with 1 N NaHCO3 (80 µL) and 150 µL of l-FDLA (10 mg/mL in acetone). The solution was stirred at 45 °C for 1 h, cooled to room temperature (rt), quenched with 1 N HCl (80 µL), and dried under vacuum. Similarly, the standard l- and d-lysines were derivatized separately. The derivatives of the hydrolysate of compound 1 and the standard amino acids l- and d-lysines were subjected to LC-MS analysis with a Phenomenex Luna C18 reversed-phase column (250 × 4.6 mm, 5 µm) at a flow rate of 1.0 mL/min and with a linear gradient of H2O (containing 0.1% formic acid) and MeOH (90:10 to 0:100 over 30 min and a hold at 100% MeOH for 5 min). The absolute configuration of the amino acid in 1 was determined by comparing the retention times of the l- and d-lysine derivatives, which were identified by LC/MS. The retention times for the l-FDLA derivatives of the hydrolysate of compound 1 and the standard l- and d-lysines are 22.2, 23.0, and 22.2 min, respectively.
Alcohol 2
Sodium methoxide (500 µL, 0.5 N) in MeOH was added to 1 (3.0 mg), and the solution was stirred for 19 h at rt. The reaction was then quenched via the addition of H+ form Dowex resin, leading to a pH of 4–5; the resin was subsequently removed by filtration. The reaction product was purified by reversed-phase HPLC (10–60% MeOH–H2O with H2O containing 0.1% acetic acid over 30 min, 4.0 mg/mL) using a Phenomenex Luna C18 column (250 × 10 mm, 5 µm), yielding diester 2 (1.5 mg, tR 22.6 min). 1H NMR data of 2 (CDCl3–CD3OD, 4:1, 600 MHz) δH 7.37 (1H, d, J = 7.9 Hz), 4.30 (1H, m), 4.04 (1H, m), 3.57 (3H, s), 3.53 (3H, s), 3.46 (2H, m), 2.48 (2H, m), 2.46 (2H, m), 2.40 (2H, m), 1.66 (1H, m), 1.55 (1H, m), 1.54 (1H, m), 1.47 (1H, m), 1.20 (2H, m), 1.08 (3H, d, J = 6.2 Hz); HRESIMS [M + Na]+ m/z 399.1730 (calcd for C16H28N2O8Na, 399.1738).
C-9/C-9′/C-9″ Absolute Configuration of 1
Employing a previously reported methodology,9 [Rh2(OCOCF3)4] (2.0 mg) was dissolved into CH2Cl2 (200 µL). To the rhodium solution was then added 2 (0.5 mg), and the mixture was briefly agitated to ensure visual homogeneity. The first ECD spectrum was recorded immediately, and monitoring continued until such time as no further changes in readout could be ascertained; this stationary phase was achieved after approximately 10 min, at which point the Rh-2 complexation process was believed complete. The inherent ECD was subtracted.9 The E band observed at ca. 350 nm in the induced ECD spectrum was correlated to the absolute configuration of the C-9/C-9′/ C-9″ secondary alcohol on the basis of data for the Rh-2 complex.9
Chrome Azurol S Assay.12
Hexadecyltrimethylammonium bromide (CTAB) (21.9 mg) was dissolved in 25 mL of H2O at 35 °C. To this solution were added 1.5 mL of 1 mM iron(III) chloride solution (prepared by dissolving anhydrous FeCl3 in a 10 mM aqueous HCl solution) and 7.5 mL of a 2 mM aqueous CAS solution at rt. In a separate Erlenmeyer flask, 9.76 g of 2-(N-morpholino)ethanesulfonic acid (MES) was diluted in 50 mL of water, and a 50% KOH solution was used to adjust the pH of this solution to 5.6. Then the premade CTAB–CAS–Fe(III) solution was poured into this MES buffer while stirring, and H2O was added to make 100 mL to finish the preparation of the modified CAS assay solution. CTAB–CAS–Fe(III) with MES buffer solution (100 µL) was added to each well of the 96-well microplate. Each well was then treated with 100 µL of the dilute solution of thalassosamide or deferoxamine mesylate in H2O to achieve final concentrations ranging from 1.28 mM to 5 µM. After incubation at 37 °C for 3 h, the resulting color changes were observed by visual inspection, or the corresponding absorption changes were recorded.
Antibacterial Assay
Thalassosamide (1) was tested for antibacterial activity against P. aeruginosa (ATCC #27853) and E. coli (ATCC #25922), and MICs were determined using a dilution antimicrobial susceptibility test for aerobic bacteria.19 Thalassosamide (1) was dissolved in DMSO and serially diluted to 10 concentrations (0.25–128 µg/mL) in a 96-well plate. Gentamicin was used as a positive control and exhibited an MIC of 4 µg/mL against P. aeruginosa and E. coli. Thalassosamide and gentamicin were tested in triplicate. On each plate, there were six untreated media controls. The plates were incubated at 35 °C for 18 h. The MIC was determined as the lowest concentration that inhibited visible growth of bacteria.
In Vivo Antibacterial Assays
Thalassosamide (1) was tested for in vivo antibacterial activity in a murine model involving thigh infection with P. aeruginosa (ATCC #275853). Efforts to assess in vivo activity of 1 in murine models of thigh infection treatment were carried out as previously described.18 The P. aeruginosa pathogen was grown, subcultured, and quantified using Mueller-Hinton broth (MHB) and agar (Difco Laboratories). Animals were maintained in accordance with American Association for Accreditation of Laboratory Animal Care (AAALAC) criteria, and all animal studies were approved by the Animal Research Committees of the William S. Middleton Memorial VA Hospital and the University of Wisconsin. Following 1 h of infection mice were subjected to intraperitoneal administration of 1 (at doses of either 80 or 160 mg/kg). At time points ranging from 0 → 6 h following treatment with 1, mouse groups of two treated and two untreated specimens were euthanized and thigh tissues processed as previously described for CFU determinations.18
Supplementary Material
Acknowledgments
This work was supported by funding from the University of Wisconsin–Madison School of Pharmacy. This work was also funded by the U19 AI109673, NIH. We thank the Analytical Instrumentation Center (AIC) at the University of Wisconsin–Madison School of Pharmacy for the facilities to acquire spectroscopic data, especially MS data. This study made use of the National Magnetic Resonance Facility at Madison (NMRFAM), which is supported by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS).
Footnotes
ASSOCIATED CONTENT
- Spectroscopic data including 1D and 2D NMR, HRMS spectra of compounds 1 and 2, and MS/MS spectrum of compound 1 are provided as are the results of HTP screenings of WMMC 317 and DFT/DP4 calculations applied to determining the configuration of 1 (PDF)
The authors declare no competing financial interest.
References
- 1.Fair RJ, Tor Y. Perspect. Med. Chem. 2014;6:25–64. doi: 10.4137/PMC.S14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Rajan S, Saiman L. Semin. Respir. Infect. 2002;17:47–56. doi: 10.1053/srin.2002.31690. [DOI] [PubMed] [Google Scholar]; (b) Richards MJ, Edwards JR, Culver DH, Gaynes RP. Crit. Care Med. 1999;27:887–892. doi: 10.1097/00003246-199905000-00020. [DOI] [PubMed] [Google Scholar]; (c) Church D, Elsayed S, Reid O, Winston B, Lindsay R. Clin. Microbiol. Rev. 2006;19:403–434. doi: 10.1128/CMR.19.2.403-434.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antibiotic Resistance Threats in the United States. Centers for Disease Control and Prevention; Atlanta: 2013. [accessed December 21, 2015]. http://www.cdc.gov/drugresistance/threat-report-2013/index.html. [Google Scholar]
- 4.Fothergill JL, Winstanley C, James CE. Expert Rev. Anti-Infect. Ther. 2012;10:219–235. doi: 10.1586/eri.11.168. [DOI] [PubMed] [Google Scholar]
- 5.Newman DJ, Cragg GM. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 6.Cordell GA, Shin YG. Pure Appl. Chem. 1999;71:1089–1094. [Google Scholar]
- 7.Hou Y, Braun DR, Michel CR, Klassen JL, Adnani N, Wyche TP, Bugni TS. Anal. Chem. 2012;84:4277–4283. doi: 10.1021/ac202623g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marfey P. Carlsberg Res. Commun. 1984;49:591–596. [Google Scholar]
- 9.(a) Frelek J, Szczepek WJ. Tetrahedron: Asymmetry. 1999;10:1507–1520. [Google Scholar]; (b) Gerards M, Snatzke G. Tetrahedron: Asymmetry. 1990;1:221–236. [Google Scholar]; (c) Zhang F, Li L, Niu S, Si Y, Guo L, Jiang X, Che Y. J. Nat. Prod. 2012;75:230–237. doi: 10.1021/np2009362. [DOI] [PubMed] [Google Scholar]
- 10.Smith SG, Goodman JM. J. Org. Chem. 2009;74:4597–4607. doi: 10.1021/jo900408d. [DOI] [PubMed] [Google Scholar]
- 11.(a) Karplus M. J. Am. Chem. Soc. 1963;85:2870–2871. [Google Scholar]; (b) Wyche TP, Hou Y, Vazquez-Rivera E, Braun D, Bugni TS. J. Nat. Prod. 2012;75:735–740. doi: 10.1021/np300016r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexander DB, Zuberer DA. Biol. Fertil. Soils. 1991;12:39–45. [Google Scholar]
- 13.Thompson MG, Corey BW, Si Y, Craft DW, Zurawski VZ. Antimicrob. Agents Chemother. 2012;56:5419–5421. doi: 10.1128/AAC.01197-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreau-Marquis S, O’Toole GA, Stanton BA. Am. J. Respir. Cell Mol. Biol. 2009;41:305–313. doi: 10.1165/rcmb.2008-0299OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Toole GA. J. Visualized Exp. 2011;47:e2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh D, Strangman WK, Kauffman CA, Jensen PR, Fenical W. Org. Lett. 2007;9:1525–1528. doi: 10.1021/ol070294u. [DOI] [PubMed] [Google Scholar]
- 17.Um S, Pyee Y, Kim E, Lee SK, Shin J, Oh D. Mar. Drugs. 2013;11:611–622. doi: 10.3390/md11030611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, Adnani N, Vazquez-Rivera E, Braun DR, Tonelli M, Andes DR, Bugni TS. J. Org. Chem. 2015;80:8713–8719. doi: 10.1021/acs.joc.5b01486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. 7. NCCLS; Villanova, PA, USA: 2006. Approved standard M7-A7. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.